
Monocytes and Macrophages in Spondyloarthritis: Functional Roles and Effects of Current Therapies

 , and
, and 
Abstract
1. Introduction
2. Genetic Factors
3. Epigenetics and Environmental Factors
4. Etiopathogenesis
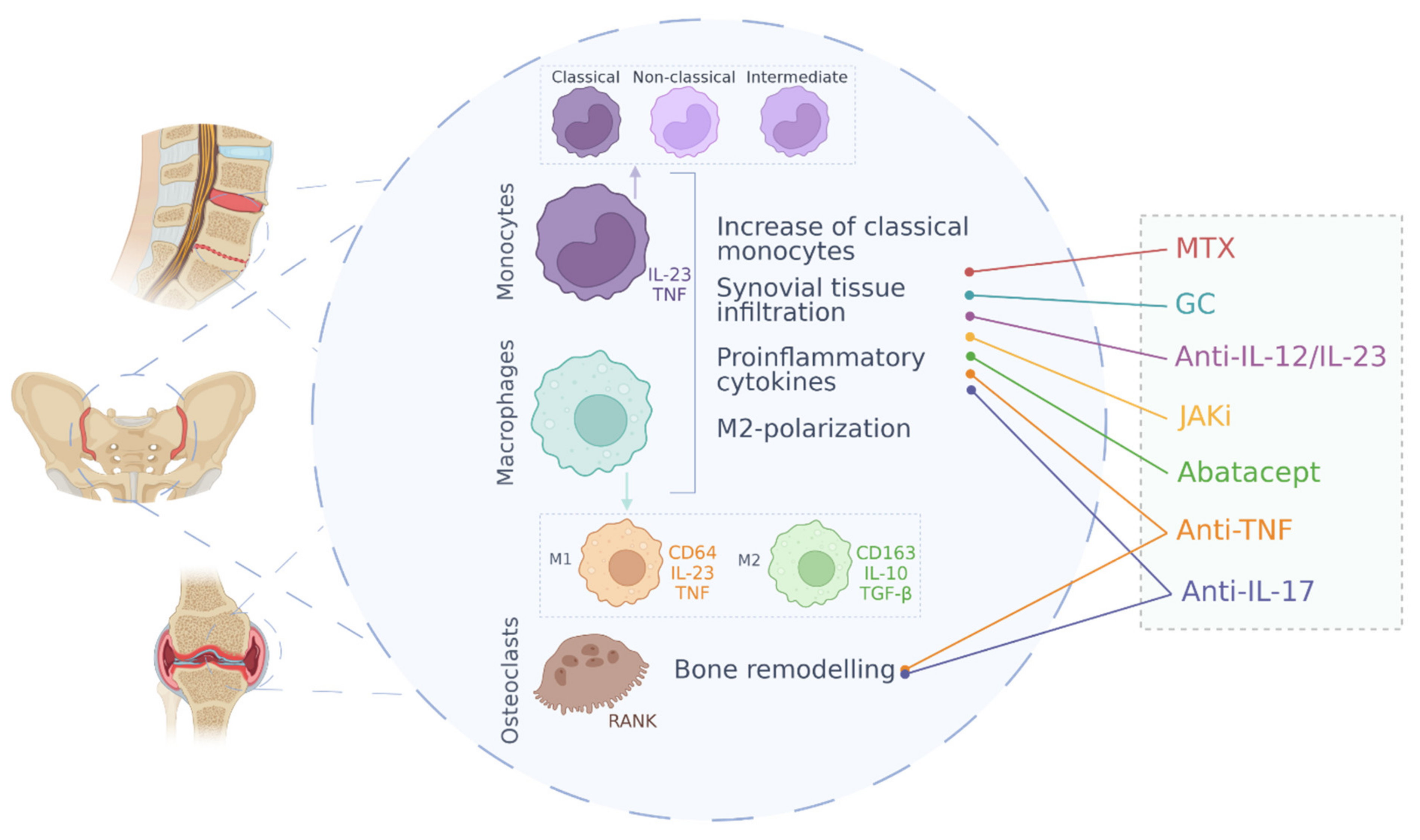
5. Cell Pathology: Monocytes, Macrophages and Osteoclasts in Spondyloarthritis
5.1. Monocytes
5.2. Macrophages
5.3. Osteoclasts
6. SpA Treatments and Effect on Monocyte/Macrophage Function
6.1. NSAIDs
6.2. Glucocorticoids
6.3. Non-Biological Disease-Modifying Anti-Rheumatic Drugs
6.4. Anti-TNF Treatments
6.5. Anti-IL-17 Treatments
6.6. Anti-IL-12/Anti-IL-23 Therapy
6.7. JAK Inhibitors
6.8. Other Biological Therapies
6.8.1. CTLA4-Ig (Abatacept)
6.8.2. IL-6 Inhibitors
6.9. Directed Therapies: From Monocytes and Macrophages to Disease Management
6.9.1. Granulocyte–Monocyte Colony Stimulating Factor (GM-CSF) Inhibition
6.9.2. Apheresis
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vanhoof, J.; Declerck, K.; Geusens, P. Prevalence of Rheumatic Diseases in a Rheumatological Outpatient Practice. Ann. Rheum. Dis. 2002, 61, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Sieper, J. Ankylosing Spondylitis. Lancet 2007, 369, 1379–1390. [Google Scholar] [CrossRef]
- Capelusnik, D.; Ramiro, S.; Schneeberger, E.E.; Citera, G. Peripheral Arthritis and Higher Disease Activity Lead to More Functional Impairment in Axial Spondyloarthritis: Longitudinal Analysis from ESPAXIA. Semin. Arthritis Rheum. 2021, 51, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Paramarta, J.E.; de Rycke, L.; Ambarus, C.A.; Tak, P.P.; Baeten, D. Undifferentiated Spondyloarthritis vs Ankylosing Spondylitis and Psoriatic Arthritis: A Real-Life Prospective Cohort Study of Clinical Presentation and Response to Treatment. Rheumatology 2013, 52, 1873–1878. [Google Scholar] [CrossRef]
- Boel, A.; López-Medina, C.; van der Heijde, D.; van Gaalen, F.A. Age at Onset in Axial Spondyloarthritis around the World: Data from the International ASAS-PERSPA Study. Ann. Rheum. Dis. 2021, 80, 735. [Google Scholar] [CrossRef]
- Van Tubergen, A. The Changing Clinical Picture and Epidemiology of Spondyloarthritis. Nat. Rev. Rheumatol. 2015, 11, 110–118. [Google Scholar] [CrossRef]
- Ehrenfeld, M. Geoepidemiology: The Environment and Spondyloarthropathies. Autoimmun. Rev. 2010, 9, A325–A329. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; Liava, C.; Daoussis, D.; Akriviadis, E.; Garyfallos, A.; Dimitroulas, T. Inflammatory Bowel Diseases and Spondyloarthropathies: From Pathogenesis to Treatment. World J. Gastroenterol. 2019, 25, 2162–2176. [Google Scholar] [CrossRef]
- Sieper, J.; Poddubnyy, D. New Evidence on the Management of Spondyloarthritis. Nat. Rev. Rheumatol. 2016, 12, 282–295. [Google Scholar] [CrossRef]
- Lories, R.J. Advances in Understanding the Pathophysiology of Spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 331–341. [Google Scholar] [CrossRef]
- Rojas-Vargas, M.; Munoz-Gomariz, E.; Escudero, A.; Font, P.; Zarco, P.; Almodovar, R.; Gratacos, J.; Mulero, J.; Juanola, X.; Montilla, C.; et al. First Signs and Symptoms of Spondyloarthritis—Data from an Inception Cohort with a Disease Course of Two Years or Less (REGISPONSER-Early). Rheumatology 2009, 48, 404–409. [Google Scholar] [CrossRef]
- Wu, I.B.; Schwartz, R.A. Reiter’s Syndrome: The Classic Triad and More. J. Am. Acad. Dermatol. 2008, 59, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Holden, W.; Orchard, T.; Wordsworth, P. Enteropathic Arthritis. Rheum. Dis. Clin. North Am. 2003, 29, 513–530. [Google Scholar] [CrossRef]
- Coates, L.C.; Kavanaugh, A.; Mease, P.J.; Soriano, E.R.; Laura Acosta-Felquer, M.; Armstrong, A.W.; Bautista-Molano, W.; Boehncke, W.-H.; Campbell, W.; Cauli, A.; et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 Treatment Recommendations for Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Carron, P.; de Craemer, A.-S.; van den Bosch, F. Peripheral Spondyloarthritis: A Neglected Entity—State of the Art. RMD Open 2020, 6, e001136. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, S.; Motreff, P.; Soubrier, M. Spondyloarthropathies: An Independent Cardiovascular Risk Factor? Jt. Bone Spine 2010, 77, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Maguire, S.; Gallagher, P.; O’Shea, F. The Negative Impact of Depression in Women with Axial Spondyloarthropathy. Jt. Bone Spine 2022, 89, 105261. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Winthrop, K.; Bohn, R.L.; Suruki, R.; Siegel, S.; Stark, J.L.; Xie, F.; Yun, H.; Chen, L.; Deodhar, A. The Annual Diagnostic Prevalence of Ankylosing Spondylitis and Axial Spondyloarthritis in the United States Using Medicare and MarketScan Databases. ACR Open Rheumatol. 2021, 3, 743–752. [Google Scholar] [CrossRef]
- Colaco, K.; Widdifield, J.; Luo, J.; Rosen, C.F.; Alhusayen, R.; Paterson, J.M.; Campbell, W.; Tu, K.; Bernatsky, S.; Gladman, D.D.; et al. Trends in Mortality and Cause-Specific Mortality among Patients with Psoriasis and Psoriatic Arthritis in Ontario, Canada. J. Am. Acad. Dermatol. 2021, 84, 1302–1309. [Google Scholar] [CrossRef]
- Rosine, N.; Miceli-Richard, C. Innate Cells: The Alternative Source of IL-17 in Axial and Peripheral Spondyloarthritis? Front. Immunol. 2021, 11, 3206. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 3234. [Google Scholar] [CrossRef] [PubMed]
- Roszkowski, L.; Ciechomska, M. Tuning Monocytes and Macrophages for Personalized Therapy and Diagnostic Challenge in Rheumatoid Arthritis. Cells 2021, 10, 1860. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Lo, M.S.; Reis, P.C.; Sullivan, K.E. New Insights into the Immunopathogenesis of Systemic Lupus Erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.; Yeremenko, N.; Tak, P.P.; Baeten, D. Pathogenesis of Spondyloarthritis. Curr. Opin. Rheumatol. 2012, 24, 351–358. [Google Scholar] [CrossRef]
- McMichael, A.; Bowness, P. HLA-B27: Natural Function and Pathogenic Role in Spondyloarthritis. Arthritis Res. 2002, 4, S153–S158. [Google Scholar] [CrossRef]
