
Macrophage-Specific MCPIP1/Regnase-1 Attenuates Kidney Ischemia-Reperfusion Injury by Shaping the Local Inflammatory Response and Tissue Regeneration


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
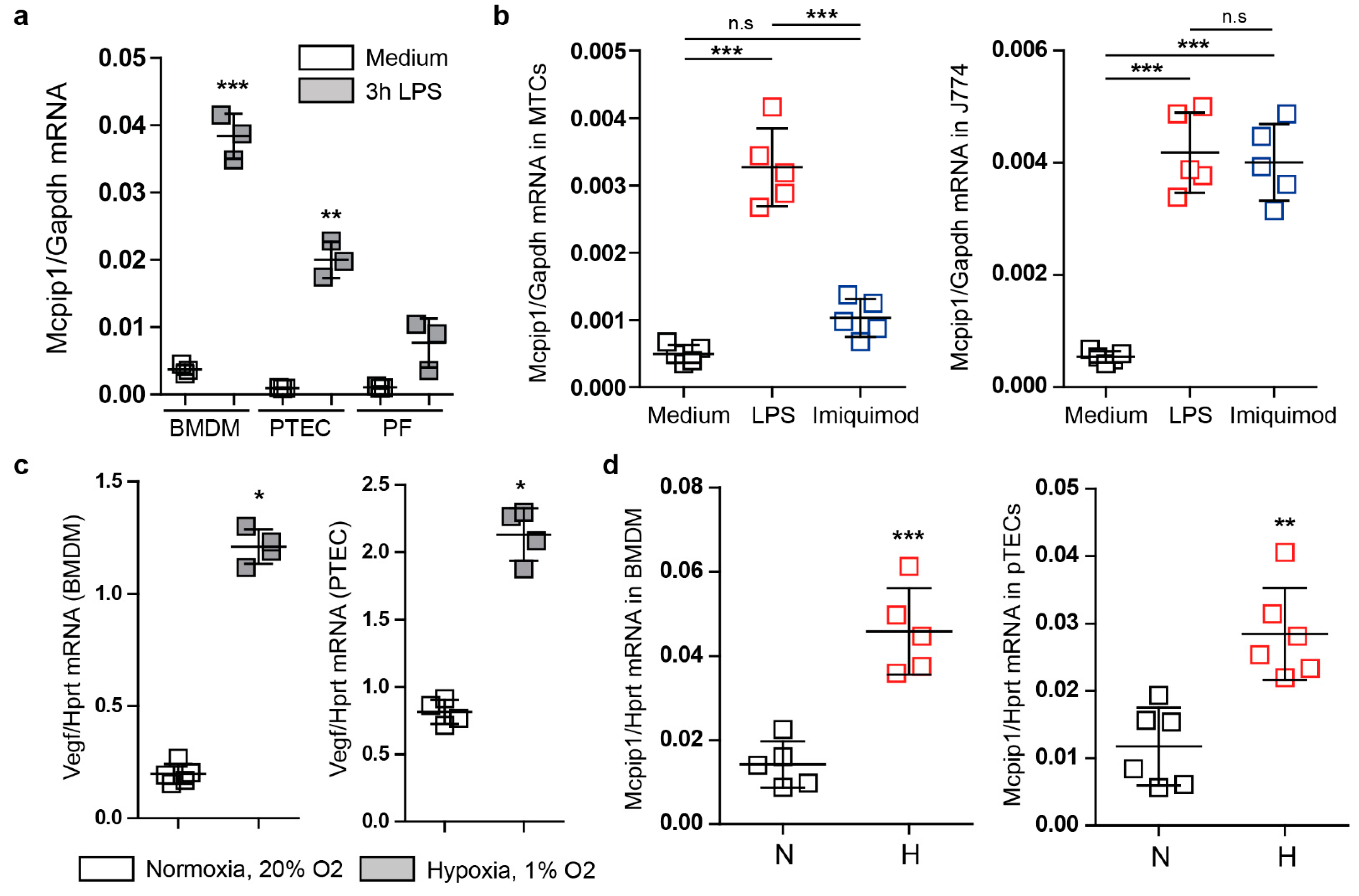
3.1. Mcpip1 Is Induced in Macrophages and Renal Tubular Cells In Vitro following Oxidative Stress
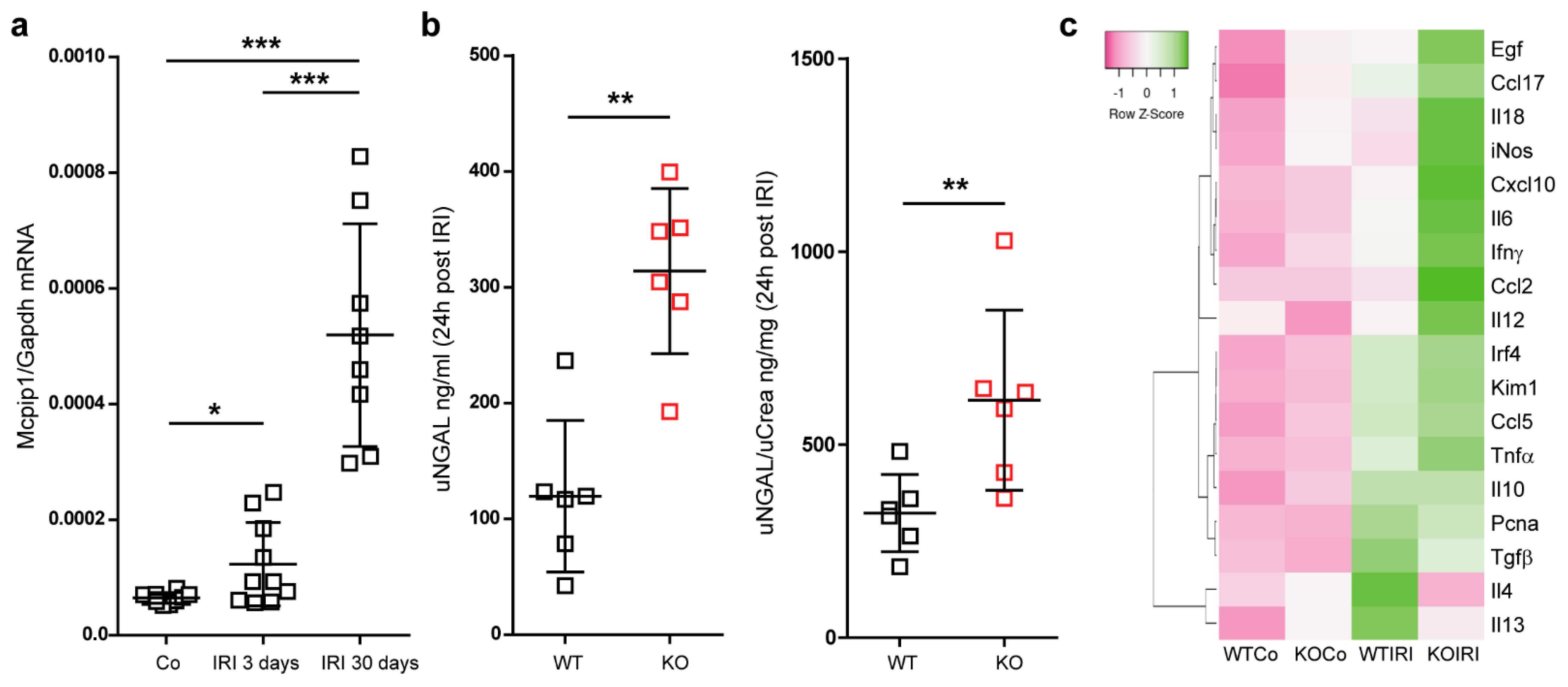
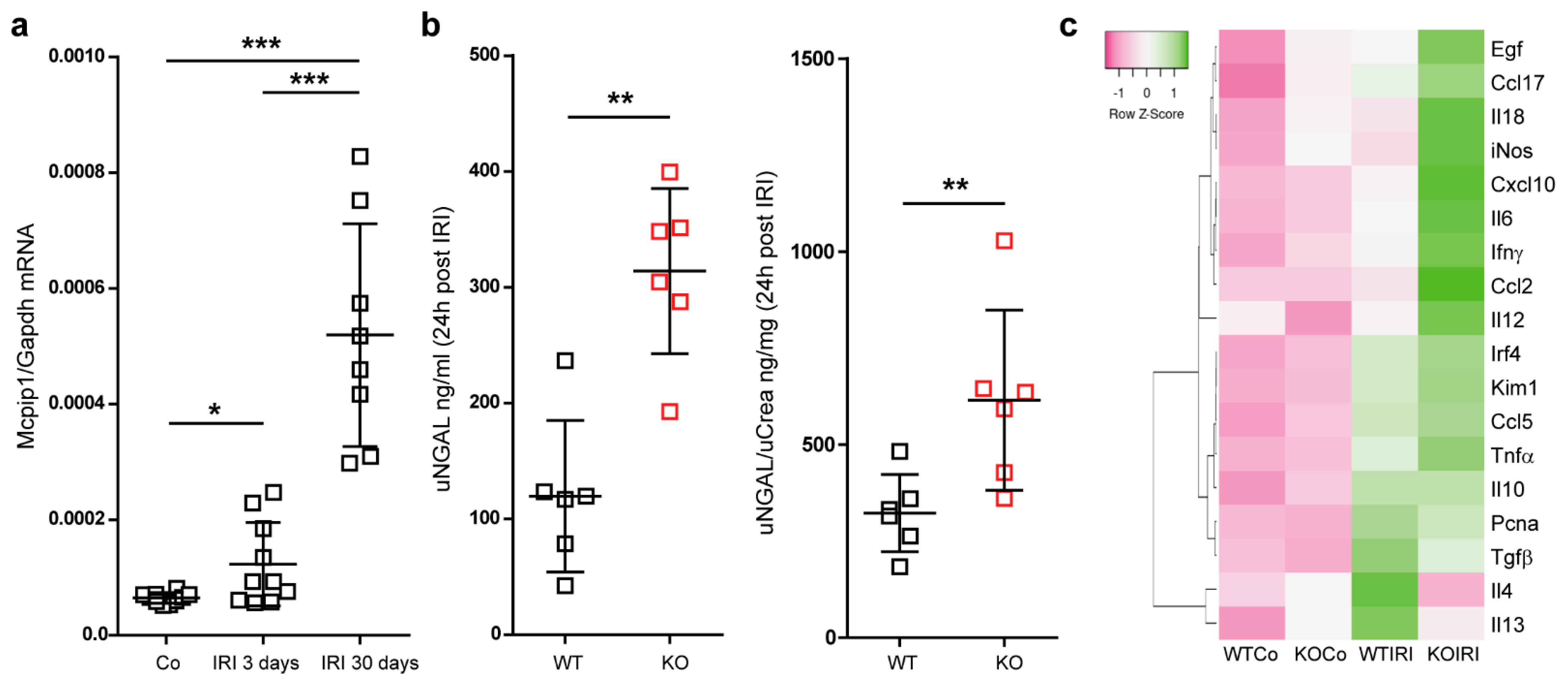
3.2. Mcpip1 Is Induced in the Kidney Tissue following IRI and Macrophage Expression of Mcpip1 Regulates Acute Inflammation upon Kidney Ischemic Damage
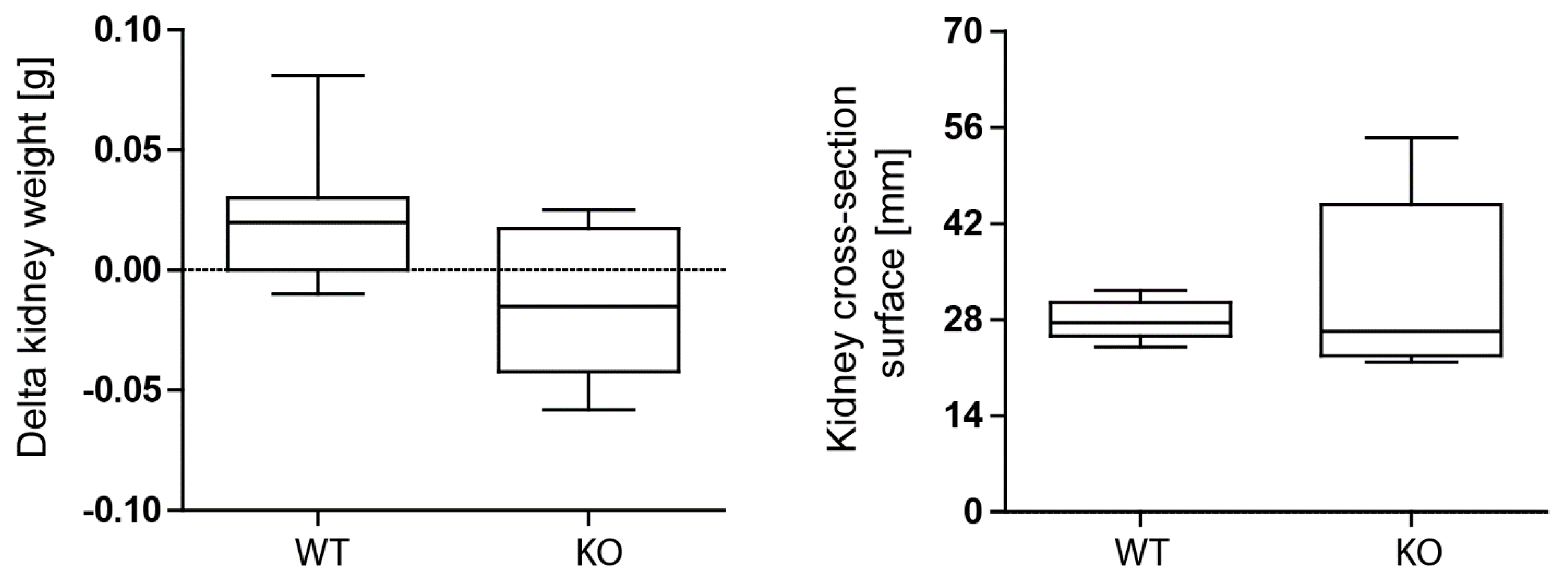
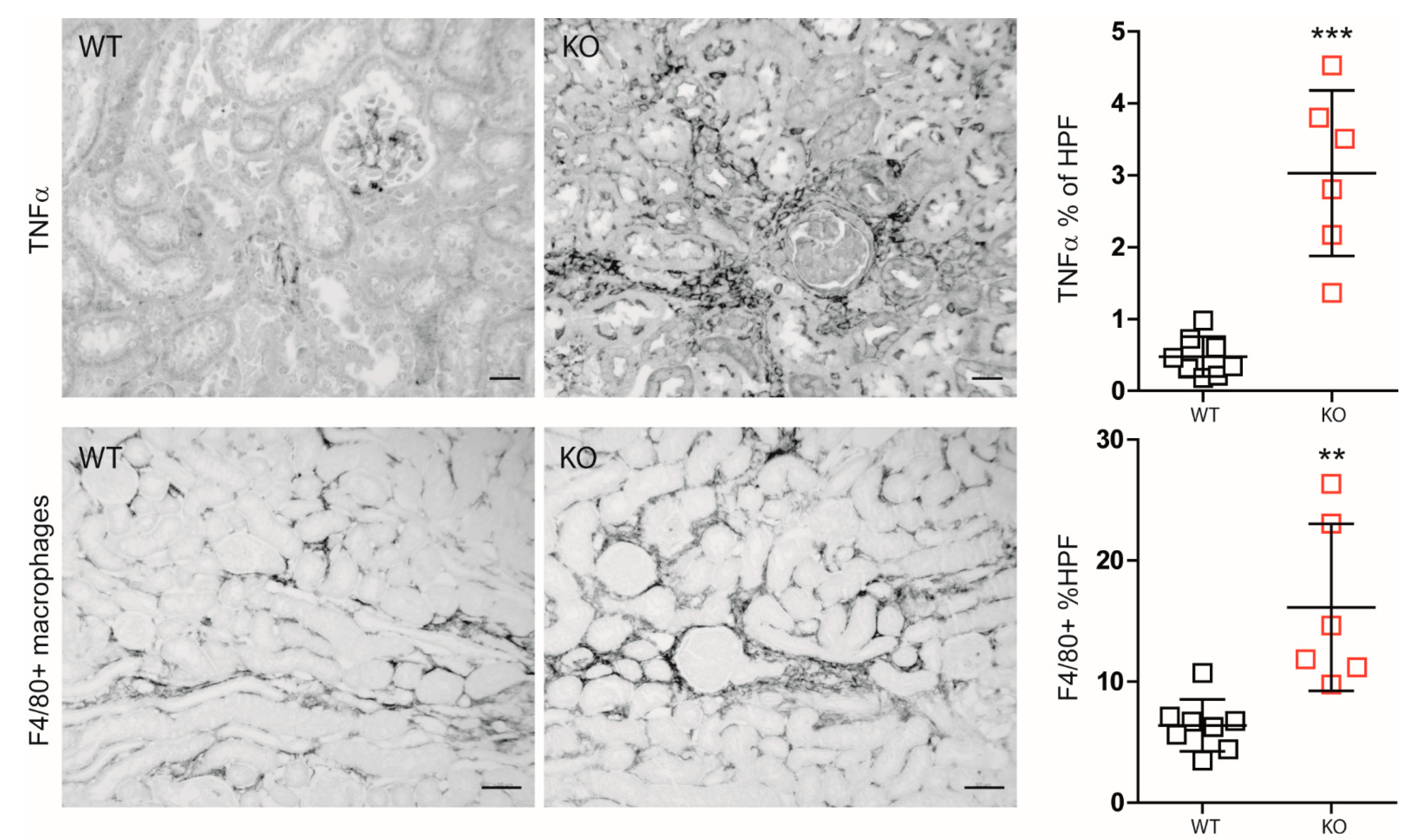
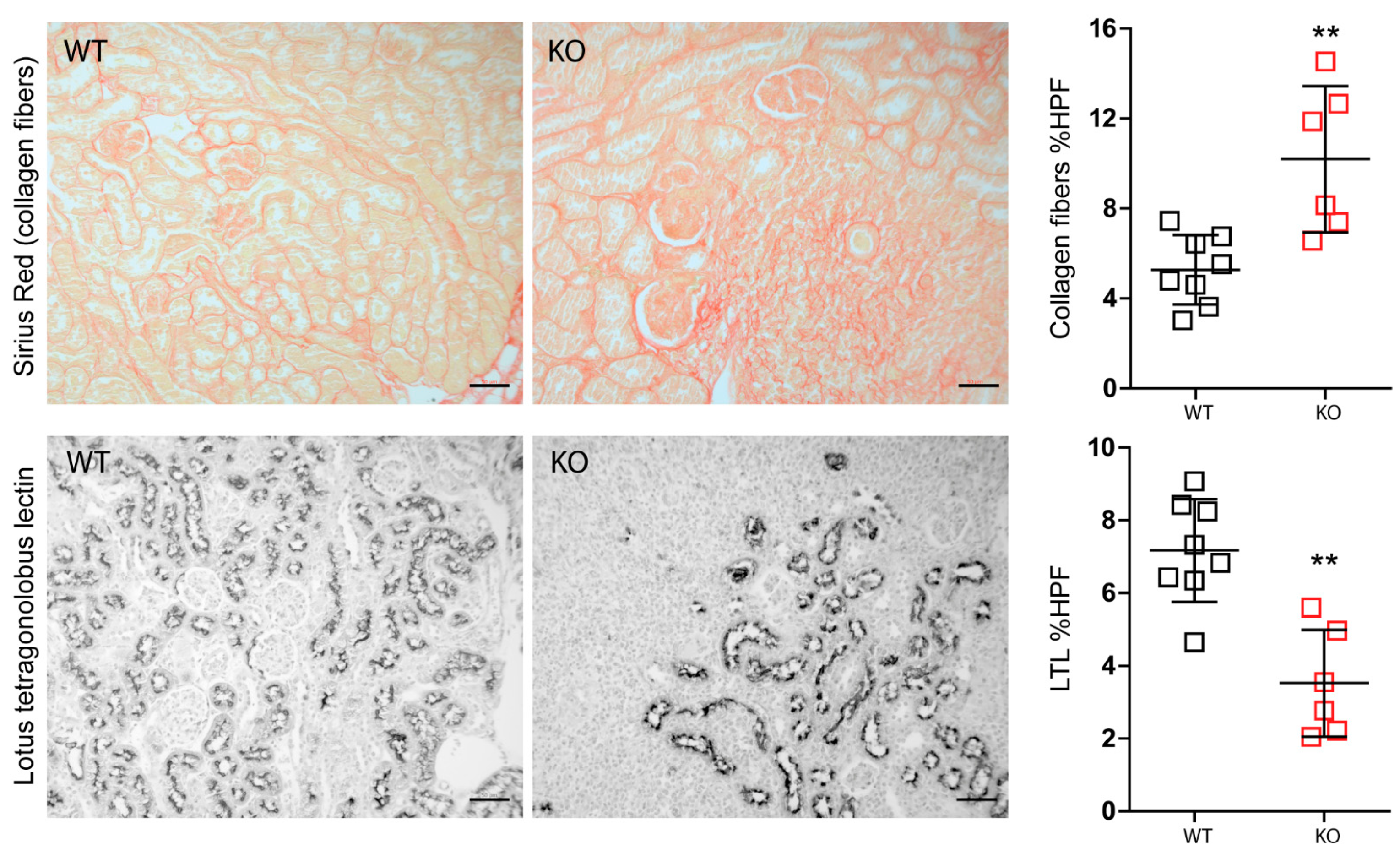
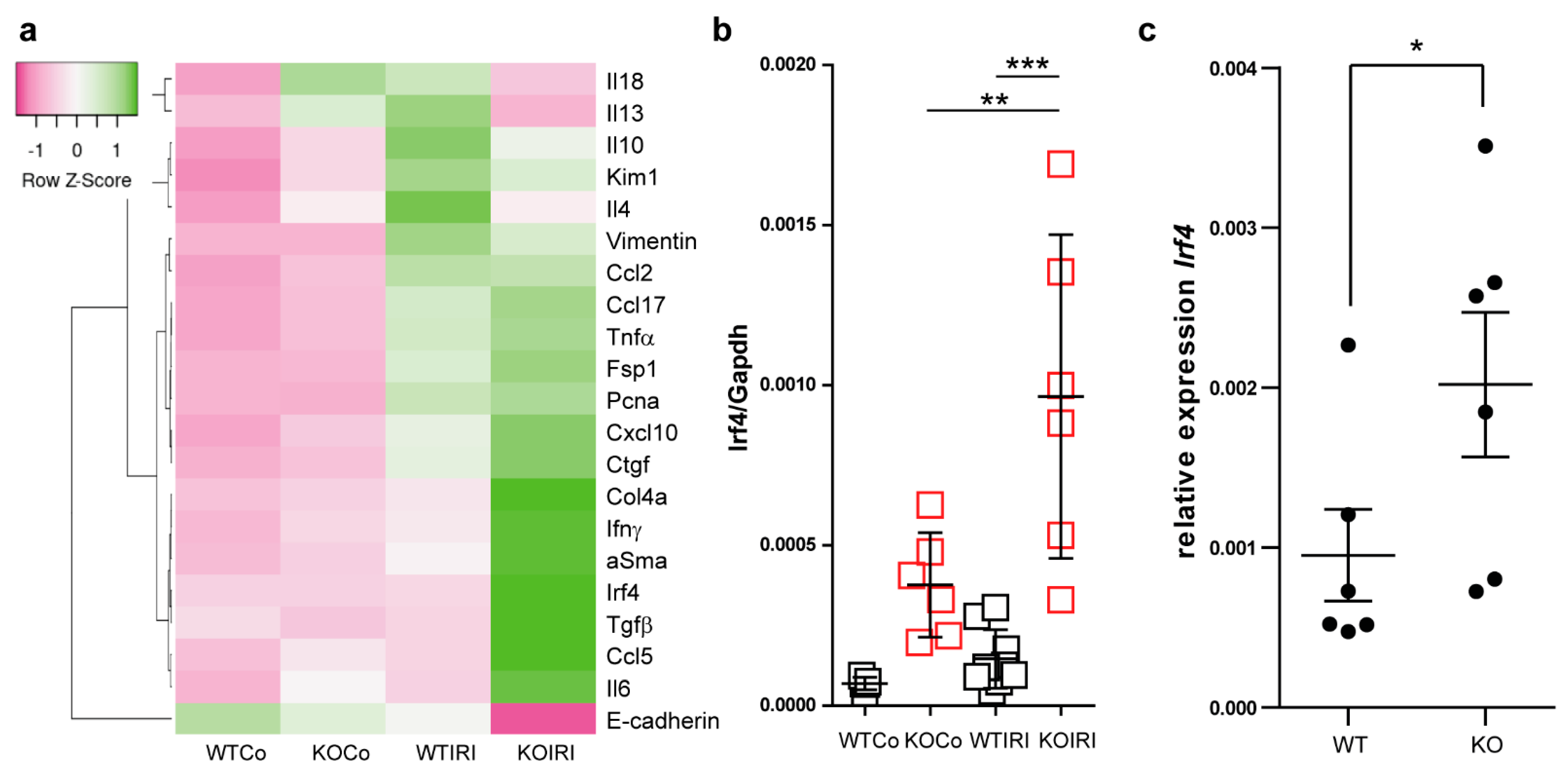
3.3. Macrophage MCPIP1 Promotes Resolution of the Chronic Inflammatory Response Associated with Aggravated Kidney Injury and Fibrosis after IRI
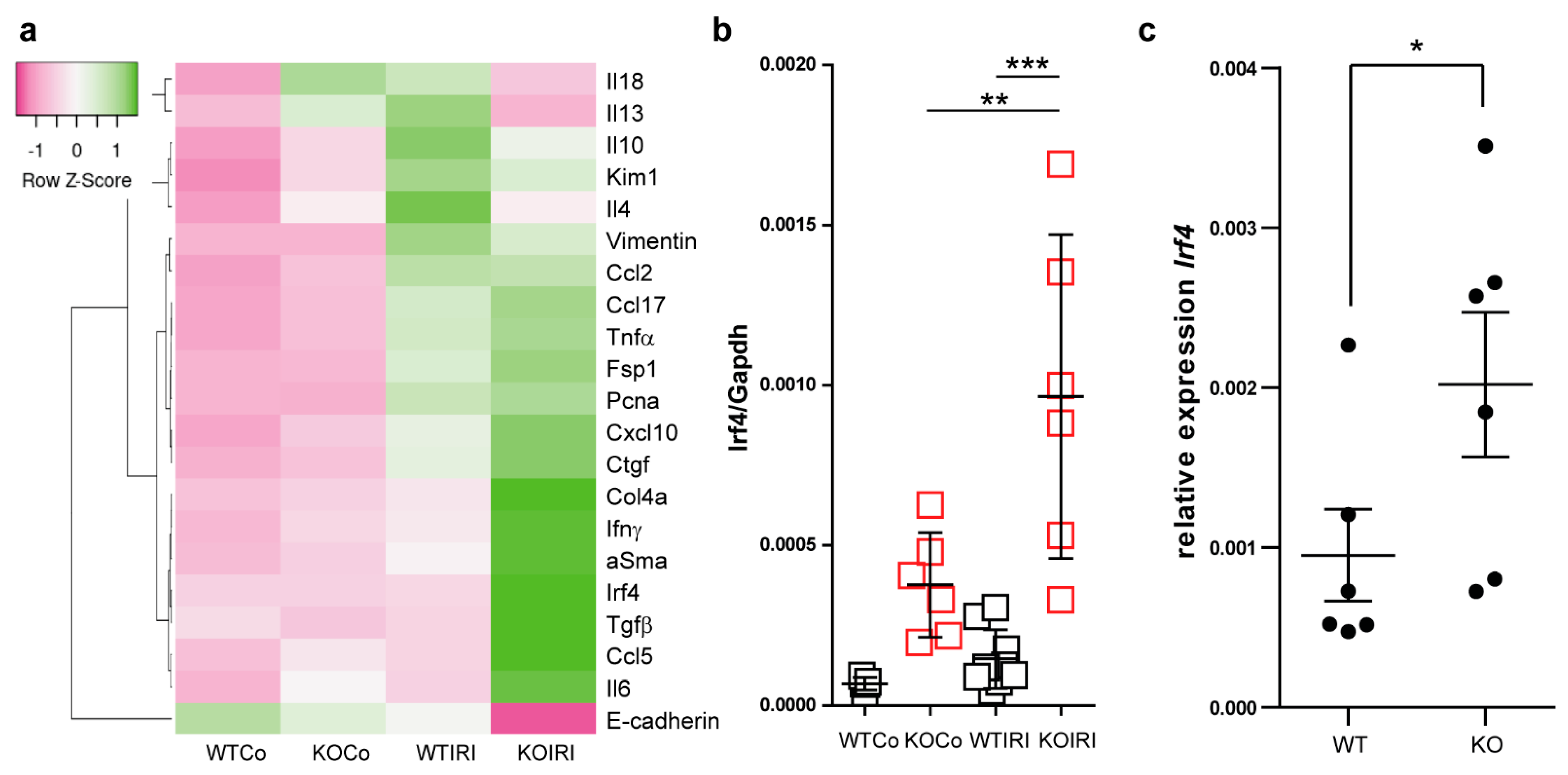
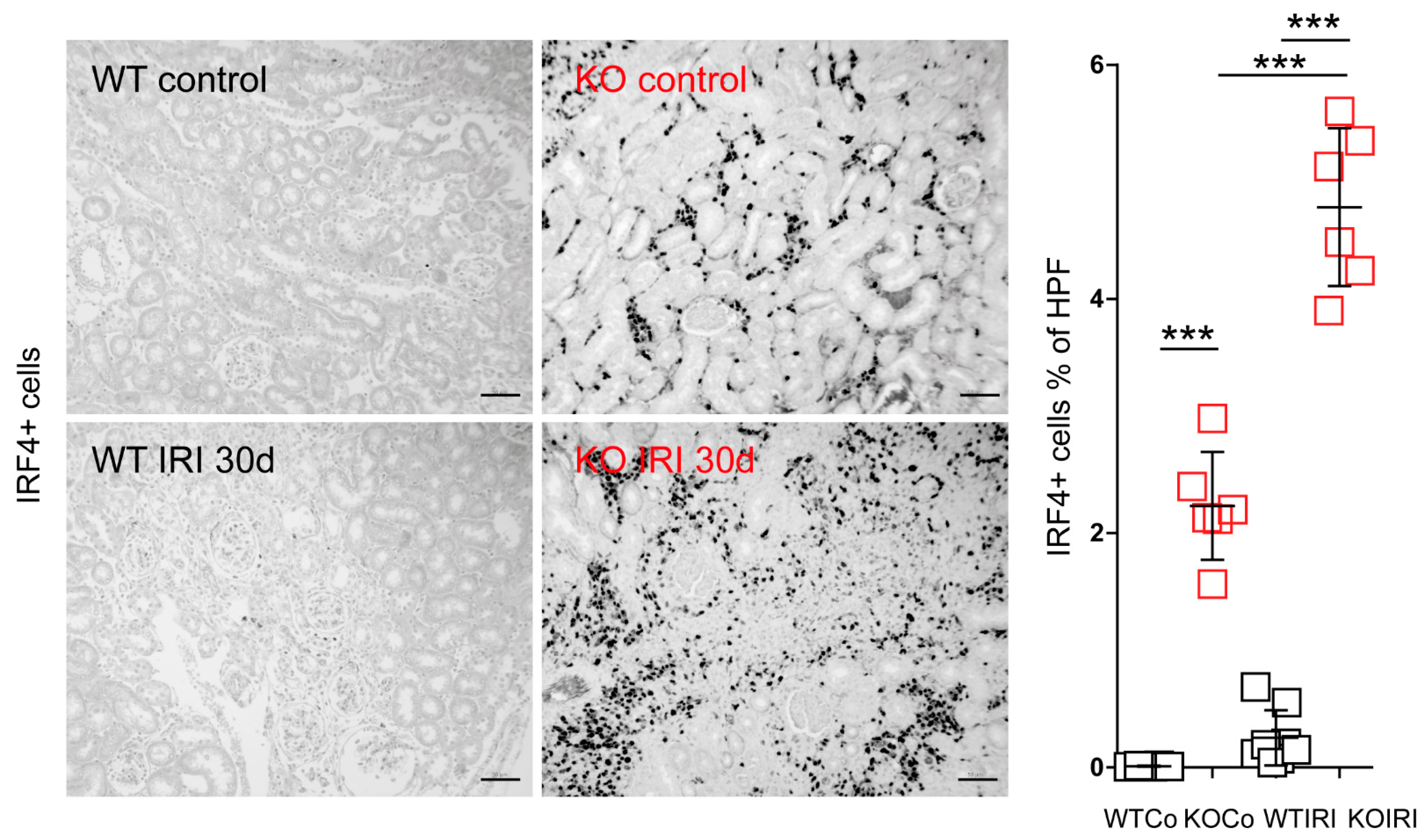
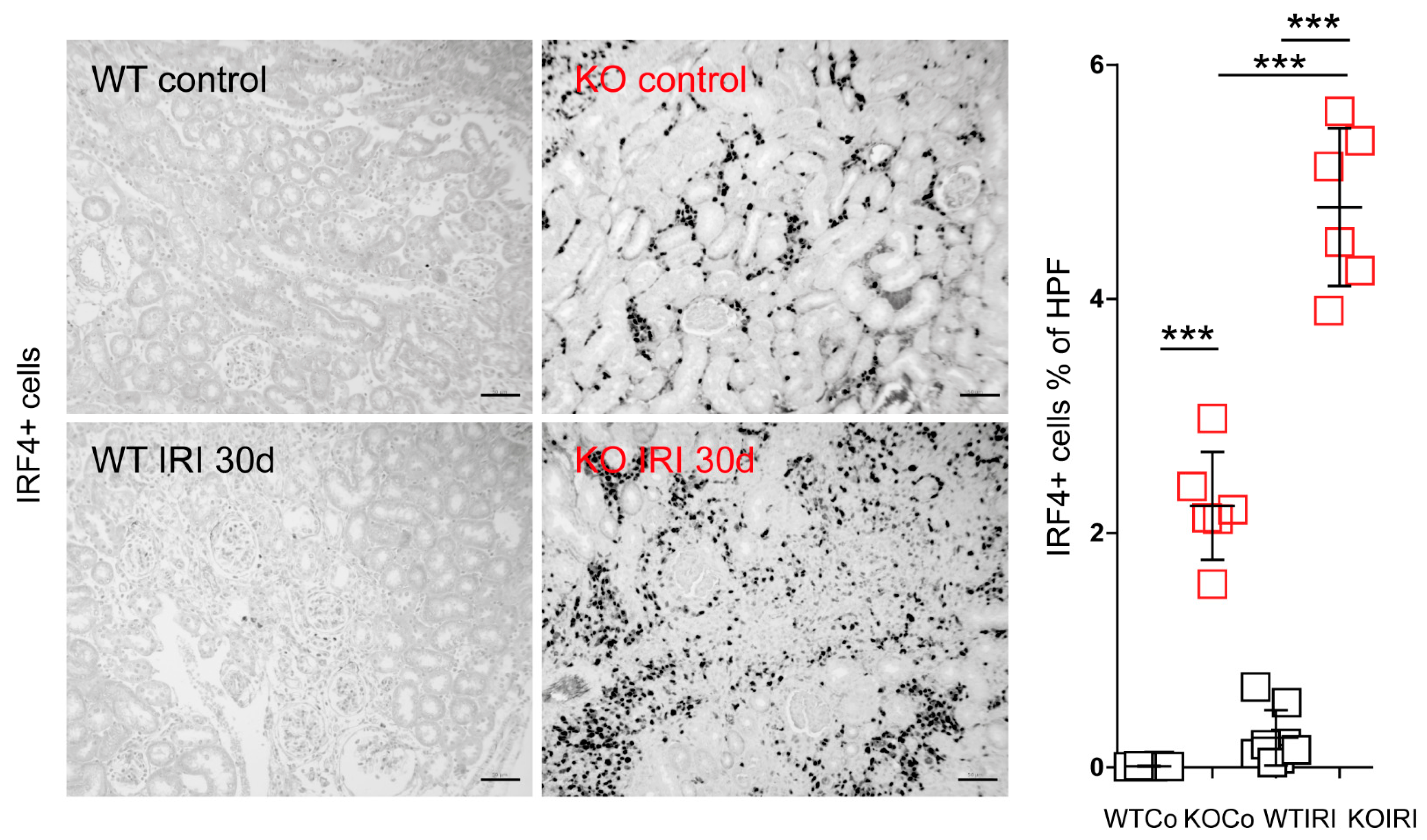
3.4. MCPIP1 Regulates the Number of IRF4+ Cells in the Kidney Postischemic Injury
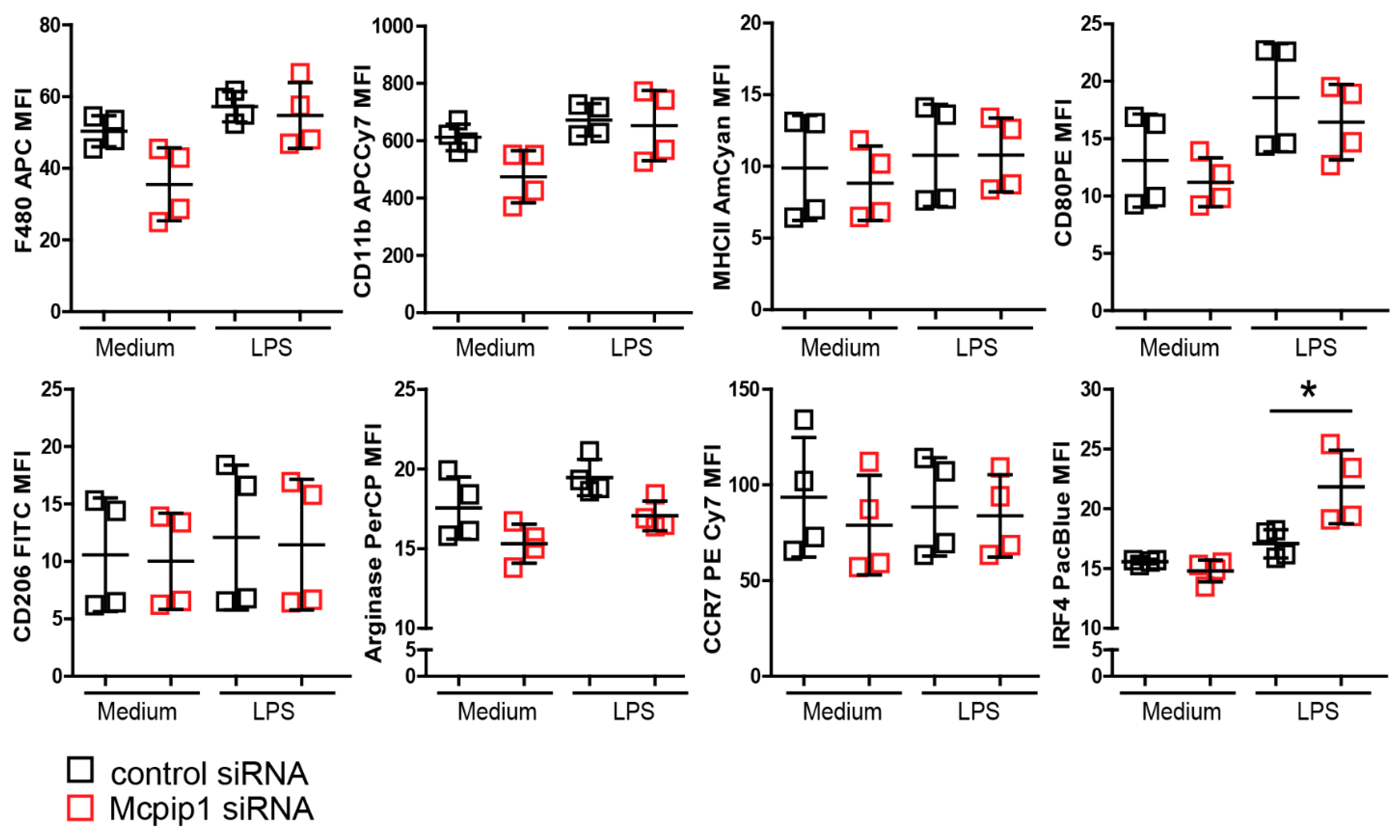
3.5. MCPIP1 Modulates the Macrophage Phenotype upon Inflammation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucaloiu, I.D.; Kirchner, H.L.; Norfolk, E.R.; Hartle, J.E., 2nd; Perkins, R.M. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012, 81, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, R.K.; Hsu, C.-Y. The Role of Acute Kidney Injury in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.C.-T.; Zhang, Y.-Y.; Chan, M.K.-K.; Lam, W.W.-Y.; Chung, J.Y.-F.; Kang, W.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. The Emerging Role of Innate Immunity in Chronic Kidney Diseases. Int. J. Mol. Sci. 2020, 21, 4018. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Zhang, Y.-G. Kidney and innate immunity. Immunol. Lett. 2017, 183, 73–78. [Google Scholar] [CrossRef]
- Anders, H.-J.; Lech, M. NOD-like and Toll-like receptors or inflammasomes contribute to kidney disease in a canonical and a non-canonical manner. Kidney Int. 2013, 84, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-edged Sword. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2019, 67, 663–681. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the maintenance of homeostasis. Cell. Mol. Immunol. 2021, 18, 579–587. [Google Scholar] [CrossRef]
