
Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging



Abstract
1. Introduction
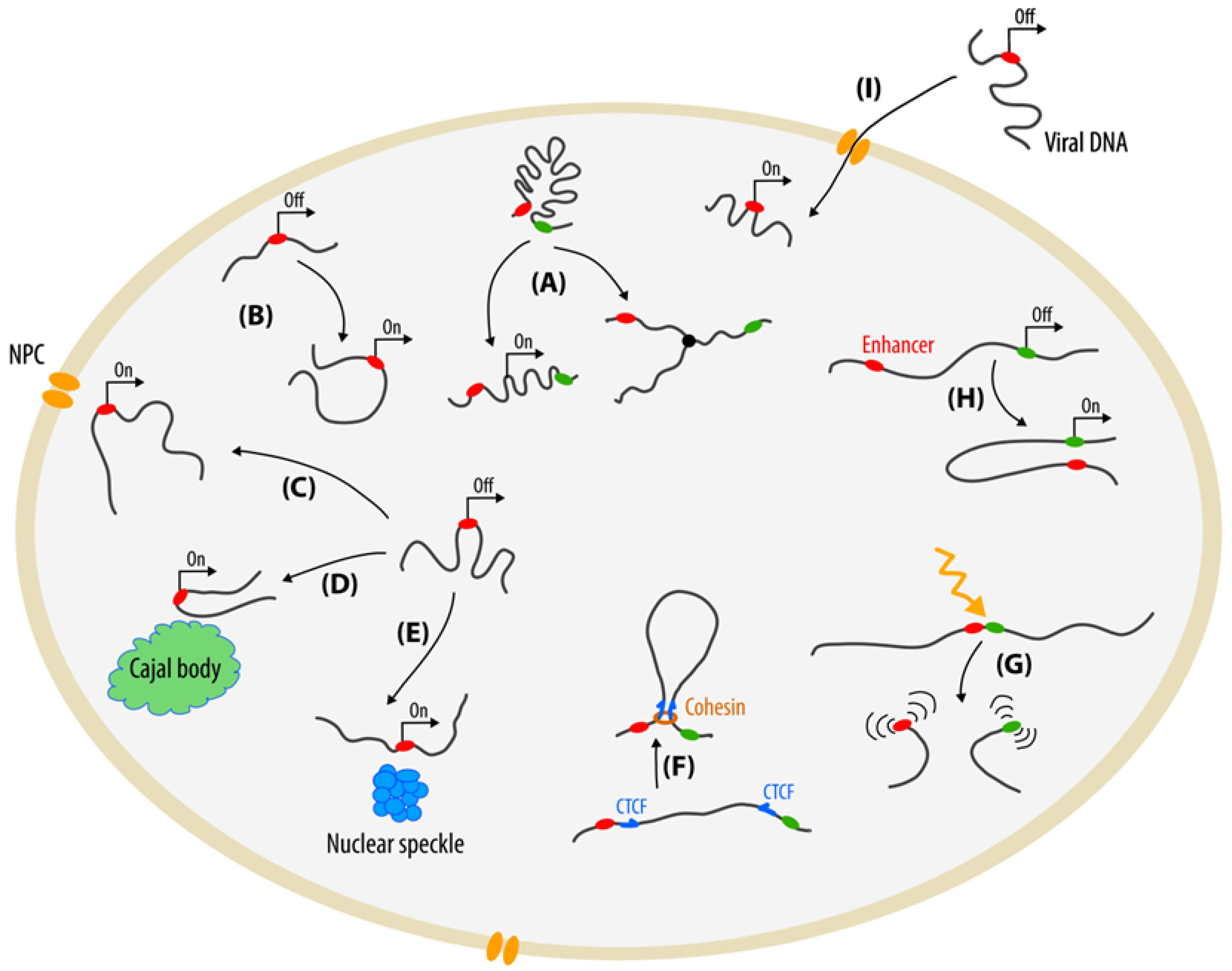