- Bodis, G.; Toth, V.; Schwarting, A. Role of Human Leukocyte Antigens (HLA) in Autoimmune Diseases. Rheumatol. Ther. 2018, 5, 5–20. [Google Scholar] [CrossRef]
- Kavadichanda, C.G.; Geng, J.; Bulusu, S.N.; Negi, V.S.; Raghavan, M. Spondyloarthritis and the Human Leukocyte Antigen (HLA)-B*27 Connection. Front. Immunol. 2021, 12, 497. [Google Scholar] [CrossRef]
- Jeanty, C.; Sourisce, A.; Noteuil, A.; Jah, N.; Wielgosik, A.; Fert, I.; Breban, M.; André, C. HLA-B27 Subtype Oligomerization and Intracellular Accumulation Patterns Correlate with Predisposition to Spondyloarthritis. Arthritis Rheumatol. 2014, 66, 2113–2123. [Google Scholar] [CrossRef]
- Prajzlerová, K.; Grobelná, K.; Pavelka, K.; Šenolt, L.; Filková, M. An Update on Biomarkers in Axial Spondyloarthritis. Autoimmun. Rev. 2016, 15, 501–509. [Google Scholar] [CrossRef]
- Apps, R.; Qi, Y.; Carlson, J.M.; Chen, H.; Gao, X.; Thomas, R.; Yuki, Y.; del Prete, G.Q.; Goulder, P.; Brumme, Z.L.; et al. Influence of HLA-C Expression Level on HIV Control. Science 2013, 340, 87–91. [Google Scholar] [CrossRef]
- Chandran, V.; Bull, S.B.; Pellett, F.J.; Ayearst, R.; Pollock, R.A.; Gladman, D.D. Killer-Cell Immunoglobulin-like Receptor Gene Polymorphisms and Susceptibility to Psoriatic Arthritis. Rheumatology 2014, 53, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.J.; Bridges, S.L.; Ahmed, S. HLA-C: An Accomplice in Rheumatic Diseases. ACR Open Rheumatol. 2019, 1, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Stuart, P.E.; Tejasvi, T.; Shaiq, P.A.; Kullavanijaya, P.; Qamar, R.; Raja, G.K.; Li, Y.; Voorhees, J.J.; Abecasis, G.R.; Elder, J.T.; et al. A Single SNP Surrogate for Genotyping HLA-C*06:02 in Diverse Populations. J. Investig. Dermatol. 2015, 135, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Pulit, S.L.; Leo, P.J.; Pointon, J.J.; Robinson, P.C.; Weisman, M.H.; Ward, M.; Gensler, L.S.; Zhou, X.; Garchon, H.J.; et al. Major Histocompatibility Complex Associations of Ankylosing Spondylitis Are Complex and Involve Further Epistasis with ERAP1. Nat. Commun. 2015, 6, 7146. [Google Scholar] [CrossRef] [PubMed]
- Costantino, F.; Talpin, A.; Evnouchidou, I.; Kadi, A.; Leboime, A.; Said-Nahal, R.; Bonilla, N.; Letourneur, F.; Leturcq, T.; Ka, Z.; et al. ERAP1 Gene Expression Is Influenced by Nonsynonymous Polymorphisms Associated with Predisposition to Spondyloarthritis. Arthritis Rheumatol. 2015, 67, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Costantino, F.; Breban, M.; Garchon, H.J. Genetics and Functional Genomics of Spondyloarthritis. Front. Immunol. 2018, 9, 2933. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D. The Genetic Basis of Spondyloarthritis. Ann. Rheum. Dis. 2011, 70, i44–i50. [Google Scholar] [CrossRef]
- International Genetics of Ankylosing Spondylitis Consortium (IGAS). Identification of Multiple Risk Variants for Ankylosing Spondylitis through High-Density Genotyping of Immune-Related Loci. Nat. Genet. 2013, 45, 730–738. [Google Scholar] [CrossRef]
- Vecellio, M.; Roberts, A.R.; Cohen, C.J.; Cortes, A.; Knight, J.C.; Bowness, P.; Wordsworth, B.P. The Genetic Association of RUNX3 with Ankylosing Spondylitis Can Be Explained by Allele-Specific Effects on IRF4 Recruitment that Alter Gene Expression. Ann. Rheum. Dis. 2016, 75, 1534–1540. [Google Scholar] [CrossRef]
- Roberts, A.R.; Vecellio, M.; Chen, L.; Ridley, A.; Cortes, A.; Knight, J.C.; Bowness, P.; Cohen, C.J.; Wordsworth, B.P. An Ankylosing Spondylitis-Associated Genetic Variant in the IL23R-IL12RB2 Intergenic Region Modulates Enhancer Activity and Is Associated with Increased Th1-Cell Differentiation. Ann. Rheum. Dis. 2016, 75, 2150–2156. [Google Scholar] [CrossRef]
- Fogel, O.; Bugge Tinggaard, A.; Fagny, M.; Sigrist, N.; Roche, E.; Leclere, L.; Deleuze, J.F.; Batteux, F.; Dougados, M.; Miceli-Richard, C.; et al. Deregulation of MicroRNA Expression in Monocytes and CD4+ T Lymphocytes from Patients with Axial Spondyloarthritis. Arthritis Res. Ther. 2019, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sanchez, C.; Font-Ugalde, P.; Ruiz-Limon, P.; Lopez-Pedrera, C.; Castro-Villegas, M.C.; Abalos-Aguilera, M.C.; Barbarroja, N.; de la Rosa, I.A.; Lopez-Montilla, M.D.; Escudero-Contreras, A.; et al. Circulating MicroRNAs as Potential Biomarkers of Disease Activity and Structural Damage in Ankylosing Spondylitis Patients. Hum. Mol. Genet. 2018, 27, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Toussirot, E.; Abbas, W.; Khan, K.A.; Tissot, M.; Jeudy, A.; Baud, L.; Bertolini, E.; Wendling, D.; Herbein, G. Imbalance between HAT and HDAC Activities in the PBMCs of Patients with Ankylosing Spondylitis or Rheumatoid Arthritis and Influence of HDAC Inhibitors on TNF Alpha Production. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, L. Role of Histone Deacetylase 3 in Ankylosing Spondylitis via Negative Feedback Loop with MicroRNA-130a and Enhancement of Tumor Necrosis Factor-1α Expression in Peripheral Blood Mononuclear Cells. Mol. Med. Rep. 2016, 13, 35–40. [Google Scholar] [CrossRef]
- Cherqaoui, B.; Crémazy, F.; Hue, C.; Garchon, H.J.; Breban, M.; Costantino, F. Epigenetics of Spondyloarthritis. Jt. Bone Spine 2020, 87, 565–571. [Google Scholar] [CrossRef]
- Dougados, M.; Baeten, D. Spondyloarthritis. Lancet 2011, 377, 2127–2137. [Google Scholar] [CrossRef]
- Asquith, M.J.; Stauffer, P.; Davin, S.; Mitchell, C.; Lin, P.; Rosenbaum, J.T. Perturbed Mucosal Immunity and Dysbiosis Accompany Clinical Disease in a Rat Model of Spondyloarthritis. Arthritis Rheumatol. 2016, 68, 2151–2162. [Google Scholar] [CrossRef]
- Ansalone, C.; Utriainen, L.; Milling, S.; Goodyear, C.S. Role of Gut Inflammation in Altering the Monocyte Compartment and Its Osteoclastogenic Potential in HLA–B27–Transgenic Rats. Arthritis Rheumatol. 2017, 69, 1807–1815. [Google Scholar] [CrossRef]