- Lech, M.; Gröbmayr, R.; Ryu, M.; Lorenz, G.; Hartter, I.; Mulay, S.R.; Susanti, H.E.; Kobayashi, K.S.; Flavell, R.A.; Anders, H.-J. Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 2014, 25, 292–304. [Google Scholar] [CrossRef]
- Lin, S.L.; Castaño, A.P.; Nowlin, B.T.; Lupher, M.L.J.; Duffield, J.S. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J. Immunol. 2009, 183, 6733–6743. [Google Scholar] [CrossRef] [Green Version]
- Lech, M.; Gröbmayr, R.; Weidenbusch, M.; Anders, H.-J. Tissues use resident dendritic cells and macrophages to maintain homeostasis and to regain homeostasis upon tissue injury: The immunoregulatory role of changing tissue environments. Mediat. Inflamm. 2012, 2012, 951390. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterlé, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol. 2012, 23, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.-J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta 2013, 1832, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Fu, S.; Peng, W.; Rao, Z. MCP-1-induced protein-1, an immune regulator. Protein Cell 2012, 3, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Leppek, K.; Schott, J.; Reitter, S.; Poetz, F.; Hammond, M.C.; Stoecklin, G. Roquin promotes constitutive mrna decay via a conserved class of stem-loop recognition motifs. Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, H.; Takeuchi, O.; Teraguchi, S.; Matsushita, K.; Uehata, T.; Kuniyoshi, K.; Satoh, T.; Saitoh, T.; Matsushita, M.; Standley, D.M.; et al. The IκB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR–IL-1R by controlling degradation of regnase-1. Nat. Immunol. 2011, 12, 1167–1175. [Google Scholar] [CrossRef]
- Zhou, L.; Azfer, A.; Niu, J.; Graham, S.; Choudhury, M.; Adamski, F.M.; Younce, C.; Binkley, P.F.; Kolattukudy, P.E. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 2006, 98, 1177–1185. [Google Scholar] [CrossRef]
