
Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy



Abstract
:
1. Introduction
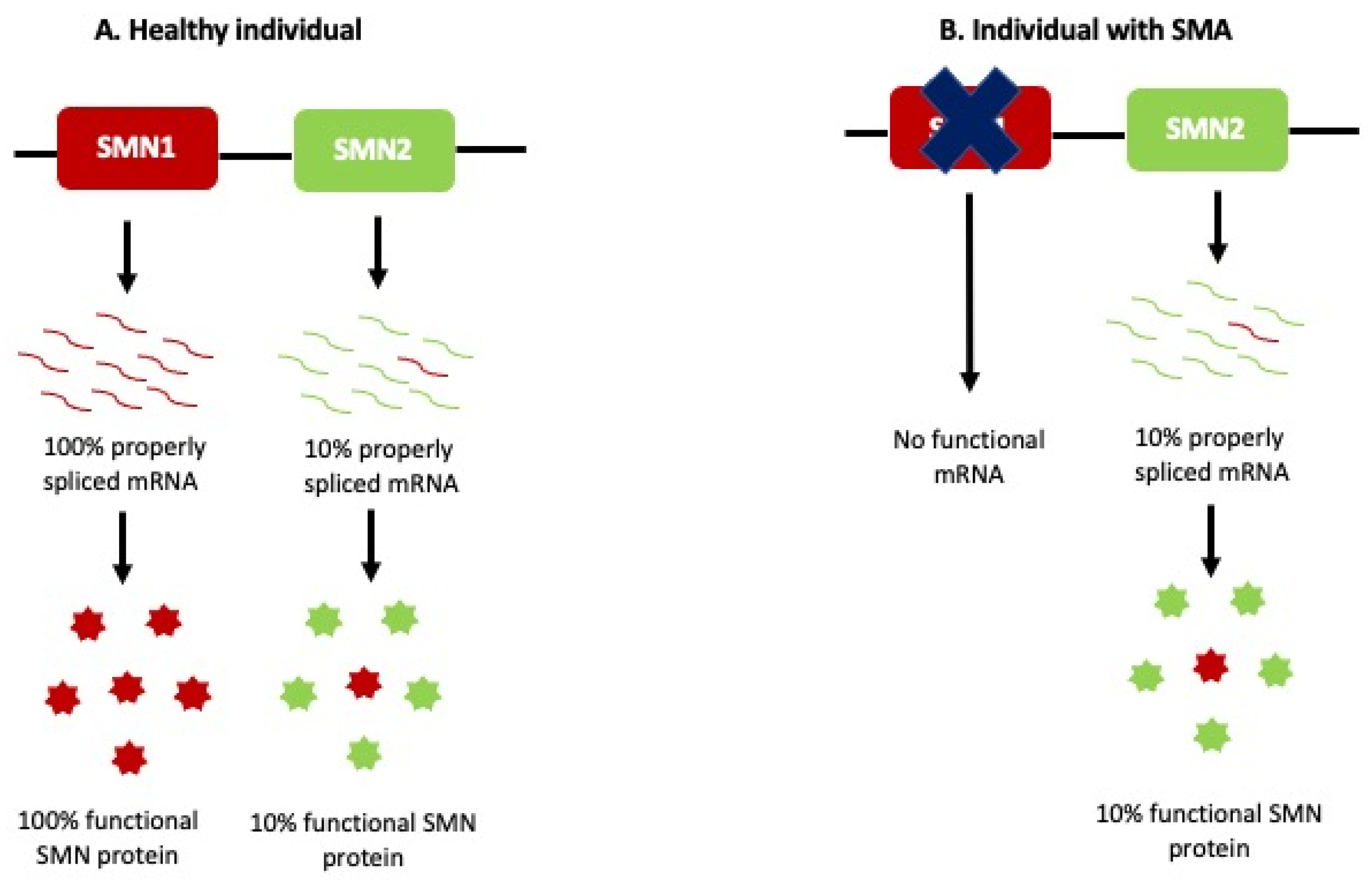
2. SMA: Background
3. SMA: Molecular Characteristics
4. Therapeutic Targets
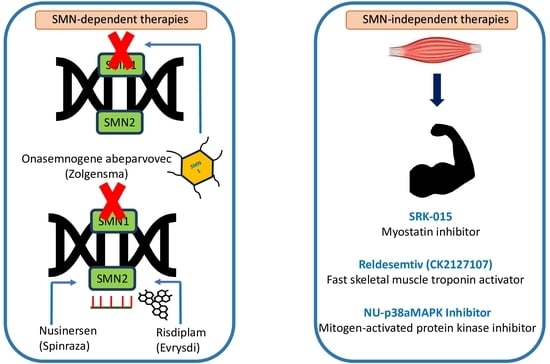
4.1. SMN-Dependent Therapies
4.1.1. Antisense Oligonucleotide (AON)-Based Treatment for SMA
4.1.2. Nusinersen—First Approved AON for SMA
4.1.3. Gene Therapy for SMA
4.1.4. Onasemnogene Abeparvovec: Approval
4.1.5. Small-Molecule Drugs
4.1.6. Risdiplam (Evrysdi): Small-Molecule Compound for SMA
4.1.7. Combination Therapies
4.2. SMN-Independent Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munsat, T.L.; Davies, K.E. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord. 1992, 2, 423–428. [Google Scholar] [CrossRef]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Son, H.W.; Yokota, T. Recent Advances and Clinical Applications of Exon Inclusion for Spinal Muscular Atrophy. Methods Mol. Biol. 2018, 1828, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Oskoui, M.; Kaufmann, P. Spinal muscular atrophy. Neurotherapeutics 2008, 5, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Schrank, B.; Gotz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [Green Version]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs 2019, 79, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Risdiplam: First Approval. Drugs 2020, 80, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubowitz, V. Ramblings in the history of spinal muscular atrophy. Neuromuscul. Disord. 2009, 19, 69–73. [Google Scholar] [CrossRef]
- Munsat, T.L. Workshop report: International SMA collaboration. Neuromuscul. Disord. 1991, 1, 81. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Melki, J.; Lefebvre, S.; Burglen, L.; Burlet, P.; Clermont, O.; Millasseau, P.; Reboullet, S.; Benichou, B.; Zeviani, M.; Paslier, D.L.; et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science 1994, 264, 1474–1477. [Google Scholar] [CrossRef]
- Rochette, C.F.; Gilbert, N.; Simard, L.R. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef]
