
Mitochondria Fusion upon SERCA Inhibition Prevents Activation of the NLRP3 Inflammasome in Human Monocytes

 ,
,  , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. THP-1 Cell Culture
2.2.2. Isolation and Culture of Primary Human Monocytes
2.2.3. Total Cell Lysates Preparation
2.2.4. Western Blotting
2.2.5. Determination of Secreted IL-1β Levels in the Cell Supernatant
2.2.6. Determination of Mitochondrial Membrane Potential Assay
2.2.7. Detection of Mitochondrial ROS Production
2.2.8. Transmission Electron Microscopy (TEM)
2.2.9. Detection of Mitochondrial Calcium Content
2.2.10. Evaluation of Cell Viability
2.2.11. Statistical Analysis
3. Results
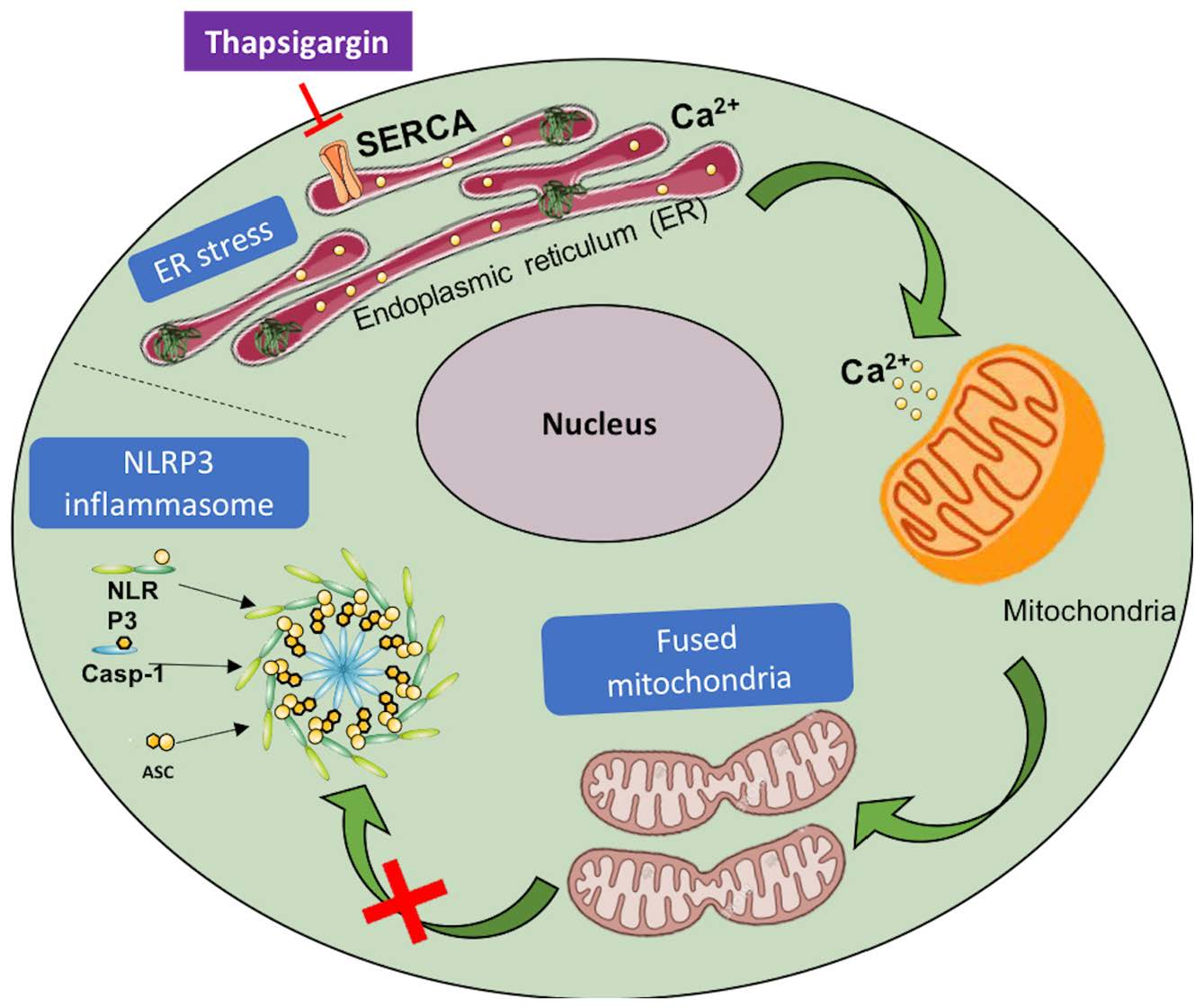
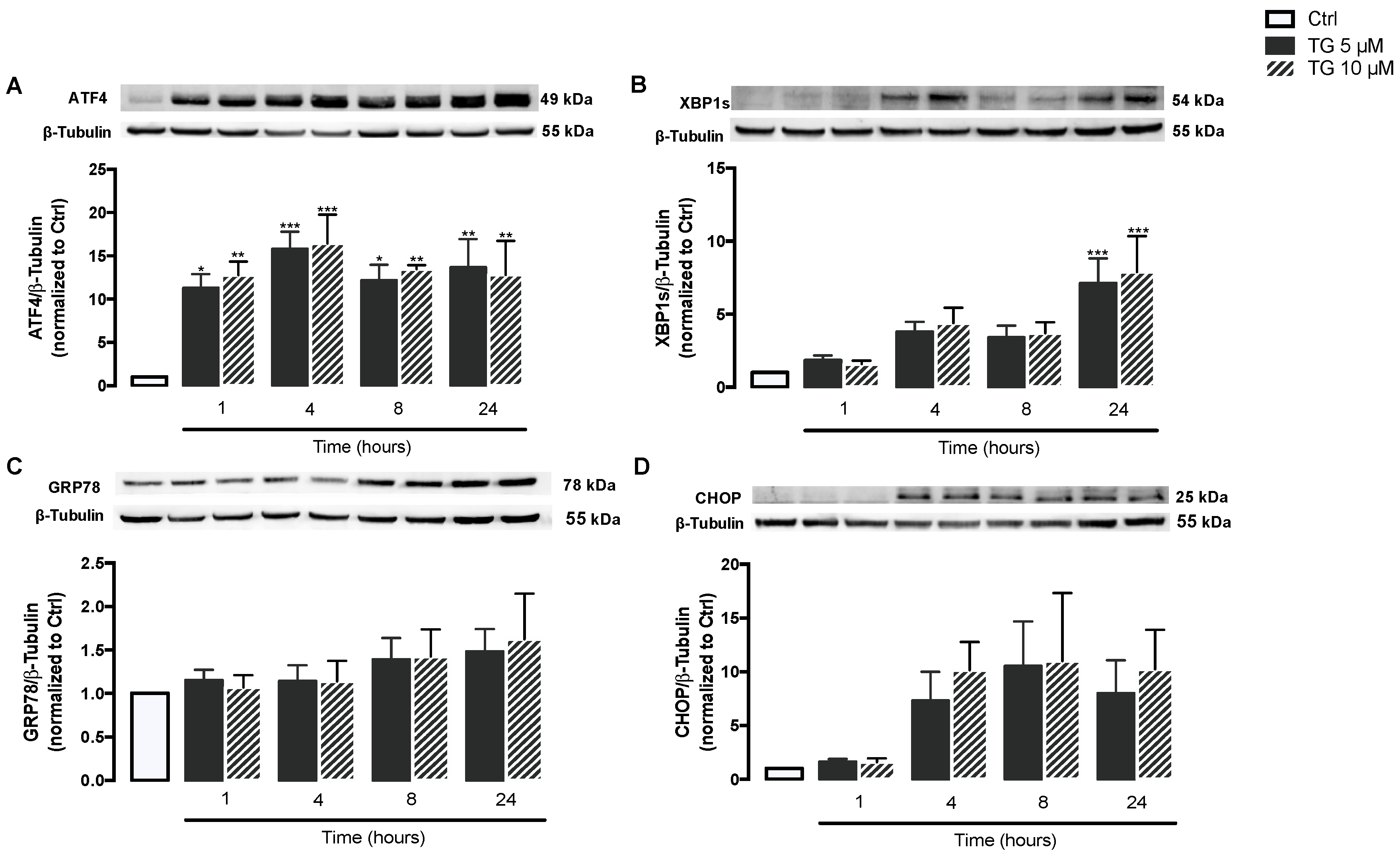
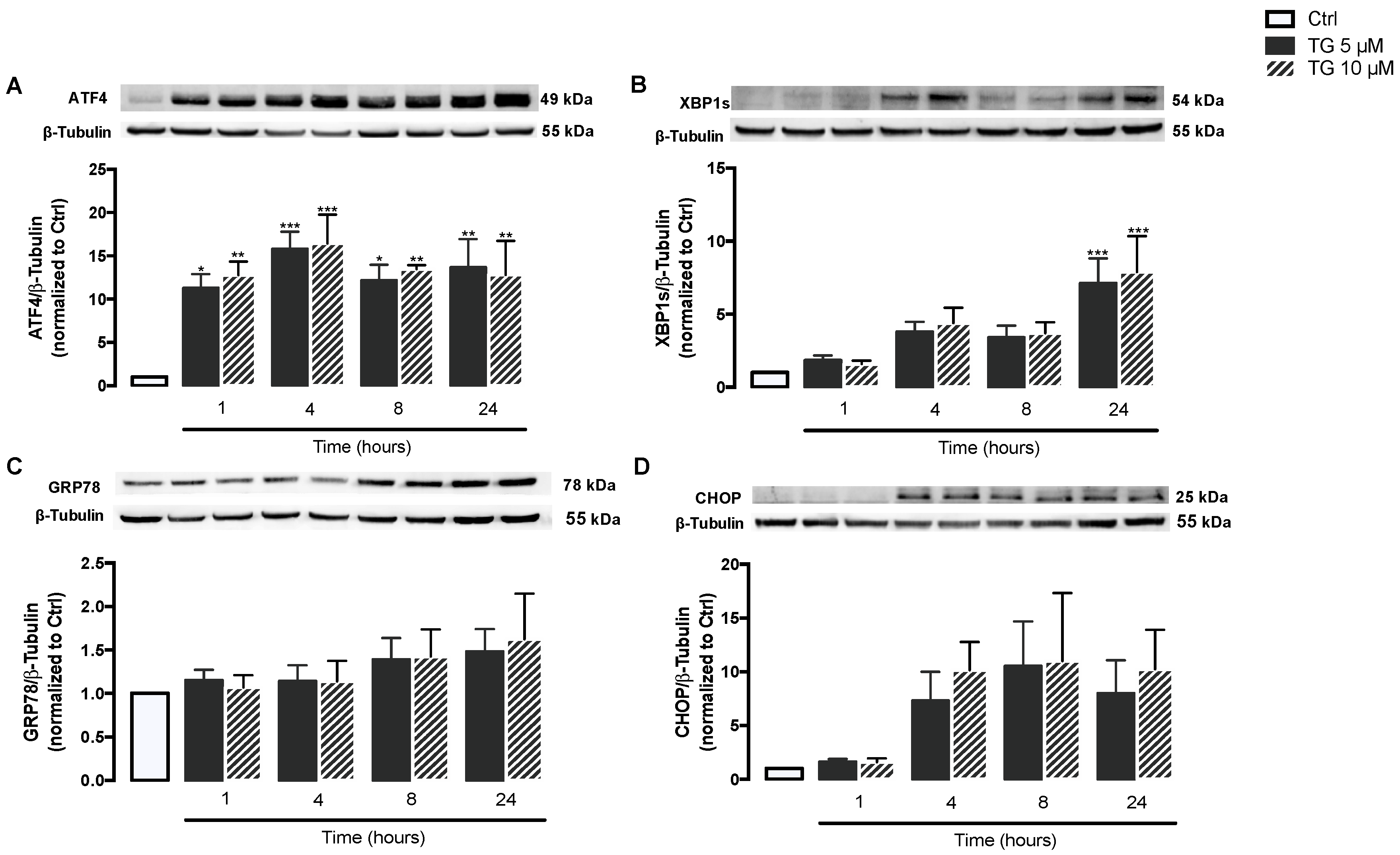
3.1. SERCA Inhibition Induces ER Stress in Human Monocytes
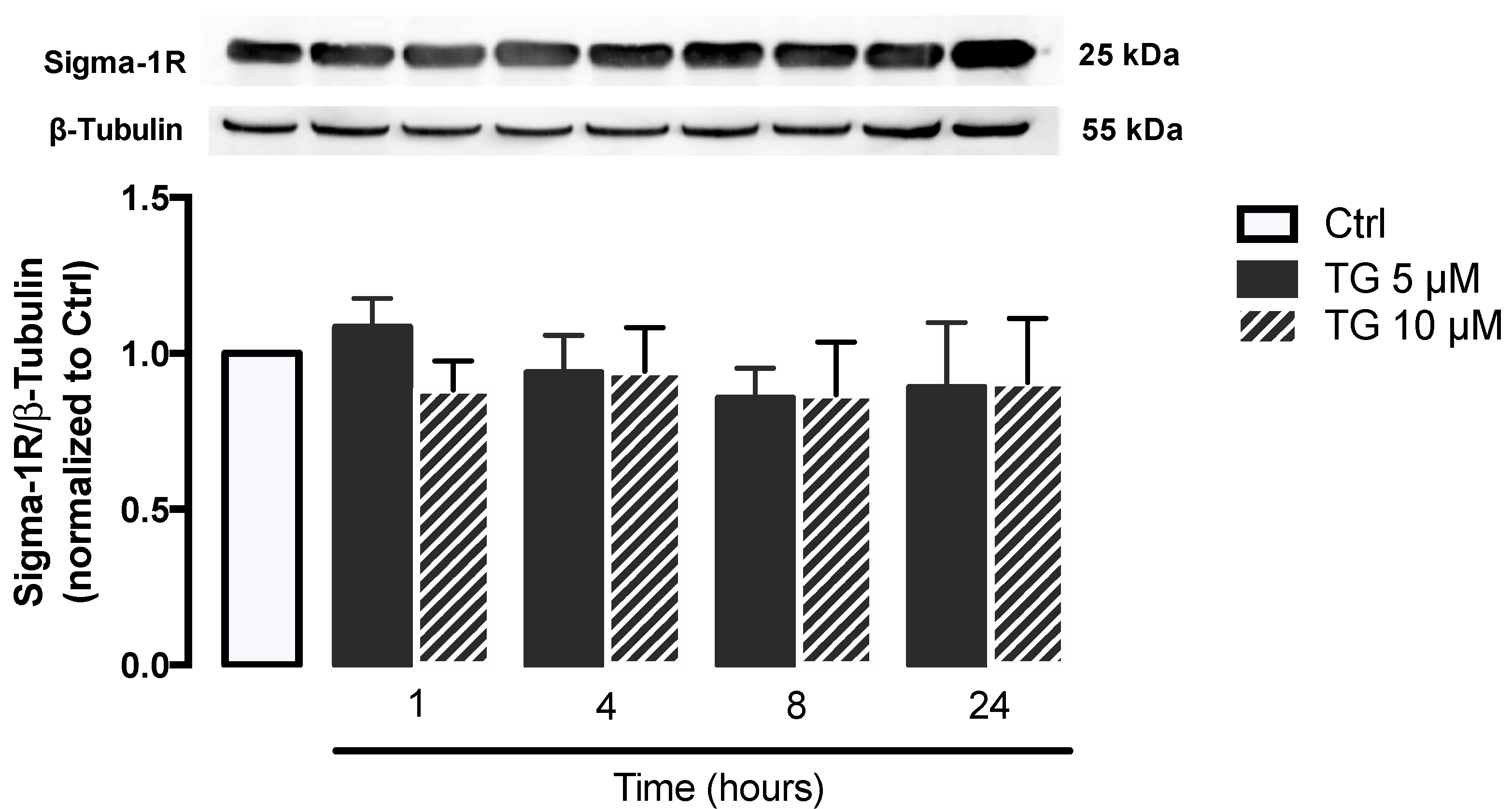