- Uehata, T.; Iwasaki, H.; Vandenbon, A.; Matsushita, K.; Hernandez-Cuellar, E.; Kuniyoshi, K.; Satoh, T.; Mino, T.; Suzuki, Y.; Standley, D.M.; et al. Malt1-induced cleavage of regnase-1 in CD4+ helper T cells regulates immune activation. Cell 2013, 153, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y.; et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell 2015, 161, 1058–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Peng, W.; Sun, Y.; Wang, X.; Xu, Y.; Li, X.; Gao, G.; Rao, Z. Structural study of MCPIP1 N-terminal conserved domain reveals a PIN-like RNase. Nucleic Acids Res. 2012, 40, 6957–6965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokogawa, M.; Tsushima, T.; Noda, N.N.; Kumeta, H.; Enokizono, Y.; Yamashita, K.; Standley, D.M.; Takeuchi, O.; Akira, S.; Inagaki, F. Structural basis for the regulation of enzymatic activity of Regnase-1 by domain-domain interactions. Sci. Rep. 2016, 6, 22324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehata, T.; Akira, S. MRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 708–713. [Google Scholar] [CrossRef]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef]
- Dobosz, E.; Wilamowski, M.; Lech, M.; Bugara, B.; Jura, J.; Potempa, J.; Koziel, J. MCPIP-1, Alias Regnase-1, Controls Epithelial Inflammation by Posttranscriptional Regulation of IL-8 Production. J. Innate Immun. 2016, 8, 564–578. [Google Scholar] [CrossRef]
- Blazusiak, E.; Florczyk, D.; Jura, J.; Potempa, J.; Koziel, J. Differential regulation by toll-like receptor agonists reveals that MCPIP1 is the potent regulator of innate immunity in bacterial and viral infections. J. Innate Immun. 2013, 5, 15–23. [Google Scholar] [CrossRef]
- Liang, J.; Wang, J.; Saad, Y.; Warble, L.; Becerra, E.; Kolattukudy, P.E. Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. J. Neuroinflamm. 2011, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-J.; Chu, J.-S.; Chien, H.-L.; Tseng, C.-H.; Ko, P.-C.; Mei, Y.-Y.; Tang, W.-C.; Kao, Y.-T.; Cheng, H.-Y.; Liang, Y.-C.; et al. MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J. Immunol. 2014, 193, 4159–4168. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.-J.; Choi, J.-A.; Lee, J.-H.; Choi, C.H.; Kim, H.-J.; Song, C.