- Lefebvre, S.; Sarret, C. Pathogenesis and therapeutic targets in spinal muscular atrophy (SMA). Arch. Pediatrics 2020, 27, S3–S7. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J 1996, 15, 3555–3565. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.; Almeida, F.; Calapez, A.; Lafarga, M.; Berciano, M.T.; Carmo-Fonseca, M. The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J. Cell Biol. 1999, 147, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, S.J.; Battle, D.J.; Dreyfuss, G. Molecular functions of the SMN complex. J. Child Neurol. 2007, 22, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Burlet, P.; Huber, C.; Bertrandy, S.; Ludosky, M.A.; Zwaenepoel, I.; Clermont, O.; Roume, J.; Delezoide, A.L.; Cartaud, J.; Munnuch, A.; et al. The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum. Mol. Genet. 1998, 7, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [Green Version]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H. The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet. 1997, 6, 1205–1214. [Google Scholar] [CrossRef]
- Boido, M.; Vercelli, A. Neuromuscular junctions as key contributors and therapeutic targets in spinal muscular atrophy. Front. Neuroanat. 2016, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Park, G.H.; Maeno-Hichiki, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Gen. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, S.; Van Alstyne, M.; Simon, C.M.; Mentis, G.Z.; Pellizzoni, L. SMN controls neuromuscular junction integrity through U7 snRNP. Biorxiv 2021. [Google Scholar] [CrossRef]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Shishimorova, M.; Cao, L.C.; Gangwani, L.; Singh, R.N. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009, 6, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottesen, E.W. ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.H.; Schray, R.C.; Patterson, C.A.; Ayitey, S.O.; Tallent, M.K.; Lutz, G.J. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J. Neurosci. 2009, 29, 7633–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Mateson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Sardone, V.; Zhou, H.; Muntoni, F.; Ferlini, A.; Falzarano, M.S. Antisense Oligonucleotide-Based Therapy for Neuromuscular Disease. Molecules 2017, 22, 563. [Google Scholar] [CrossRef] [Green Version]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Summerton, J.E. Invention and Early History of Morpholinos: From Pipe Dream to Practical Products. Methods Mol. Biol. 2017, 1565, 1–15. [Google Scholar] [CrossRef]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H.M. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene Ther. 2013, 24, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Salani, S.; Ruepp, M.D.; Rizzo, F.; Ruggieri, M.; Zanetta, C.; Brajkovic, S.; Moulton, H.M.; Muehlemann, O.; et al. Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Delta7 mouse model phenotype. Clin. Ther. 2014, 36, 340–356.e5. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today 2020, 56, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Park, J.; Ryu, J.; Kim, K.A.; Lee, H.J.; Bahn, J.H.; Han, K.; Choi, E.Y.; Lee, K.S.; Kwon, H.Y.; Choi, S.Y. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J. Gen. Virol 2002, 83 (Pt 5), 1173–1181. [Google Scholar] [CrossRef]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J.A. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates with Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol. Ther. Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, C.A.; Wood, M.J. Cell penetrating peptide delivery of splice directing oligonucleotides as a treatment for Duchenne muscular dystrophy. Curr. Pharm. Des. 2013, 19, 2948–2962. [Google Scholar] [CrossRef] [PubMed]
- Boisguerin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Jarver, P.; O’Donovan, L.; Gait, M.J. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2014, 24, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Betts, C.; Saleh, A.F.; Ivanova, G.D.; Lee, H.; Seow, Y.; Kim, D.; Gait, M.J.; Wood, M.J.A. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol. Ther. 2010, 18, 819–827. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Merritt, T.; Seow, Y.; Ashraf, S.; Wang, Q.; Boutilier, J.; Wood, M.J.A. Functional rescue of dystrophin-deficient mdx mice by a chimeric peptide-PMO. Mol. Ther. 2010, 18, 1822–1829. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Southwell, A.L.; Kordasiewicz, H.B.; Langbehn, D.; Skotte, N.H.; Parsons, M.P.; Villanueva, E.B.; Caron, N.S.; Ostergaard, M.E.; Anderson, L.M.; Xie, Y.; et al. Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci. Transl. Med. 2018, 10, eaar3959. [Google Scholar] [CrossRef] [Green Version]