- Manasson, J.; Wallach, D.S.; Guggino, G.; Stapylton, M.; Badri, M.H.; Solomon, G.; Reddy, S.M.; Coras, R.; Aksenov, A.A.; Jones, D.R.; et al. Interleukin-17 Inhibition in Spondyloarthritis Is Associated with Subclinical Gut Microbiome Perturbations and a Distinctive Interleukin-25–Driven Intestinal Inflammation. Arthritis Rheumatol. 2020, 72, 645–657. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Zhang, P.; Song, C.; Pan, F.; Li, G.; Peng, L.; Yang, Y.; Wei, Z.; Huang, F. Gut Microbiota Changes in Patients with Spondyloarthritis: A Systematic Review. Semin. Arthritis Rheum. 2021, in press. [Google Scholar] [CrossRef]
- Cambré, I.; Gaublomme, D.; Burssens, A.; Jacques, P.; Schryvers, N.; De Muynck, A.; Meuris, L.; Lambrecht, S.; Carter, S.; de Bleser, P.; et al. Mechanical Strain Determines the Site-Specific Localization of Inflammation and Tissue Damage in Arthritis. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharip, A.; Kunz, J. Understanding the Pathogenesis of Spondyloarthritis. Biomolecules 2020, 10, 1461. [Google Scholar] [CrossRef] [PubMed]
- Stoll, M.L. Interactions of the Innate and Adaptive Arms of the Immune System in the Pathogenesis of Spondyloarthritis. Clin. Exp. Rheumatol. 2011, 29, 322–330. [Google Scholar] [PubMed]
- Navid, F.; Layh-Schmitt, G.; Sikora, K.A.; Cougnoux, A.; Colbert, R.A. The Role of Autophagy in the Degradation of Misfolded HLA-B27 Heavy Chains. Arthritis Rheumatol. 2018, 70, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Rysnik, O.; McHugh, K.; van Duivenvoorde, L.; van Tok, M.; Guggino, G.; Taurog, J.; Kollnberger, S.; Ciccia, F.; Baeten, D.; Bowness, P. Non-Conventional Forms of HLA-B27 Are Expressed in Spondyloarthritis Joints and Gut Tissue. J. Autoimmun. 2016, 70, 12–21. [Google Scholar] [CrossRef]
- Generali, E.; Bose, T.; Selmi, C.; Voncken, J.W.; Damoiseaux, J.G.M.C. Nature versus Nurture in the Spectrum of Rheumatic Diseases: Classification of Spondyloarthritis as Autoimmune or Autoinflammatory. Autoimmun. Rev. 2018, 17, 935–941. [Google Scholar] [CrossRef]
- Crotti, T.N.; Smith, M.D.; Weedon, H.; Ahern, M.J.; Findlay, D.M.; Kraan, M.; Tak, P.P.; Haynes, D.R. Receptor Activator NF-KappaB Ligand (RANKL) Expression in Synovial Tissue from Patients with Rheumatoid Arthritis, Spondyloarthropathy, Osteoarthritis, and from Normal Patients: Semiquantitative and Quantitative Analysis. Ann. Rheum. Dis. 2002, 61, 1047–1054. [Google Scholar] [CrossRef]
- Wong, K.L.; Tai, J.J.Y.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene Expression Profiling Reveals the Defining Features of the Classical, Intermediate, and Nonclassical Human Monocyte Subsets. Blood 2011, 118, 16–31. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of Monocytes and Dendritic Cells in Blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Surdacki, A.; Sulicka, J.; Korkosz, M.; Mikoałjczyk, T.; Telesinśka-Jasiówka, D.; Klimek, E.; Kierzkowska, I.; Guzik, T.; Grodzicki, T.K. Blood Monocyte Heterogeneity and Markers of Endothelial Activation in Ankylosing Spondylitis. J. Rheumatol. 2014, 41, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Zeng, M.; Thomas, R.; Ranganathan, V.; Rahman, A.; Alessandro, R.; Rizzo, A.; Saieva, L.; Macaluso, F.; et al. Proinflammatory CX3CR1+CD59+Tumor Necrosis Factor–Like Molecule 1A+Interleukin-23+ Monocytes Are Expanded in Patients with Ankylosing Spondylitis and Modulate Innate Lymphoid Cell 3 Immune Functions. Arthritis Rheumatol. 2018, 70, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Edelmann, M.; diGleria, K.; Kollnberger, S.; Kramer, H.; McGowan, S.; McHugh, K.; Taylor, S.; Kessler, B.; Bowness, P. Ankylosing Spondylitis Monocytes Show Upregulation of Proteins Involved in Inflammation and the Ubiquitin Proteasome Pathway. Ann. Rheum. Dis. 2009, 68, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.; Wu, P.; Sieper, J.; Syrbe, U. In Vivo Pre-Activation of Monocytes in Patients with Axial Spondyloarthritis. Arthritis Res. Ther. 2015, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, L.; Vandooren, B.; Kruithof, E.; de Keyser, F.; Veys, E.M.; Baeten, D. Tumor Necrosis Factor α Blockade Treatment Down-Modulates the Increased Systemic and Local Expression of Toll-like Receptor 2 and Toll-like Receptor 4 in Spondylarthropathy. Arthritis Rheum. 2005, 52, 2146–2158. [Google Scholar] [CrossRef]
- Aochi, S.; Tsuji, K.; Sakaguchi, M.; Huh, N.; Tsuda, T.; Yamanishi, K.; Komine, M.; Iwatsuki, K. Markedly Elevated Serum Levels of Calcium-Binding S100A8/A9 Proteins in Psoriatic Arthritis Are Due to Activated Monocytes/Macrophages. J. Am. Acad. Dermatol. 2011, 64, 879–887. [Google Scholar] [CrossRef]
- Huang, Y.; Deng, W.; Zheng, S.; Feng, F.; Huang, Z.; Huang, Q.; Guo, X.; Huang, Z.; Huang, X.; Pan, X.; et al. Relationship between Monocytes to Lymphocytes Ratio and Axial Spondyloarthritis. Int. Immunopharmacol. 2018, 57, 43–46. [Google Scholar] [CrossRef]
- Gulino, G.R.; Van Mechelen, M.; Lories, R. Cellular and Molecular Diversity in Spondyloarthritis. Semin. Immunol. 2021, 101521. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Culemann, S.; Grüneboom, A.; Nicolás-Ávila, J.Á.; Weidner, D.; Lämmle, K.F.; Rothe, T.; Quintana, J.A.; Kirchner, P.; Krljanac, B.; Eberhardt, M.; et al. Locally Renewing Resident Synovial Macrophages Provide a Protective Barrier for the Joint. Nature 2019, 572, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Krausz, S.; van Eijk, M.; Hamann, J.; Radstake, T.R.D.J.; Reedquist, K.A.; Tak, P.P.; Baeten, D.L.P. Systematic Validation of Specific Phenotypic Markers for in Vitro Polarized Human Macrophages. J. Immunol. Methods 2012, 375, 196–206. [Google Scholar] [CrossRef]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 Polarization and Rheumatoid Arthritis: A Systematic Review. Autoimmun. Rev. 2019, 18, 102397. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-Cell Transcriptomics and Mass Cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef]
- Ambarus, C.A.; Noordenbos, T.; de Hair, M.J.H.; Tak, P.P.; Baeten, D.L.P. Intimal Lining Layer Macrophages but Not Synovial Sublining Macrophages Display an IL-10 Polarized-like Phenotype in Chronic Synovitis. Arthritis Res. Ther. 2012, 14, R74. [Google Scholar] [CrossRef]