-H. Mycobacterium tuberculosis 38-kDa antigen induces endoplasmic reticulum stress-mediated apoptosis via toll-like receptor 2/4. Apoptosis 2015, 20, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qiu, C.; Miao, R.; Zhou, J.; Lee, A.; Liu, B.; Lester, S.N.; Fu, W.; Zhu, L.; Zhang, L.; et al. MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 19083–19088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobosz, E.; Lorenz, G.; Ribeiro, A.; Wurf, V.; Wadowska, M.; Kotlinowski, J.; Schmaderer, C.; Potempa, J.; Fu, M.; Koziel, J.; et al. Murine myeloid cell MCPIP1 suppresses autoimmunity by regulating B-cell expansion and differentiation. Dis. Model. Mech. 2021, 14, dmm047589. [Google Scholar] [CrossRef] [PubMed]
- Dobosz, E.; Wadowska, M.; Kaminska, M.; Wilamowski, M.; Honarpisheh, M.; Bryzek, D.; Potempa, J.; Jura, J.; Lech, M.; Koziel, J. MCPIP-1 Restricts Inflammation via Promoting Apoptosis of Neutrophils. Front. Immunol. 2021, 12, 627922. [Google Scholar] [CrossRef]
- Xiaoming, A.; Wenbo, J.; Jinyi, W.; Bin, W.; Chunyang, H.; Qi, C.; Lianbao, K. Macrophage Regnase-1 Deletion Deteriorates Liver Ischemia/Reperfusion Injury through Regulation of Macrophage Polarization. Front. Physiol. 2020, 11, 582347. [Google Scholar] [CrossRef]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef]
- Gantier, M.P.; Tong, S.; Behlke, M.A.; Xu, D.; Phipps, S.; Foster, P.S.; Williams, B.R.G. TLR7 is involved in sequence-specific sensing of single-stranded RNAs in human macrophages. J. Immunol. 2008, 180, 2117–2124. [Google Scholar] [CrossRef]
- Vesentini, N.; Barsanti, C.; Martino, A.; Kusmic, C.; Ripoli, A.; Rossi, A.; L’Abbate, A. Selection of reference genes in different myocardial regions of an in vivo ischemia/reperfusion rat model for normalization of antioxidant gene expression. BMC Res. Notes 2012, 5, 124. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Miao, R.; Zhou, Z.; Wang, T.; Liu, J.; Liu, G.; Chen, Y.E.; Xin, H.-B.; Zhang, J.; Fu, M. MCPIP1 Negatively Regulates Toll-like Receptor 4 Signaling and Protects Mice from LPS-induced Septic Shock. Cell. Signal. 2013, 25, 1228. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Zheng, E.; Sareli, C.; Kolattukudy, P.E.; Niu, J. Monocyte Chemotactic Protein-Induced Protein 1 (MCPIP-1): A Key Player of Host Defense and Immune Regulation. Front. Immunol. 2021, 12, 727861. [Google Scholar] [CrossRef]