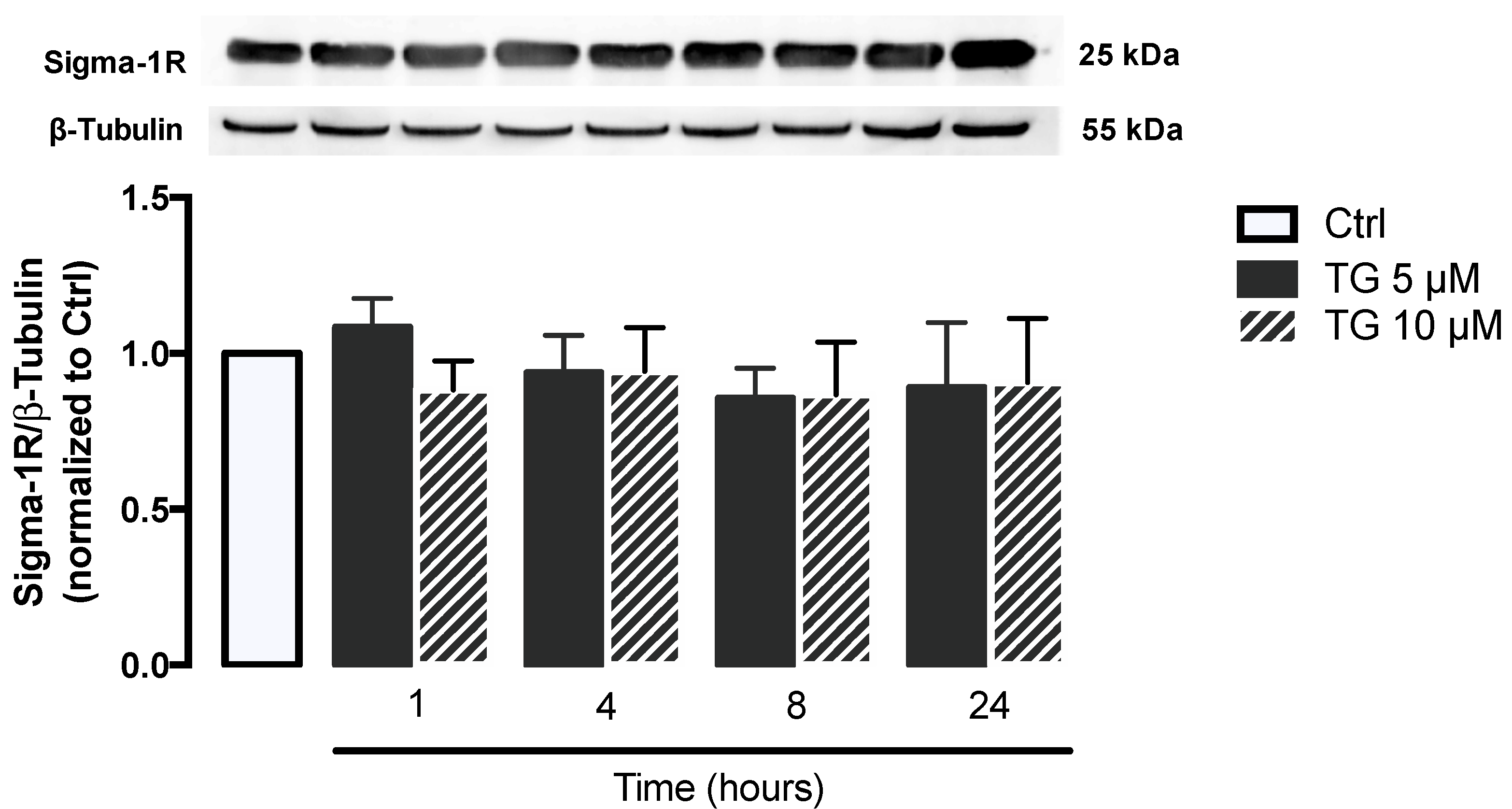
3.2. SERCA Inhibition Is Not Able to Upregulate ER Stress Response Strategies
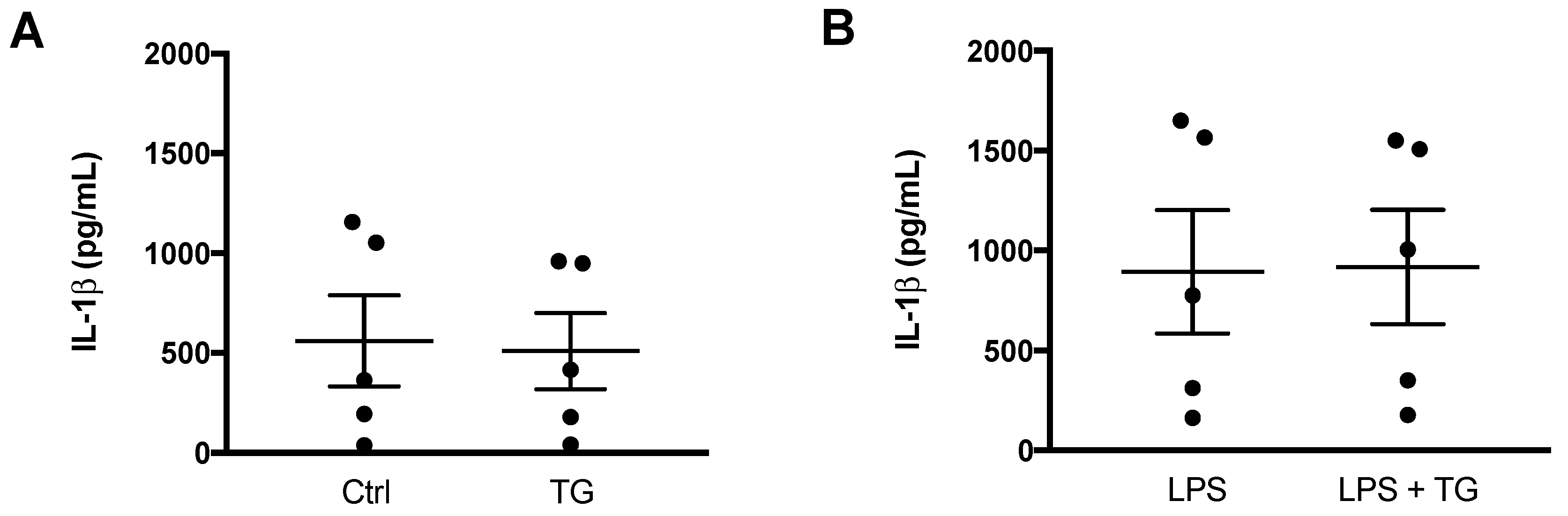
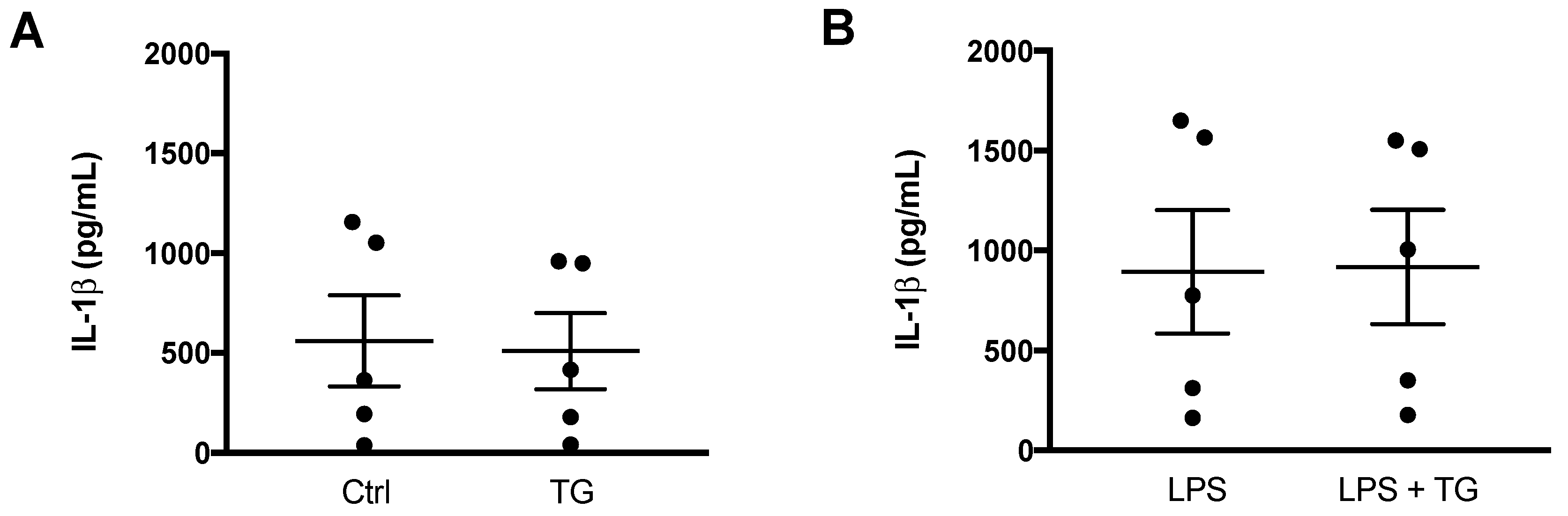
3.3. SERCA Inhibition Fails to Promote IL-1β Secretion in Human Monocytes
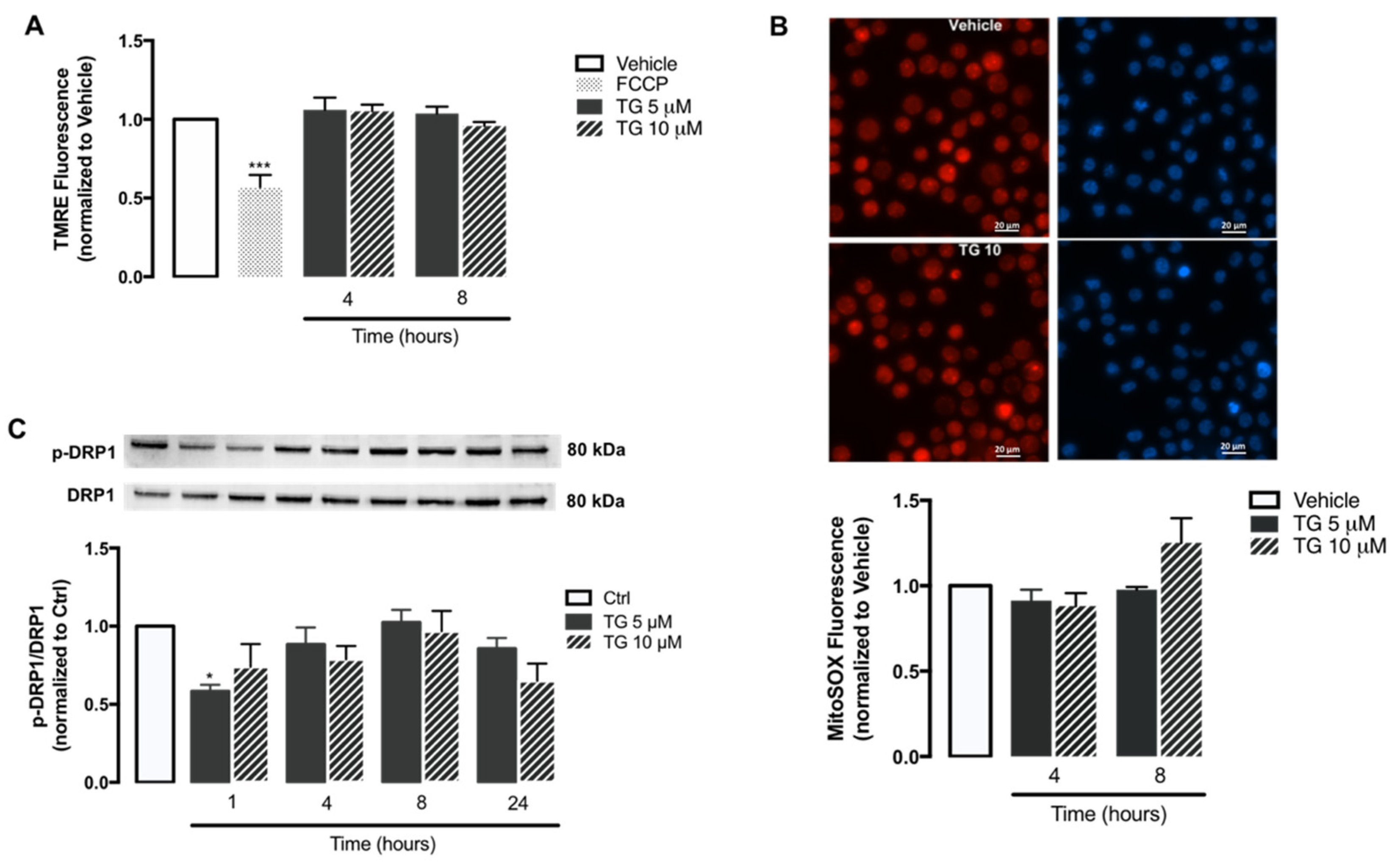
3.4. NLRP3 Inflammasome-Associated Triggers Are Not Promoted by SERCA Inhibition
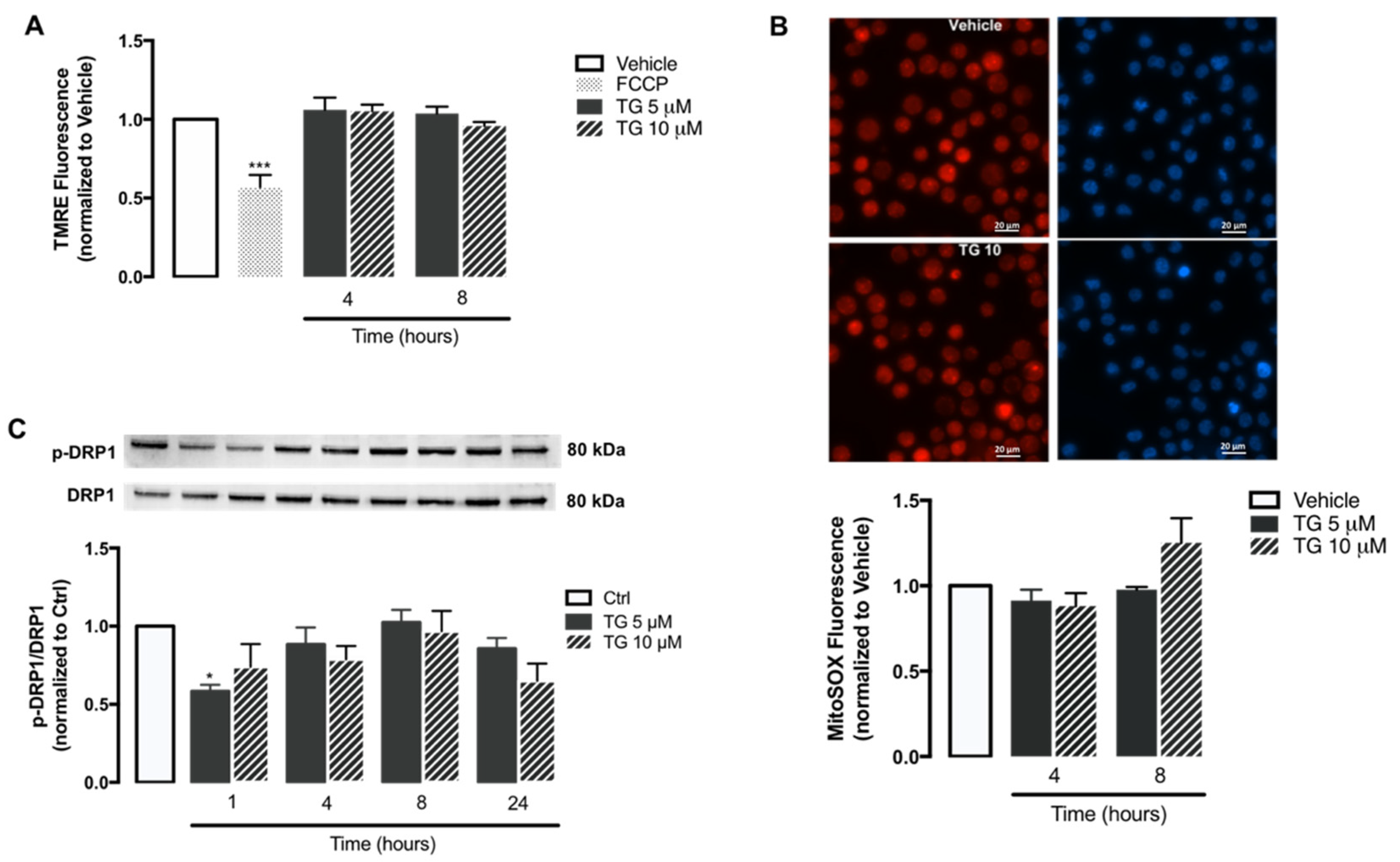
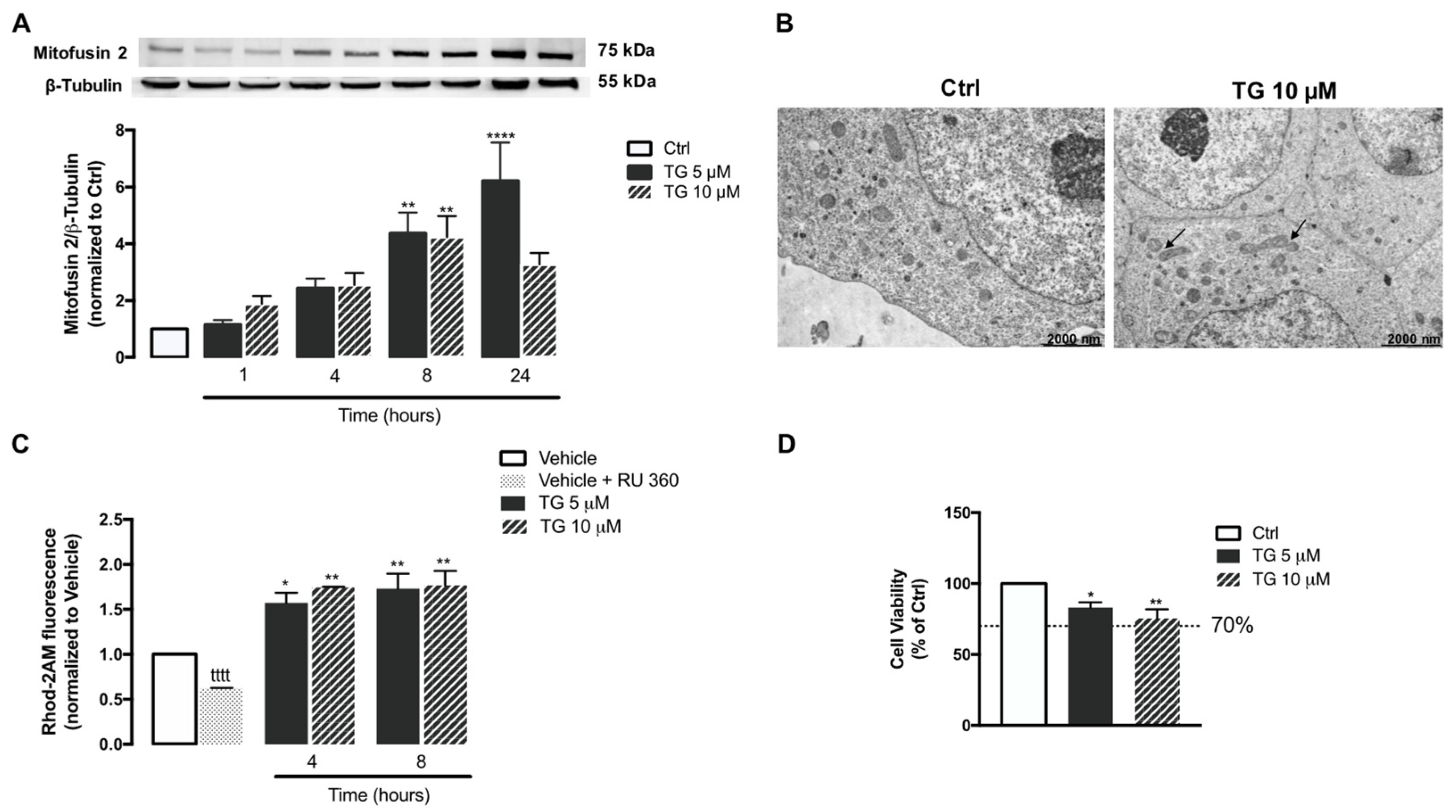
3.5. SERCA Inhibition Increases Mitochondrial Fusion and Ca2+ Content
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, E.; Teodoro, T.; Volchuk, A. Endoplasmic Reticulum Stress: Signaling the Unfolded Protein Response. Physiology 2007, 22, 193–201. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug Interactions With the Ca2+-ATPase From Sarco(Endo)Plasmic Reticulum (SERCA). Front. Mol. Biosci. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Ortiz, R.; Espinoza-Fonseca, L.M. Linking Biochemical and Structural States of SERCA: Achievements, Challenges, and New Opportunities. Int. J. Mol. Sci. 2020, 21, 4146. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin—From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. In Methods in Enzymology; Elsevier BV: Amsterdam, The Netherlands, 2011; Volume 490, pp. 71–92. [Google Scholar]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Karagoz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, 033886. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.-P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Roldan, J.L.; Ossa, F.; Schnell, J.R. Characterization of the Human Sigma-1 Receptor Chaperone Domain Structure and Binding Immunoglobulin Protein (BiP) Interactions. J. Biol. Chem. 2013, 288, 21448–21457. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Holley, C.L.; Schroder, K. The rOX-stars of inflammation: Links between the inflammasome and mitochondrial meltdown. Clin. Transl. Immunol. 2020, 9, e01109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; McIntyre, R.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [Green Version]
- Kowald, A.; Kirkwood, T.B.L. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc. Natl. Acad. Sci. USA 2011, 108, 10237–10242. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Won, J.-H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.-W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Reynolds, M.B.; Passalacqua, K.D.; Sexton, J.Z.; Abuaita, B.H.; O’Riordan, M.X.D. The Mitochondrial Fission Regulator DRP1 Controls Post-Transcriptional Regulation of TNF-α. Front. Cell. Infect. Microbiol. 2021, 10, 1–14. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4063562-11. [Google Scholar] [CrossRef] [PubMed]