- Baeten, D.; Demetter, P.; Cuvelier, C.A.; Kruithof, E.; van Damme, N.; de Vos, M.; Veys, E.M.; de Keyser, F. Macrophages Expressing the Scavenger Receptor CD163: A Link between Immune Alterations of the Gut and Synovial Inflammation in Spondyloarthropathy. J. Pathol. 2002, 196, 343–350. [Google Scholar] [CrossRef]
- Rudwaleit, M.; Baeten, D. Ankylosing Spondylitis and Bowel Disease. Best Practice & Research Clinical Rheumatology 2006, 20, 451–471. [Google Scholar] [CrossRef]
- Alivernini, S.; Bruno, D.; Tolusso, B.; Bui, L.; Petricca, L.; Gigante, M.R.; Birra, D.; Fedele, A.L.; Peluso, G.; Federico, F.; et al. Differential Synovial Tissue Biomarkers among Psoriatic Arthritis and Rheumatoid Factor/Anti-Citrulline Antibody-Negative Rheumatoid Arthritis. Arthritis Res. Ther. 2019, 21, 116. [Google Scholar] [CrossRef]
- Vandooren, B.; Noordenbos, T.; Ambarus, C.; Krausz, S.; Cantaert, T.; Yeremenko, N.; Boumans, M.; Lutter, R.; Tak, P.P.; Baeten, D. Absence of a Classically Activated Macrophage Cytokine Signature in Peripheral Spondylarthritis, Including Psoriatic Arthritis. Arthritis Rheum. 2009, 60, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Fearon, U. What Makes Psoriatic and Rheumatoid Arthritis so Different? RMD Open 2015, 1, e000025. [Google Scholar] [CrossRef] [PubMed]
- García Pérez, S.; Malvar Fernández, B.; Tak, P.P.; Baeten, D.L.; Reedquist, K.A. THU0517 Tie2 Signaling Induces A Pro-Inflammatory Phenotype in Rheumatoid Arthritis and Psoriatic Arthritis Macrophages. Ann. Rheum. Dis. 2014, 73, 362. [Google Scholar] [CrossRef]
- Kabala, P.A.; Malvar-Fernández, B.; Lopes, A.P.; Carvalheiro, T.; Hartgring, S.A.Y.; Tang, M.W.; Conde, C.; Baeten, D.L.; Sleeman, M.; Tak, P.P.; et al. Promotion of Macrophage Activation by Tie2 in the Context of the Inflamed Synovia of Rheumatoid Arthritis and Psoriatic Arthritis Patients. Rheumatology 2020, 59, 426–438. [Google Scholar] [CrossRef]
- Fearon, U.; Griosios, K.; Fraser, A.; Reece, R.; Emery, P.; Jones, P.F.; Veale, D.J. Angiopoietins, Growth Factors, and Vascular Morphology in Early Arthritis. J. Rheumatol. 2003, 30, 260–268. [Google Scholar]
- Van de Sande, M.G.H.; de Launay, D.; de Hair, M.J.H.; García, S.; van de Sande, G.P.M.; Wijbrandts, C.A.; Gerlag, D.M.; Reedquist, K.A.; Tak, P.P. Local Synovial Engagement of Angiogenic TIE-2 Is Associated with the Development of Persistent Erosive Rheumatoid Arthritis in Patients with Early Arthritis. Arthritis Rheum. 2013, 65, 3073–3083. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 Stimulates the Production and Expression of Proinflammatory Cytokines, IL-Beta and TNF-Alpha, by Human Macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Taniguchi, N.; Kawakami, Y.; Maruyama, I.; Lotz, M. HMGB Proteins and Arthritis. Hum. Cell 2018, 31, 1–9. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 7–10. [Google Scholar] [CrossRef]
- Franco, G.C.N.; Kajiya, M.; Nakanishi, T.; Ohta, K.; Pedro, L.; Groppo, F.C.; Ernst, C.W.O.; Boyesen, J.L.; Bartlett, J.D.; Stashenko, P.; et al. Inhibition of Matrix Metalloproteinase-9 Activity by Doxycycline Ameliorates RANK Ligand-Induced Osteoclast Differentiation in Vitro and in Vivo. Exp. Cell Res. 2012, 317, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Schwarz, E.M. Mechanisms of Bone Resorption and New Bone Formation in Spondyloarthropathies. Curr. Rheumatol. Rep. 2002, 4, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Colbert, R.A. The Interleukin-23/Interleukin-17 Axis in Spondyloarthritis Pathogenesis: Th17 and Beyond. Arthritis Rheumatol. 2014, 66, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E.; Chao, C.; Geissler, R.; Laface, D.; Blumenschein, W.; Iwakura, Y.; McClanahan, T.; Bowman, E.P. Interleukin-17A Upregulates Receptor Activator of NF-ΚB on Osteoclast Precursors. Arthritis Res. Ther. 2010, 12, R29. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Schett, G. Effects of the IL-23–IL-17 Pathway on Bone in Spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 631–640. [Google Scholar] [CrossRef]
- Korkosz, M.; Czepiel, M.; Guła, Z.; Stec, M.; Wȩglarczyk, K.; Rutkowska-Zapała, M.; Gruca, A.; Lenart, M.; Baran, J.; Gasowski, J.; et al. Sera of Patients with Axial Spondyloarthritis (AxSpA) Enhance Osteoclastogenic Potential of Monocytes Isolated from Healthy Individuals. BMC Musculoskelet. Disord. 2018, 19, 1–9. [Google Scholar] [CrossRef]
- Dalbeth, N.; Pool, B.; Smith, T.; Callon, K.E.; Lobo, M.; Taylor, W.J.; Jones, P.B.; Cornish, J.; McQueen, F.M. Circulating Mediators of Bone Remodeling in Psoriatic Arthritis: Implications for Disordered Osteoclastogenesis and Bone Erosion. Arthritis Res. Ther. 2010, 12, R164. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Hartkamp, L.M.; Malvar-Fernandez, B.; van Es, I.E.; Lin, H.; Wong, J.; Long, L.; Zanghi, J.A.; Rankin, A.L.; Masteller, E.L.; et al. Colony-Stimulating Factor (CSF) 1 Receptor Blockade Reduces Inflammation in Human and Murine Models of Rheumatoid Arthritis. Arthritis Res. Ther. 2016, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Ramiro, S.; Landewé, R.; Baraliakos, X.; van den Bosch, F.; Sepriano, A.; Regel, A.; Ciurea, A.; Dagfinrud, H.; Dougados, M.; et al. 2016 Update of the ASAS-EULAR Management Recommendations for Axial Spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 978–991. [Google Scholar] [CrossRef]
- Singh, J.A.; Guyatt, G.; Ogdie, A.; Gladman, D.D.; Deal, C.; Deodhar, A.; Dubreuil, M.; Dunham, J.; Husni, M.E.; Kenny, S.; et al. 2018 American College of Rheumatology/National Psoriasis Foundation Guideline for the Treatment of Psoriatic Arthritis. Arthritis Rheumatol. 2019, 71, 5–32. [Google Scholar] [CrossRef]
- Ward, M.M.; Deodhar, A.; Gensler, L.S.; Dubreuil, M.; Yu, D.; Khan, M.A.; Haroon, N.; Borenstein, D.; Wang, R.; Biehl, A.; et al. 2019 Update of the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network Recommendations for the Treatment of Ankylosing Spondylitis and Nonradiographic Axial Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 1599–1613. [Google Scholar] [CrossRef]