- Aslesh, T.; Yokota, T. Development of Antisense Oligonucleotide Gapmers for the Treatment of Huntington’s Disease. Methods Mol. Biol. 2020, 2176, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kalbfuss, B.; Mabon, S.A.; Misteli, T. Correction of alternative splicing of tau in frontotemporal dementia and parkinsonism linked to chromosome. J. Biol. Chem. 2001, 276, 42986–42993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Goodkey, K.; Aslesh, T.; Maruyama, R.; Yokota, T. Nusinersen in the Treatment of Spinal Muscular Atrophy. Methods Mol. Biol. 2018, 1828, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; Vivo, D.C.D.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; Vivo, D.C.D.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; Vivo, D.C.D.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: Final report of a phase 2, open-label, multicentre, dose-escalation study. Lancet Child Adolesc. Health 2021, 5, 491–500. [Google Scholar] [CrossRef]
- Sheikh, O.; Yokota, T. Restoring Protein Expression in Neuromuscular Conditions: A Review Assessing the Current State of Exon Skipping/Inclusion and Gene Therapies for Duchenne Muscular Dystrophy and Spinal Muscular Atrophy. BioDrugs 2021, 35, 389–399. [Google Scholar] [CrossRef]
- Wurster, C.D.; Winter, B.; Wollinsky, K.; Ludolph, A.C.; Uzelac, Z.; Witzel, S.; Schocke, M.; Schneider, R.; Kocak, T. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J. Neurol. 2019, 266, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Sansone, V.A.; Pirola, A.; Albamonte, E.; Pane, M.; Lizio, A.; D’Amico, A.; Catteruccia, M.; Cutrera, R.; Bruno, C.; Pedemonte, M.; et al. Respiratory Needs in Patients with Type 1 Spinal Muscular Atrophy Treated with Nusinersen. J. Pediatrics 2020, 219, 223–228 e224. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, E.; Marais, T.; Chatauret, N.; Benkhelifa-Ziyyat, S.; Duque, S.; Ravassard, P.; Carcenac, R.; Astord, S.; Pereira de Moura, A.; Voit, T.; et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011, 20, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Ferraiuolo, L.; Schmelzer, L.; Braun, L.; McGovern, V.; Likhite, S.; Michels, O.; Govoni, A.; Fitzgerald, J.; Morales, P.; et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: A dose-response study in mice and nonhuman primates. Mol. Ther. 2015, 23, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Armbruster, N.; Lattanzi, A.; Jeavons, M.; Wittenberghe, L.V.; Gjata, B.; Marais, T.; Martin, S.; Vignaud, A.; Voit, T.; Mavilio, F.; et al. Efficacy and biodistribution analysis of intracerebroventricular administration of an optimized scAAV9-SMN1 vector in a mouse model of spinal muscular atrophy. Mol. Ther. Methods Clin. Dev. 2016, 3, 16060. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants With SMA Type 1 Receiving Single-Dose Gene R.Replacement Therapy. Pediatrics Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Al-Zaidy, S.; Pickard, A.S.; Kotha, K.; Alfano, L.N.; Lowes, L.; Paul, G.; Church, K.; Lehman, K.; Sproule, D.M.; Dabbous, O.; et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatrics Pulmonol. 2019, 54, 179–185. [Google Scholar] [CrossRef]
- Day, J.W.; Chiriboga, C.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Iannaccone, S.T.; Kuntz, N.L.; Pena, L.D.M.; Schultz, M.; et al. AVXS-101 gene replacement therapy (GRT) for spinal muscular atrophy type 1 (SMA1): Pivotal phase 3 study (STR1VE) update. Am. J. Respir. Crit. Care Med. 2019, 199, A2789. [Google Scholar]
- Agency, E.M. Zolgensma: European Public Assessment Report. 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/zolgensma-epar-public-assessment-report_en.pdf (accessed on 28 November 2021).
- Van Alstyne, M.; Tattoli, I.; Delestree, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat. Neurosci. 2021, 24, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Woll, M.G. Compounds for Treating Spinal Muscular Atrophy. WO2013101974A1, 4 July 2013. [Google Scholar]
- Lee, C.-S.; Karp, G.M.; Koyama, H.; Ratni, H. Compounds for Treating Spinal Muscular Atrophy. WO2013130689A1, 6 September 2013. [Google Scholar]
- Ratni, H.; Karp, G.M.; Weetall, M.; Naryshkin, N.A.; Paushkin, S.V.; Chen, K.S.; McCarthy, K.D.; Qi, H.; Turpoff, A.; Woll, M.G.; et al. Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine To Treat Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6086–6100. [Google Scholar] [CrossRef]