- Kemmner, S.; Bachmann, Q.; Steiger, S.; Lorenz, G.; Honarpisheh, M.; Foresto-Neto, O.; Wang, S.; Carbajo-Lozoya, J.; Alt, V.; Schulte, C.; et al. STAT1 regulates macrophage number and phenotype and prevents renal fibrosis after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2019, 316, F277–F291. [Google Scholar] [CrossRef]
- Lorenz, G.; Moschovaki-Filippidou, F.; Würf, V.; Metzger, P.; Steiger, S.; Batz, F.; Carbajo-Lozoya, J.; Koziel, J.; Schnurr, M.; Cohen, C.D.; et al. IFN Regulatory Factor 4 Controls Post-ischemic Inflammation and Prevents Chronic Kidney Disease. Front. Immunol. 2019, 10, 2162. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef]
- Mittrücker, H.W.; Matsuyama, T.; Grossman, A.; Kündig, T.M.; Potter, J.; Shahinian, A.; Wakeham, A.; Patterson, B.; Ohashi, P.S.; Mak, T.W. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 1997, 275, 540–543. [Google Scholar] [CrossRef]
- Suzuki, S.; Honma, K.; Matsuyama, T.; Suzuki, K.; Toriyama, K.; Akitoyo, I.; Yamamoto, K.; Suematsu, T.; Nakamura, M.; Yui, K.; et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc. Natl. Acad. Sci. USA 2004, 101, 8981–8986. [Google Scholar] [CrossRef] [Green Version]
- Vander Lugt, B.; Khan, A.A.; Hackney, J.A.; Agrawal, S.; Lesch, J.; Zhou, M.; Lee, W.P.; Park, S.; Xu, M.; DeVoss, J.; et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol. 2014, 15, 161–167. [Google Scholar] [CrossRef]
- Ke, X.; Chen, C.; Song, Y.; Cai, Q.; Li, J.; Tang, Y.; Han, X.; Qu, W.; Chen, A.; Wang, H.; et al. Hypoxia modifies the polarization of macrophages and their inflammatory microenvironment, and inhibits malignant behavior in cancer cells. Oncol. Lett. 2019, 18, 5871–5878. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Liang, J.; Wang, J.; Azfer, A.; Song, W.; Tromp, G.; Kolattukudy, P.E.; Fu, M. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J. Biol. Chem. 2008, 283, 6337–6346. [Google Scholar] [CrossRef] [Green Version]
- Kotlinowski, J.; Hutsch, T.; Czyzynska-Cichon, I.; Wadowska, M.; Pydyn, N.; Jasztal, A.; Kij, A.; Dobosz, E.; Lech, M.; Miekus, K.; et al. Deletion of Mcpip1 in Mcpip1fl/flAlbCre mice recapitulates the phenotype of human primary biliary cholangitis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166086. [Google Scholar] [CrossRef]