- Gossec, L.; Baraliakos, X.; Kerschbaumer, A.; de Wit, M.; McInnes, I.; Dougados, M.; Primdahl, J.; McGonagle, D.G.; Aletaha, D.; Balanescu, A.; et al. EULAR Recommendations for the Management of Psoriatic Arthritis with Pharmacological Therapies: 2019 Update. Ann. Rheum. Dis. 2020, 79, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y. Immunomodulatory Effect of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) at the Clinically Available Doses. Arch. Pharmacal Res. 2007, 30, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Belge, K.-U.; Frankenberger, M.; Mack, M.; Berki, T.; Ziegler-Heitbrock, L. Mechanism of Glucocorticoid-Induced Depletion of Human CD14+ CD16+ Monocytes. J. Leukoc. Biol. 2003, 74, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Wijngaarden, S.; van Roon, J.A.G.; van de Winkel, J.G.J.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Down-Regulation of Activating Fcγ Receptors on Monocytes of Patients with Rheumatoid Arthritis upon Methotrexate Treatment. Rheumatology 2005, 44, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Zwicker, M.; Loetscher, P. Effects of Methotrexate on Differentiation of Monocytes and Production of Cytokine Inhibitors by Monocytes. Arthritis Rheum. 1998, 41, 2032–2038. [Google Scholar] [CrossRef]
- Obeng, J.A.; Amoruso, A.; Camaschella, G.L.E.; Sola, D.; Brunelleschi, S.; Fresu, L.G. Modulation of Human Monocyte/Macrophage Activity by Tocilizumab, Abatacept and Etanercept: An in Vitro Study. Eur. J. Pharmacol. 2016, 780, 33–37. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, W.; Tao, C.; Sun, P.; Yang, Z.; Xu, W. M2 Polarization of Monocytes in Ankylosing Spondylitis and Relationship with Inflammation and Structural Damage. APMIS 2017, 125, 1070–1075. [Google Scholar] [CrossRef]
- Menegatti, S.; Guillemot, V.; Latis, E.; Yahia-Cherbal, H.; Mittermüller, D.; Rouilly, V.; Mascia, E.; Rosine, N.; Koturan, S.; Millot, G.A.; et al. Immune Response Profiling of Patients with Spondyloarthritis Reveals Signalling Networks Mediating TNF-Blocker Function in Vivo. Ann. Rheum. Dis. 2021, 80, 475–486. [Google Scholar] [CrossRef]
- Garcia-Montoya, L.; Marzo-Ortega, H. The Role of Secukinumab in the Treatment of Psoriatic Arthritis and Ankylosing Spondylitis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 169–180. [Google Scholar] [CrossRef]
- Patel, D.D.; Lee, D.M.; Kolbinger, F.; Antoni, C. Effect of IL-17A Blockade with Secukinumab in Autoimmune Diseases. Ann. Rheum. Dis. 2013, 72, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mens, L.J.J.; Sande, M.G.H.; Menegatti, S.; Chen, S.; Blijdorp, I.C.J.; Jong, H.M.; Fluri, I.A.; Latuhihin, T.E.; Kuijk, A.W.R.; Rogge, L.; et al. Brief Report: Interleukin-17 Blockade with Secukinumab in Peripheral Spondyloarthritis Impacts Synovial Immunopathology without Compromising Systemic Immune Responses. Arthritis Rheumatol. 2018, 70, 1994–2002. [Google Scholar] [CrossRef] [PubMed]
- Fiechter, R.H.; de Jong, H.M.; van Mens, L.J.J.; Fluri, I.A.; Tas, S.W.; Baeten, D.L.P.; Yeremenko, N.G.; van de Sande, M.G.H. IL-12p40/IL-23p40 Blockade with Ustekinumab Decreases the Synovial Inflammatory Infiltrate through Modulation of Multiple Signaling Pathways Including MAPK-ERK and Wnt. Front. Immunol. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- McInnes, I.B.; Szekanecz, Z.; McGonagle, D.; Maksymowych, W.P.; Pfeil, A.; Lippe, R.; Song, I.-H.; Lertratanakul, A.; Sornasse, T.; Biljan, A.; et al. A Review of JAK–STAT Signalling in the Pathogenesis of Spondyloarthritis and the Role of JAK Inhibition. Rheumatology 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Ferner, E.; Savitskaya, A.; Bluml, S.; Steiner, C.-W.; Rath, E.; Smolen, J.S.; Scheinecker, C. Effects of Abatacept on Monocytes in Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2010, 69, A67. [Google Scholar] [CrossRef]
- Maseda, D.; Johnson, E.M.; Nyhoff, L.E.; Baron, B.; Kojima, F.; Wilhelm, A.J.; Ward, M.R.; Woodward, J.G.; Brand, D.D.; Crofford, L.J. MPGES1-Dependent Prostaglandin E2 (PGE2) Controls Antigen-Specific Th17 and Th1 Responses by Regulating T Autocrine and Paracrine PGE2 Production. J. Immunol. 2018, 200, 725–736. [Google Scholar] [CrossRef]
- Poddubnyy, D.; Rudwaleit, M.; Haibel, H.; Listing, J.; Märker-Hermann, E.; Zeidler, H.; Braun, J.; Sieper, J. Effect of Non-Steroidal Anti-Inflammatory Drugs on Radiographic Spinal Progression in Patients with Axial Spondyloarthritis: Results from the German Spondyloarthritis Inception Cohort. Ann. Rheum. Dis. 2012, 71, 1616–1622. [Google Scholar] [CrossRef]
- Kroon, F.P.; van der Burg, L.R.; Ramiro, S.; Landewé, R.B.; Buchbinder, R.; Falzon, L.; van der Heijde, D. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) for Axial Spondyloarthritis (Ankylosing Spondylitis and Non-Radiographic Axial Spondyloarthritis). Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Dhir, V.; Mishra, D.; Samanta, J. Glucocorticoids in Spondyloarthritis—Systematic Review and Real-World Analysis. Rheumatology 2021, 60, 4463–4475. [Google Scholar] [CrossRef]
- Desgeorges, T.; Caratti, G.; Mounier, R.; Tuckermann, J.; Chazaud, B. Glucocorticoids Shape Macrophage Phenotype for Tissue Repair. Front. Immunol. 2019, 10, 1591. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- Gijón, M.A.; Leslie, C.C. Regulation of Arachidonic Acid Release and Cytosolic Phospholipase A2 Activation. J. Leukoc. Biol. 1999, 65, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; McEachin, R.C.; States, D.J. Computationally Identifying Novel NF-ΚB-Regulated Immune Genes in the Human Genome. Genome Res. 2003, 13, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, N.N.; Jovanovic, F.; Voronov, D.; Candido, K.D. Do Corticosteroids Still Have a Place in the Treatment of Chronic Pain? Front. Pharmacol. 2018, 9, 1229. [Google Scholar] [CrossRef]