2. Whole Chromatin Visualization
3. Tagging of Individual Chromatin Loci
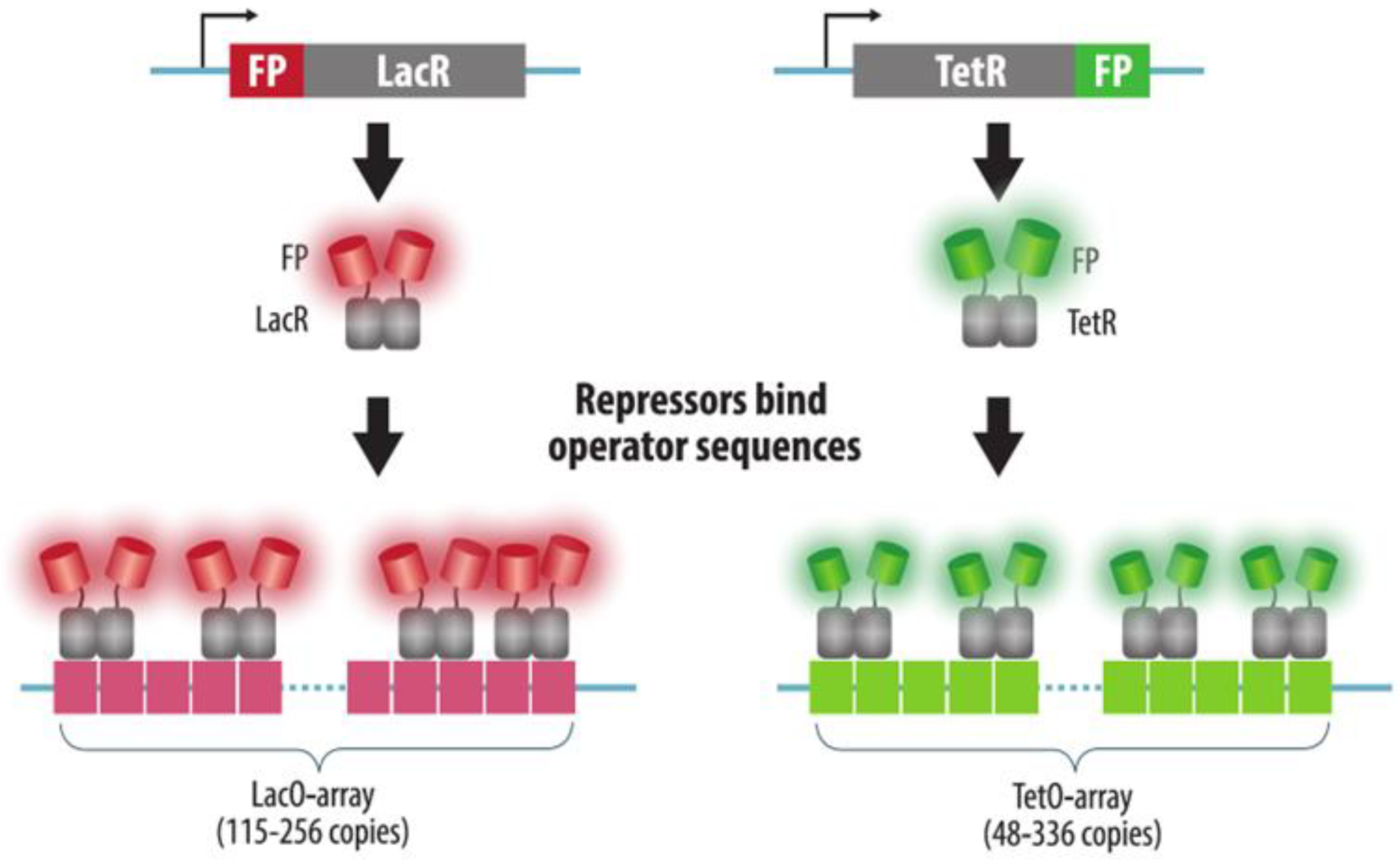
3.1. Fluorescent Repressor-Operator Systems