- So, J.-S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Jiang, C.C.; Avery-Kiejda, K.; Wang, Y.F.; Thorne, R.F.; Zhang, X.D.; Hersey, P. Thapsigargin sensitizes human melanoma cells to TRAIL-induced apoptosis by up-regulation of TRAIL-R2 through the unfolded protein response. Carcinogenesis 2007, 28, 2328–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.K.A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Yano, H.; Bonifazi, A.; Xu, M.; Guthrie, D.A.; Schneck, S.N.; Abramyan, A.M.; Fant, A.D.; Hong, W.C.; Newman, A.H.; Shi, L. Pharmacological profiling of sigma 1 receptor ligands by novel receptor homomer assays. Neuropharmacology 2018, 133, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. The unfolded protein response: The dawn of a new field. Proc. Jpn. Acad. Ser. B 2015, 91, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 Receptor Chaperone at the ER-Mitochondrion Interface Mediates the Mitochondrion-ER-Nucleus Signaling for Cellular Survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Ainbinder, A.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 2015, 57, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabouny, R.; Fraunberger, E.; Geoffrion, M.; Ng, A.C.-H.; Baird, S.D.; Screaton, R.A.; Milne, R.; McBride, H.; Shutt, T. The Keap1–Nrf2 Stress Response Pathway Promotes Mitochondrial Hyperfusion Through Degradation of the Mitochondrial Fission Protein Drp1. Antioxid. Redox Signal. 2017, 27, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Menezes-Filho, S.L.; Assali, E.A.; Gonçalves, I.G.; Cabral-Costa, J.V.; Abreu, P.; Miller, N.; Nolasco, P.; Laurindo, F.R.M.; Bruni-Cardoso, A.; et