- Sasse, S.K.; Gruca, M.; Allen, M.A.; Kadiyala, V.; Song, T.; Gally, F.; Gupta, A.; Pufall, M.A.; Dowell, R.D.; Gerber, A.N. Nascent Transcript Analysis of Glucocorticoid Crosstalk with TNF Defines Primary and Cooperative Inflammatory Repression. Genome Res. 2019, 29, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Mozo, L.; Suarez, A.; Gutierrez, C. Glucocorticoids Up-Regulate Constitutive Interleukin-10 Production by Human Monocytes. Clin. Exp. Allergy 2004, 34, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Udalova, I.A.; Mantovani, A.; Feldmann, M. Macrophage Heterogeneity in the Context of Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2016, 12, 472–485. [Google Scholar] [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Role of Cytokines in Intervertebral Disc Degeneration: Pain and Disc Content. Nat. Rev. Rheumatol. 2014, 10, 44–56. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Aune, T.M. Methotrexate and Its Mechanisms of Action in Inflammatory Arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Sitkovsky, M. Adenosine and Adenosine Receptors in the Pathogenesis and Treatment of Rheumatic Diseases. Nat. Rev. Rheumatol. 2017, 13, 41–51. [Google Scholar] [CrossRef]
- Olsen, N.J.; Spurlock, C.F.; Aune, T.M. Methotrexate Induces Production of IL-1 and IL-6 in the Monocytic Cell Line U937. Arthritis Res. Ther. 2014, 16, R17. [Google Scholar] [CrossRef] [PubMed]
- Kane, D.; Gogarty, M.; O’Leary, J.; Silva, I.; Bermingham, N.; Bresnihan, B.; FitzGerald, O. Reduction of Synovial Sublining Layer Inflammation and Proinflammatory Cytokine Expression in Psoriatic Arthritis Treated with Methotrexate. Arthritis Rheum. 2004, 50, 3286–3295. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Menegatti, S.; Bianchi, E.; Rogge, L. Anti-TNF Therapy in Spondyloarthritis and Related Diseases, Impact on the Immune System and Prediction of Treatment Responses. Front. Immunol. 2019, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Kruithof, E.; van den Bosch, F.; Demetter, P.; van Damme, N.; Cuvelier, C.; de Vos, M.; Mielants, H.; Veys, E.M.; de Keyser, F. Immunomodulatory Effects of Anti-Tumor Necrosis Factor? Therapy on Synovium in Spondylarthropathy: Histologic Findings in Eight Patients from an Open-Label Pilot Study. Arthritis Rheum. 2001, 44, 186–195. [Google Scholar] [CrossRef]
- Van Kuijk, A.W.R.; Gerlag, D.M.; Vos, K.; Wolbink, G.; De Groot, M.; De Rie, M.A.; Zwinderman, A.H.; Dijkmans, B.A.C.; Tak, P.P. A Prospective, Randomised, Placebo-Controlled Study to Identify Biomarkers Associated with Active Treatment in Psoriatic Arthritis: Effects of Adalimumab Treatment on Synovial Tissue. Ann. Rheum. Dis. 2009, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Cañete, J.D.; Pablos, J.L.; Sanmartí, R.; Mallofré, C.; Marsal, S.; Maymó, J.; Gratacós, J.; Mezquita, J.; Mezquita, C.; Cid, M.C. Antiangiogenic Effects of Anti-Tumor Necrosis Factor α Therapy with Infliximab in Psoriatic Arthritis. Arthritis Rheum. 2004, 50, 1636–1641. [Google Scholar] [CrossRef]
- Kruithof, E.; Baeten, D.; van den Bosch, F.; Mielants, H.; Veys, E.M.; de Keyser, F. Histological Evidence that Infliximab Treatment Leads to Downregulation of Inflammation and Tissue Remodelling of the Synovial Membrane in Spondyloarthropathy. Ann. Rheum. Dis. 2005, 64, 529–536. [Google Scholar] [CrossRef]
- Batko, B.; Schramm-Luc, A.; Skiba, D.; Mikolajczyk, T.; Siedlinski, M. TNF-α Inhibitors Decrease Classical CD14hiCD16− Monocyte Subsets in Highly Active, Conventional Treatment Refractory Rheumatoid Arthritis and Ankylosing Spondylitis. Int. J. Mol. Sci. 2019, 20, 291. [Google Scholar] [CrossRef]
- Bloemendaal, F.M.; Koelink, P.J.; Van Schie, K.A.; Rispens, T.; Peters, C.P.; Buskens, C.J.; Van Der Bilt, J.D.; Bemelman, W.A.; Korf, H.; Sabino, J.G.; et al. TNF-Anti-TNF Immune Complexes Inhibit IL-12/IL-23 Secretion by Inflammatory Macrophages via an Fc-Dependent Mechanism. J. Crohn’s Colitis 2018, 12, 1122–1130. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Sebald, H.-J.; Villiger, P.M.; Hofstetter, W.; Seitz, M. Infliximab Inhibits Bone Resorption by Circulating Osteoclast Precursor Cells in Patients with Rheumatoid Arthritis and Ankylosing Spondylitis. Ann. Rheum. Dis. 2007, 67, 620–624. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Wildenberg, M.E.; Duijvestein, M.; Verhaar, A.P.; Van Den Brink, G.R.; Hommes, D.W. AntiTumor Necrosis Factor-α Antibodies Induce Regulatory Macrophages in an Fc Region-Dependent Manner. Gastroenterology 2011, 140, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; Van Der Heijde, D.; Landewé, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a Human Anti-Interleukin-17A Monoclonal Antibody, in Patients with Psoriatic Arthritis (FUTURE 2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef]
- Mease, P.J.; Smolen, J.S.; Behrens, F.; Nash, P.; Liu Leage, S.; Li, L.; Tahir, H.; Gooderham, M.; Krishnan, E.; Liu-Seifert, H.; et al. A Head-to-Head Comparison of the Efficacy and Safety of Ixekizumab and Adalimumab in Biological-Naïve Patients with Active Psoriatic Arthritis: 24-Week Results of a Randomised, Open-Label, Blinded-Assessor Trial. Ann. Rheum. Dis. 2020, 79, 123–131. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Cheng-Chung Wei, J.; Dougados, M.; Mease, P.; Deodhar, A.; Maksymowych, W.P.; Van den Bosch, F.; Sieper, J.; Tomita, T.; Landewé, R.; et al. Ixekizumab, an Interleukin-17A Antagonist in the Treatment of Ankylosing Spondylitis or Radiographic Axial Spondyloarthritis in Patients Previously Untreated with Biological Disease-Modifying Anti-Rheumatic Drugs (COAST-V): 16 Week Results of a Phase 3 Ra. Lancet 2018, 392, 2441–2451. [Google Scholar] [CrossRef]
- Wasilewska, A.; Winiarska, M.; Olszewska, M.; Rudnicka, L. Interleukin-17 Inhibitors. A New Era in Treatment of Psoriasis and Other Skin Diseases. Postepy Dermatol. I Alergol. 2016, 33, 247–252. [Google Scholar] [CrossRef]