- Calder, A.N.; Androphy, E.J.; Hodgetts, K.J. Small Molecules in Development for the Treatment of Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 10067–10083. [Google Scholar] [CrossRef] [Green Version]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; Berg, L.H.V.D.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2020, 1, CD006282. [Google Scholar] [CrossRef]
- Tiziano, F.D.; Lomastro, R.; Abiusi, E.; Pasanisi, M.B.; Pietro, L.D.; Fiori, S.; Baranello, G.; Angelini, C.; Soraru, G.; Gaiani, A.; et al. Longitudinal evaluation of SMN levels as biomarker for spinal muscular atrophy: Results of a phase IIb double-blind study of salbutamol. J. Med. Genet. 2019, 56, 293–300. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Servais, L.B.G.; Masson, R.; Mazukiewicz-Beldzinska, M.; Rose, K.; Vlodavets, D.; Xiong, H.; Zanotelli, E.; El-Khairi, M.; Fuerst-Recktenwald, S.; Gerber, M.; et al. FIREFISH Part 2: Efficacy and safety of risdiplam (RG7916) in infants with type 1 spinal muscular atrophy (SMA). Neurology 2020, 94 (Suppl. S15), 1302. [Google Scholar]
- Kakazu, J.; Walker, N.L.; Babin, K.C.; Trettin, K.A.; Lee, C.; Sutker, P.B.; Kaye, A.M.; Kaye, A.D. Risdiplam for the use of spinal muscular atrophy. Orthop. Rev. Pavia 2021, 13, 25579. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Collins, E.; Lewis, L.; Guntrum, D.; Eichinger, K.; Voter, K.; Abdel-Hamid, H.Z.; Ciafaloni, E. Combination therapy with nusinersen and AVXS-101 in SMA type. Neurology 2019, 93, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Rao, V.K.; Arya, K.; Kuntz, N.L.; DiDonato, C.J.; Napchan-Pomerantz, G.; Agarwal, A.; Stefans, V.; Katsuno, M.; Veerapandiyan, A. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve 2020, 62, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, S.E.; Curry, C.; Gross, B.; Rubin, A.I.; Linn, R.; Yum, S.W.; Kichula, E.A. Clinical Course in a Patient With Spinal Muscular Atrophy Type 0 Treated With Nusinersen and Onasemnogene Abeparvovec. J. Child Neurol. 2020, 35, 717–723. [Google Scholar] [CrossRef]
- Reldesemtiv (Formerly CK-2127107). Available online: https://smanewstoday.com/ck-2127107-ck-107/ (accessed on 25 June 2021).
- The Role of p38 MAPK Activation in Spinal Muscular Atrophy (Pellizzoni, L); National Institute of Health (NIH), Columbia University (N.Y.): New York, NY, USA, 2018.