al. Mitochondrial morphology regulates organellar Ca2+uptake and changes cellular Ca2+homeostasis. FASEB J. 2019, 33, 13176–13188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Z.-H.; Chen, M.-H.; Yang, J.; Leng, J.; Cao, G.-S.; Xin, G.-Z.; Liu, L.-F.; Kou, J.-P.; Liu, B.-L.; et al. Inhibition of Mitochondrial Fission and NOX2 Expression Prevent NLRP3 Inflammasome Activation in the Endothelium: The Role of Corosolic Acid Action in the Amelioration of Endothelial Dysfunction. Antioxid. Redox Signal. 2016, 24, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. In Macrophage Activation—Biology and Disease; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, A.C.; Madeira, N.; Morais, S.; Macedo, A.; Cruz, M.T.; Pereira, C.M.F. Mitochondria Fusion upon SERCA Inhibition Prevents Activation of the NLRP3 Inflammasome in Human Monocytes. Cells 2022, 11, 433. https://doi.org/10.3390/cells11030433
Pereira AC, Madeira N, Morais S, Macedo A, Cruz MT, Pereira CMF. Mitochondria Fusion upon SERCA Inhibition Prevents Activation of the NLRP3 Inflammasome in Human Monocytes. Cells. 2022; 11(3):433. https://doi.org/10.3390/cells11030433
Chicago/Turabian StylePereira, Ana Catarina, Nuno Madeira, Sofia Morais, António Macedo, Maria Teresa Cruz, and Cláudia M. F. Pereira. 2022. "Mitochondria Fusion upon SERCA Inhibition Prevents Activation of the NLRP3 Inflammasome in Human Monocytes" Cells 11, no. 3: 433. https://doi.org/10.3390/cells11030433
APA StylePereira, A. C., Madeira, N., Morais, S., Macedo, A., Cruz, M. T., & Pereira, C. M. F. (2022). Mitochondria Fusion upon SERCA Inhibition Prevents Activation of the NLRP3 Inflammasome in Human Monocytes. Cells, 11(3), 433. https://doi.org/10.3390/cells11030433