- Chiricozzi, A.; De Simone, C.; Fossati, B.; Peris, K. Emerging Treatment Options for the Treatment of Moderate to Severe Plaque Psoriasis and Psoriatic Arthritis: Evaluating Bimekizumab and Its Therapeutic Potential. Psoriasis Targets Ther. 2019, 9, 73–74. [Google Scholar] [CrossRef]
- Rafael-Vidal, C.; Pérez, N.; Altabás, I.; Garcia, S.; Pego-Reigosa, J.M. Blocking IL-17: A Promising Strategy in the Treatment of Systemic Rheumatic Diseases. Int. J. Mol. Sci. 2020, 21, 7100. [Google Scholar] [CrossRef]
- Barin, J.G.; Baldeviano, G.C.; Talor, M.V.; Wu, L.; Ong, S.; Quader, F.; Chen, P.; Zheng, D.; Caturegli, P.; Rose, N.R.; et al. Macrophages Participate in IL-17-Mediated Inflammation. Eur. J. Immunol. 2012, 42, 726–736. [Google Scholar] [CrossRef]
- Bastos, K.R.B.; Marinho, C.R.F.; Barboza, R.; Russo, M.; Álvarez, J.M.; D’Império Lima, M.R. What Kind of Message Does IL-12/IL-23 Bring to Macrophages and Dendritic Cells? Microbes Infect. 2004, 6, 630–636. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, B.S.; Kastelein, R.A.; Cua, D.J. Understanding the IL-23–IL-17 Immune Pathway. Trends Immunol. 2006, 27, 17–23. [Google Scholar] [CrossRef]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and Mechanism of Ustekinumab: A Human Monoclonal Antibody Targeting Interleukin-12 and Interleukin-23 for Treatment of Immune-Mediated Disorders. mAbs 2011, 3, 535–545. [Google Scholar] [CrossRef]
- Spinelli, F.R.; Colbert, R.A.; Gadina, M. JAK1: Number One in the Family; Number One in Inflammation? Rheumatology 2021, 60, ii3–ii10. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling via the JAK/STAT Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Malyshev, I.; Malyshev, Y. Current Concept and Update of the Macrophage Plasticity Concept: Intracellular Mechanisms of Reprogramming and M3 Macrophage “Switch” Phenotype. BioMed Res. Int. 2015, 2015, 1–22. [Google Scholar] [CrossRef]
- EMA announcement Tofacitinib for Psoriatic Arthritis (PsA) Is Now Approved by the European Commission. Rheumatology 2018, 57, e55. [CrossRef][Green Version]
- Deodhar, A.; Sliwinska-Stanczyk, P.; Xu, H.; Baraliakos, X.; Gensler, L.S.; Fleishaker, D.; Wang, L.; Wu, J.; Menon, S.; Wang, C.; et al. Tofacitinib for the Treatment of Ankylosing Spondylitis: A Phase III, Randomised, Double-Blind, Placebo-Controlled Study. Ann. Rheum. Dis. 2021, 80, 1004–1013. [Google Scholar] [CrossRef]
- Olivieri, I.; D’Angelo, S.; Mennillo, G.A.; Pistone, G.; Scarano, E.; Padula, A. Abatacept in Spondyloarthritis Refractory to Tumour Necrosis Factor α Inhibition: Figure 1. Ann. Rheum. Dis. 2009, 68, 151–152. [Google Scholar] [CrossRef]
- Bonelli, M.; Ferner, E.; Göschl, L.; Blüml, S.; Hladik, A.; Karonitsch, T.; Kiener, H.P.; Byrne, R.; Niederreiter, B.; Steiner, C.W.; et al. Abatacept (CTLA-4IG) Treatment Reduces the Migratory Capacity of Monocytes in Patients with Rheumatoid Arthritis. Arthritis Rheum. 2013, 65, 599–607. [Google Scholar] [CrossRef]
- Wenink, M.H.; Santegoets, K.C.M.; Platt, A.M.; van den Berg, W.B.; van Riel, P.L.C.M.; Garside, P.; Radstake, T.R.D.J.; McInnes, I.B. Abatacept Modulates Proinflammatory Macrophage Responses upon Cytokine-Activated T Cell and Toll-like Receptor Ligand Stimulation. Ann. Rheum. Dis. 2012, 71, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Soldano, S.; Gotelli, E.; Montagna, P.; Campitiello, R.; Paolino, S.; Pizzorni, C.; Sulli, A.; Smith, V.; Tardito, S. CTLA4-Ig Treatment Induces M1–M2 Shift in Cultured Monocyte-Derived Macrophages from Healthy Subjects and Rheumatoid Arthritis Patients. Arthritis Res. Ther. 2021, 23, 306. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; Porter-Brown, B.; Thompson, L.; Harari, O.; Dougados, M. Assessment of Short-Term Symptomatic Efficacy of Tocilizumab in Ankylosing Spondylitis: Results of Randomised, Placebo-Controlled Trials. Ann. Rheum. Dis. 2014, 73, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; Braun, J.; Kay, J.; Badalamenti, S.; Radin, A.R.; Jiao, L.; Fiore, S.; Momtahen, T.; Yancopoulos, G.D.; Stahl, N.; et al. Sarilumab for the Treatment of Ankylosing Spondylitis: Results of a Phase II, Randomised, Double-Blind, Placebo-Controlled Study (ALIGN). Ann. Rheum. Dis. 2015, 74, 1051–1057. [Google Scholar] [CrossRef]
- Merashli, M.; de Marco, G.; Podgorski, M.; McGonagle, D.; Marzo-Ortega, H. Evidence of Response to IL-6 Inhibition in Some Cases of Refractory Spondyloarthritis-Associated Peripheral Synovitis. Ann. Rheum. Dis. 2016, 75, 1418–1420. [Google Scholar] [CrossRef]
- De Wilde, K.; Martens, A.; Lambrecht, S.; Jacques, P.; Drennan, M.B.; Debusschere, K.; Govindarajan, S.; Coudenys, J.; Verheugen, E.; Windels, F.; et al. A20 Inhibition of STAT1 Expression in Myeloid Cells: A Novel Endogenous Regulatory Mechanism Preventing Development of Enthesitis. Ann. Rheum. Dis. 2017, 76, 585–592. [Google Scholar] [CrossRef]
- Al-Mossawi, M.H.; Chen, L.; Fang, H.; Ridley, A.; de Wit, J.; Yager, N.; Hammitzsch, A.; Pulyakhina, I.; Fairfax, B.P.; Simone, D.; et al. Unique Transcriptome Signatures and GM-CSF Expression in Lymphocytes from Patients with Spondyloarthritis. Nat. Commun. 2017, 8, 1510. [Google Scholar] [CrossRef]
- Blijdorp, I.C.J.; Menegatti, S.; van Mens, L.J.J.; van de Sande, M.G.H.; Chen, S.; Hreggvidsdottir, H.S.; Noordenbos, T.; Latuhihin, T.E.; Bernink, J.H.; Spits, H.; et al. Expansion of Interleukin-22– and Granulocyte–Macrophage Colony-Stimulating Factor–Expressing, but Not Interleukin-17A–Expressing, Group 3 Innate Lymphoid Cells in the Inflamed Joints of Patients With Spondyloarthritis. Arthritis Rheumatol. 2019, 71, 392–402. [Google Scholar] [CrossRef]
- Regan-Komito, D.; Swann, J.W.; Demetriou, P.; Cohen, E.S.; Horwood, N.J.; Sansom, S.N.; Griseri, T. GM-CSF Drives Dysregulated Hematopoietic Stem Cell Activity and Pathogenic Extramedullary Myelopoiesis in Experimental Spondyloarthritis. Nat. Commun. 2020, 11, 155. [Google Scholar] [CrossRef]