- Günthner, R.; Kumar, V.R.S.; Lorenz, G.; Anders, H.-J.; Lech, M. Pattern-recognition receptor signaling regulator mRNA expression in humans and mice, and in transient inflammation or progressive fibrosis. Int. J. Mol. Sci. 2013, 14, 18124–18147. [Google Scholar] [CrossRef]
- Steiger, S.; Kumar, S.V.; Honarpisheh, M.; Lorenz, G.; Günthner, R.; Romoli, S.; Gröbmayr, R.; Susanti, H.-E.; Potempa, J.; Koziel, J.; et al. Immunomodulatory Molecule IRAK-M Balances Macrophage Polarization and Determines Macrophage Responses during Renal Fibrosis. J. Immunol. 2017, 199, 1440–1452. [Google Scholar] [CrossRef]
- Moschovaki-Filippidou, F.; Steiger, S.; Lorenz, G.; Schmaderer, C.; Ribeiro, A.; von Rauchhaupt, E.; Cohen, C.D.; Anders, H.-J.; Lindenmeyer, M.; Lech, M. Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice. Int. J. Mol. Sci. 2020, 21, 6978. [Google Scholar] [CrossRef]
- Baek, J.-H. The Impact of Versatile Macrophage Functions on Acute Kidney Injury and Its Outcomes. Front. Physiol. 2019, 10, 1016. [Google Scholar] [CrossRef] [Green Version]
- Ysebaert, D.K.; De Greef, K.E.; Vercauteren, S.R.; Verhulst, A.; Kockx, M.; Verpooten, G.A.; De Broe, M.E. Effect of immunosuppression on damage, leukocyte infiltration, and regeneration after severe warm ischemia/reperfusion renal injury. Kidney Int. 2003, 64, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, G.M.; Cenedeze, M.A.; Feitoza, C.Q.; Wang, P.M.H.; Bertocchi, A.P.F.; Damião, M.J.; Pinheiro, H.S.; Antunes Teixeira, V.P.; dos Reis, M.A.; Pacheco-Silva, A.; et al. The role of heme oxygenase 1 in rapamycin-induced renal dysfunction after ischemia and reperfusion injury. Kidney Int. 2006, 70, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Lieberthal, W.; Levine, J.S. The role of the mammalian target of rapamycin (mTOR) in renal disease. J. Am. Soc. Nephrol. 2009, 20, 2493–2502. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Plüddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, N.; Niu, J.; Saad, Y.; Kumar, S.; Sirakova, T.; Becerra, E.; Li, X.; Kolattukudy, P.E. Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J. Immunol. 