- Deguise, M.O.; De Repentigny, Y.; McFall, E.; Auclair, N.; Sad, S.; Kothary, R. Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum. Mol. Genet. 2017, 26, 801–819. [Google Scholar] [CrossRef] [Green Version]
- Khairallah, M.T.; Astroski, J.; Custer, S.K.; Androphy, E.J.; Franklin, C.L.; Lorson, C.L. SMN deficiency negatively impacts red pulp macrophages and spleen development in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 932–941. [Google Scholar] [CrossRef]
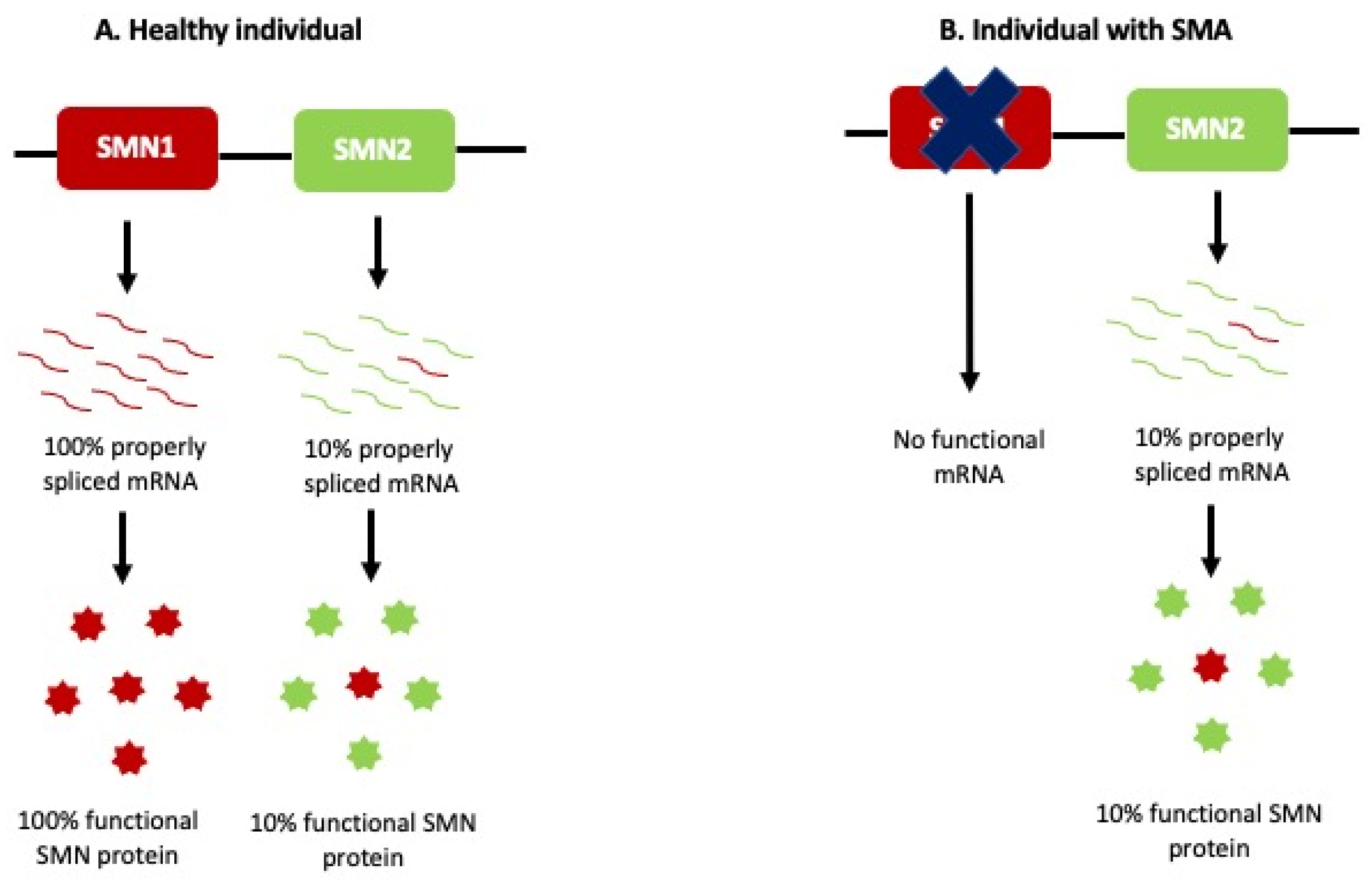

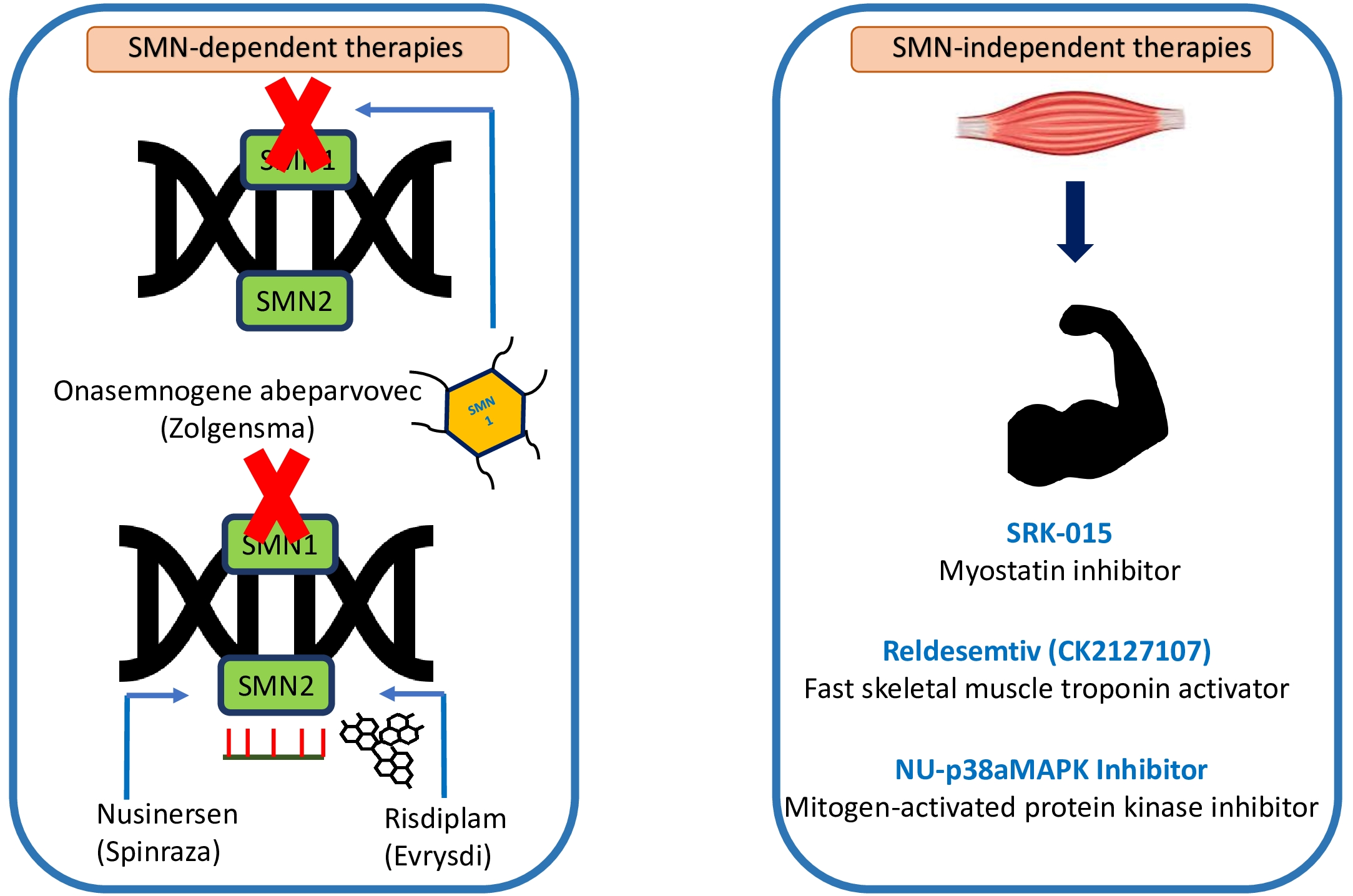{kind=link}
{kind=link}
| Treatment Name | Type | Manufacturer | Science | Status |
|---|---|---|---|---|
| Nusinersen (brand name Spinraza) | Antisense oligonucleotide | Biogen | AON with MOE chemistry targeting SMN2 ISS-N1 that promotes exon 7 inclusion | Approved by FDA in 2016 |
| Onasemnogene abeparvovec (brand name Zolgensma) | Gene therapy | Novartis, AveXis | scAAV9-SMN under the control of a CBA promoter | Approved by FDA in 2019 |
| Risdiplam (brand name Evrysdi) | Small molecule | Roche | SMN2 splicing modifier administered orally | Approved by FDA in 2020 |
| SMA Type | Age of Onset | Maximum Age of Survival | SMN2 Copy Number | Alternate Name |
|---|---|---|---|---|
| 0 | Prenatal | <1 month | 1 | - |
| 1 | 0–6 months | <2 years | 2 | Werdnig–Hoffman disease |
| 2 | <18 months | >2 years | 3,4 | Dubowitz disease |
| 3a (juvenile onset) | 18 months–3 years | Adult | 3,4 | Kugelberg–Welander disease |
| 3b | >3 years | Adult | 4 | Kugelberg–Welander disease |
| 4 | > 21 years | Adult | 4–8 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslesh, T.; Yokota, T. Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells 2022, 11, 417. https://doi.org/10.3390/cells11030417
Aslesh T, Yokota T. Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells. 2022; 11(3):417. https://doi.org/10.3390/cells11030417
Chicago/Turabian StyleAslesh, Tejal, and Toshifumi Yokota. 2022. "Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy" Cells 11, no. 3: 417. https://doi.org/10.3390/cells11030417
APA StyleAslesh, T., & Yokota, T. (2022). Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells, 11(3), 417. https://doi.org/10.3390/cells11030417