- Shi, H.; Chen, L.; Ridley, A.; Zaarour, N.; Brough, I.; Caucci, C.; Smith, J.E.; Bowness, P. GM-CSF Primes Proinflammatory Monocyte Responses in Ankylosing Spondylitis. Front. Immunol. 2020, 11, 1520. [Google Scholar] [CrossRef]
- Cook, A.D.; Hamilton, J.A. Investigational Therapies Targeting the Granulocyte Macrophage Colony-Stimulating Factor Receptor-α in Rheumatoid Arthritis: Focus on Mavrilimumab. Ther. Adv. Musculoskelet. Dis. 2018, 10, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.D.; Braine, E.L.; Campbell, I.K.; Rich, M.J.; Hamilton, J.A. Blockade of Collagen-Induced Arthritis Post-Onset by Antibody to Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF): Requirement for GM-CSF in the Effector Phase of Disease. Arthritis Res. 2001, 3, 293. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.D.; Lee, M.-C.; Saleh, R.; Khiew, H.-W.; Christensen, A.D.; Achuthan, A.; Fleetwood, A.J.; Lacey, D.C.; Smith, J.E.; Förster, I.; et al. TNF and Granulocyte Macrophage-Colony Stimulating Factor Interdependence Mediates Inflammation via CCL17. JCI Insight 2018, 3, e99249. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; McInnes, I.B.; Kremer, J.; Miranda, P.; Korkosz, M.; Vencovsky, J.; Rubbert-Roth, A.; Mysler, E.; Sleeman, M.A.; Godwood, A.; et al. A Randomised Phase IIb Study of Mavrilimumab, a Novel GM–CSF Receptor Alpha Monoclonal Antibody, in the Treatment of Rheumatoid Arthritis. Ann. Rheum. Dis. 2017, 76, 1020–1030. [Google Scholar] [CrossRef]
- Huizinga, T.W.J.; Batalov, A.; Stoilov, R.; Lloyd, E.; Wagner, T.; Saurigny, D.; Souberbielle, B.; Esfandiari, E. Phase 1b Randomized, Double-Blind Study of Namilumab, an Anti-Granulocyte Macrophage Colony-Stimulating Factor Monoclonal Antibody, in Mild-to-Moderate Rheumatoid Arthritis. Arthritis Res. Ther. 2017, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Saurigny, D.; Vencovsky, J.; Takeuchi, T.; Nakamura, T.; Matsievskaia, G.; Hunt, B.; Wagner, T.; Souberbielle, B. Efficacy and Safety of Namilumab, a Human Monoclonal Antibody against Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Ligand in Patients with Rheumatoid Arthritis (RA) with Either an Inadequate Response to Background Methotrexate Therapy or an Inadequate Response or Intolerance to an Anti-TNF (Tumour Necrosis Factor) Biologic Therapy: A Randomized, Controlled Trial. Arthritis Res. Ther. 2019, 21, 101. [Google Scholar] [CrossRef]
- Behrens, F.; Tak, P.P.; Østergaard, M.; Stoilov, R.; Wiland, P.; Huizinga, T.W.; Berenfus, V.Y.; Vladeva, S.; Rech, J.; Rubbert-Roth, A.; et al. MOR103, a Human Monoclonal Antibody to Granulocyte–Macrophage Colony-Stimulating Factor, in the Treatment of Patients with Moderate Rheumatoid Arthritis: Results of a Phase Ib/IIa Randomised, Double-Blind, Placebo-Controlled, Dose-Escalation Trial. Ann. Rheum. Dis. 2015, 74, 1058–1064. [Google Scholar] [CrossRef]
- Kanekura, T.; Seishima, M.; Honma, M.; Etou, T.; Eto, H.; Okuma, K.; Okubo, Y.; Yamaguchi, Y.; Kambara, T.; Mabuchi, T.; et al. Therapeutic Depletion of Myeloid Lineage Leukocytes by Adsorptive Apheresis for Psoriatic Arthritis: Efficacy of a Non-Drug Intervention for Patients Refractory to Pharmacologics. J. Dermatol. 2017, 44, 1353–1359. [Google Scholar] [CrossRef]
- Kanekura, T. Clinical and Immunological Effects of Adsorptive Myeloid Lineage Leukocyte Apheresis in Patients with Immune Disorders. J. Dermatol. 2018, 45, 943–950. [Google Scholar] [CrossRef]


{kind=link}
| Treatment Strategy | Drug | Mechanism of Action | Main Effects on Monocytes/Macrophages |
|---|---|---|---|
| NSAIDs | Meloxicam, Ibuprofen, Meclofenamate, etc. | Cyclooxygenase activity inhibition | Decreased activation of macrophages [103] |
| Glucocorticoids | Prednisolone, methylprednisolone | Phospholipase A2 suppression; NF-κB antagonization | Increased IL-10 secretion by monocytes; CD16+ cells depletion; restrained activity of pro-inflammatory cytokines [104] |
| Non-biological DMARDs | Methotrexate | Down-regulation of monocytes and macrophages activation; production of cytokine inhibitors by monocytes and IL-1β suppression [105,106] | |
| Anti-TNF | Adalimumab, Infliximab, Certolizumab pegol, Golimumab and Etanercept | TNF antibody blocking and TNF receptor blocking | Decrease of macrophage infiltration in the synovial tissue; decrease of expression levels of metalloproteinases; differentiation of immunosuppressive macrophages [107,108,109] |
| Anti-IL-17 | Secukinumab, Ixekizumab, Bimekizumab and Afasevikumab | IL-17 antibody blocking | Decrease of macrophages infiltration and MMPs expression; reduction of IL-17-mediated osteoclastogenesis [110,111,112] |
| Anti-IL-12/IL-23 | Ustekinumab | Antibody blocking of IL-12 and IL-23 cytokines | Lower infiltration of CD68+ macrophages in the synovial sublining layer [113] |
| JAK inhibitors | Tofacitinib | JAK1 and JAK3 inhibition | Decreased production of pro-inflammatory cytokines by macrophages [114] |
| CTLA4-Ig | Abatacept | Immunoglobulin against Cytotoxic T-Lymphocyte Antigen 4 | Decrease of TNF production; regulation of migratory capacity [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Ramos, S.; Rafael-Vidal, C.; Pego-Reigosa, J.M.; García, S. Monocytes and Macrophages in Spondyloarthritis: Functional Roles and Effects of Current Therapies. Cells 2022, 11, 515. https://doi.org/10.3390/cells11030515
Martínez-Ramos S, Rafael-Vidal C, Pego-Reigosa JM, García S. Monocytes and Macrophages in Spondyloarthritis: Functional Roles and Effects of Current Therapies. Cells. 2022; 11(3):515. https://doi.org/10.3390/cells11030515
Chicago/Turabian StyleMartínez-Ramos, Sara, Carlos Rafael-Vidal, José M. Pego-Reigosa, and Samuel García. 2022. "Monocytes and Macrophages in Spondyloarthritis: Functional Roles and Effects of Current Therapies" Cells 11, no. 3: 515. https://doi.org/10.3390/cells11030515
APA StyleMartínez-Ramos, S., Rafael-Vidal, C., Pego-Reigosa, J. M., & García, S. (2022). Monocytes and Macrophages in Spondyloarthritis: Functional Roles and Effects of Current Therapies. Cells, 11(3), 515. https://doi.org/10.3390/cells11030515