2015, 194, 6011–6023. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M.; Scannon, P.J.; Vincent, J.L.; White, M.; Carroll, S.F.; Palardy, J.E.; Parejo, N.A.; Pribble, J.P.; Lemke, J.H. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J. Infect. Dis. 1999, 180, 1584–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, K.M.; Hu, D.; Brenner, S.; Zöller, J.; Heinz, G.A.; Nagel, D.; Vogel, K.U.; Rehage, N.; Warth, S.C.; Edelmann, S.L.; et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nat. Immunol. 2014, 15, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Ning, H.; Wang, Q.; Lu, W.; Chang, Y.; Wang, T.T.; Lai, J.; Kolattukudy, P.E.; Hou, R.; Hoft, D.F.; et al. Monocyte chemotactic protein–induced protein 1 controls allergic airway inflammation by suppressing IL-5–producing TH2 cells through the Notch/Gata3 pathway. J. Allergy Clin. Immunol. 2018, 142, 582–594.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achuthan, A.; Cook, A.D.; Lee, M.-C.; Saleh, R.; Khiew, H.-W.; Chang, M.W.N.; Louis, C.; Fleetwood, A.J.; Lacey, D.C.; Christensen, A.D.; et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J. Clin. Investig. 2016, 126, 3453–3466. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.-C.; Jarnicki, A.; Achuthan, A.; Fleetwood, A.J.; Anderson, G.P.; Ellson, C.; Feeney, M.; Modis, L.K.; Smith, J.E.; Hamilton, J.A.; et al. CCL17 in Inflammation and Pain. J. Immunol. 2020, 205, 213–222. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Hsu, H.; Lin, C.-C.; Pan, S.-Y.; Liu, S.-Y.; Wu, C.-F.; Tsai, P.-Z.; Liao, C.-T.; Cheng, H.-T.; Chiang, W.-C.; et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J. Pathol. 2020, 250, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-C.; Lacey, D.C.; Fleetwood, A.J.; Achuthan, A.; Hamilton, J.A.; Cook, A.D. GM-CSF- and IRF4-Dependent Signaling Can Regulate Myeloid Cell Numbers and the Macrophage Phenotype during Inflammation. J. Immunol. 2019, 202, 3033–3040. [Google Scholar] [CrossRef]
- Honarpisheh, M.; Foresto-Neto, O.; Steiger, S.; Kraft, F.; Koehler, P.; von Rauchhaupt, E.; Potempa, J.; Adamowicz, K.; Koziel, J.; Lech, M. Aristolochic acid I determine the phenotype and activation of macrophages in acute and chronic kidney disease. Sci. Rep. 2018, 8, 12169. [Google Scholar] [CrossRef]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef]
- Brüstle, A.; Heink, S.; Huber, M.; Rosenplänter, C.; Stadelmann, C.; Yu, P.; Arpaia, E.; Mak, T.W.; Kamradt, T.; Lohoff, M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 2007, 8, 958–966. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Terker, A.S.; Pan, Y.; Li, Z.; Cao, S.; Wang, Y.; Niu, A.; Wang, S.; Fan, X.; Zhang, M.-Z.; et al. Deletion of Myeloid Interferon Regulatory Factor 4 (Irf4) in Mouse Model Protects against Kidney Fibrosis after Ischemic Injury by Decreased Macrophage Recruitment and Activation. J. Am. Soc. Nephrol. 2021, 32, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J. Heterogenous Role of IRF4 in Kidney Fibrosis. J. Am. Soc. Nephrol. 2021, 32, 2971–2972. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, A.; Dobosz, E.; Krill, M.; Köhler, P.; Wadowska, M.; Steiger, S.; Schmaderer, C.; Koziel, J.; Lech, M. Macrophage-Specific MCPIP1/Regnase-1 Attenuates Kidney Ischemia-Reperfusion Injury by Shaping the Local Inflammatory Response and Tissue Regeneration. Cells 2022, 11, 397. https://doi.org/10.3390/cells11030397
Ribeiro A, Dobosz E, Krill M, Köhler P, Wadowska M, Steiger S, Schmaderer C, Koziel J, Lech M. Macrophage-Specific MCPIP1/Regnase-1 Attenuates Kidney Ischemia-Reperfusion Injury by Shaping the Local Inflammatory Response and Tissue Regeneration. Cells. 2022; 11(3):397. https://doi.org/10.3390/cells11030397
Chicago/Turabian StyleRibeiro, Andrea, Ewelina Dobosz, Moritz Krill, Paulina Köhler, Marta Wadowska, Stefanie Steiger, Christoph Schmaderer, Joanna Koziel, and Maciej Lech. 2022. "Macrophage-Specific MCPIP1/Regnase-1 Attenuates Kidney Ischemia-Reperfusion Injury by Shaping the Local Inflammatory Response and Tissue Regeneration" Cells 11, no. 3: 397. https://doi.org/10.3390/cells11030397
APA StyleRibeiro, A., Dobosz, E., Krill, M., Köhler, P., Wadowska, M., Steiger, S., Schmaderer, C., Koziel, J., & Lech, M. (2022). Macrophage-Specific MCPIP1/Regnase-1 Attenuates Kidney Ischemia-Reperfusion Injury by Shaping the Local Inflammatory Response and Tissue Regeneration. Cells, 11(3), 397. https://doi.org/10.3390/cells11030397