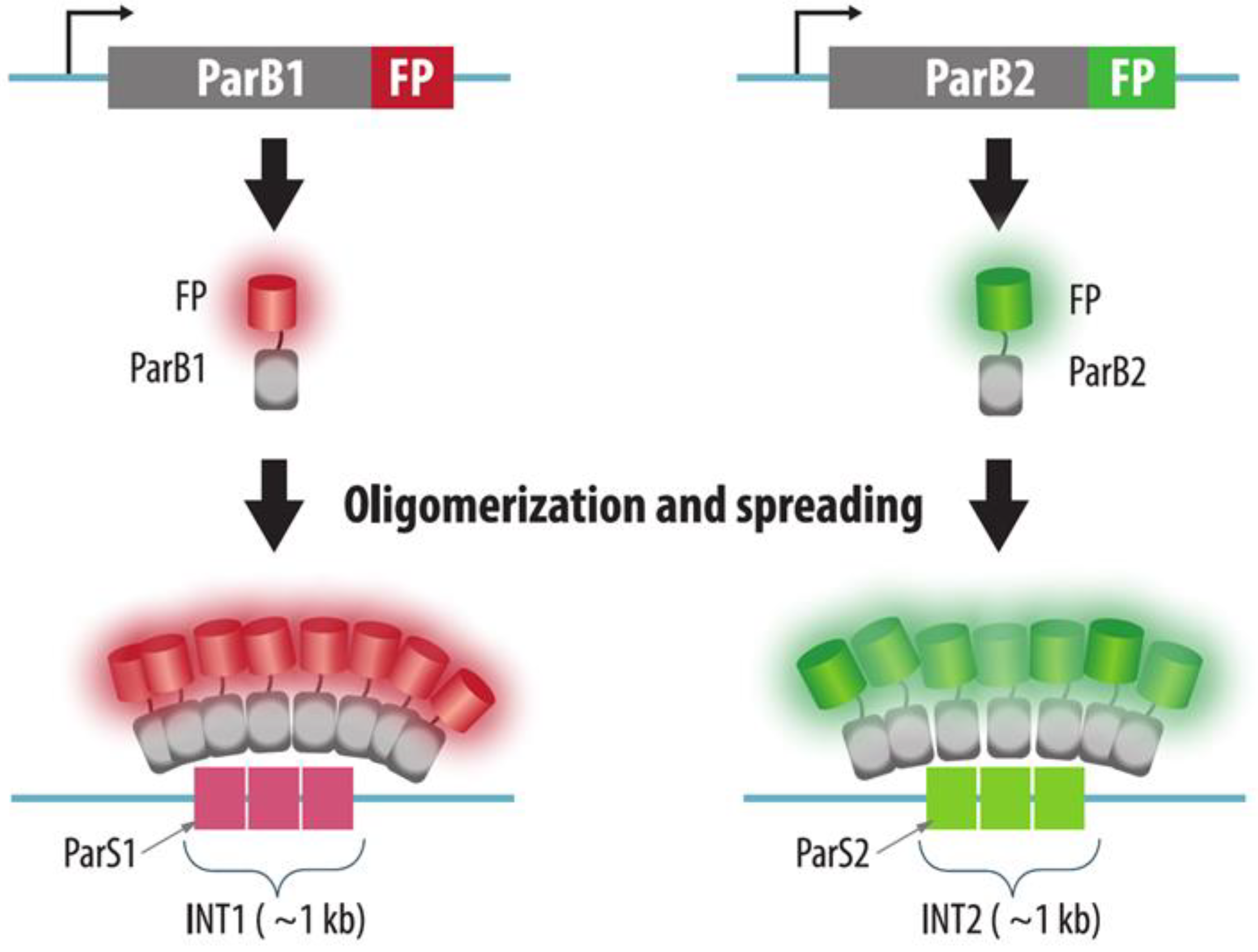
3.2. ParB-INT Systems (ANCHOR)
4. Methods Based on Programmable DNA-Binding Proteins
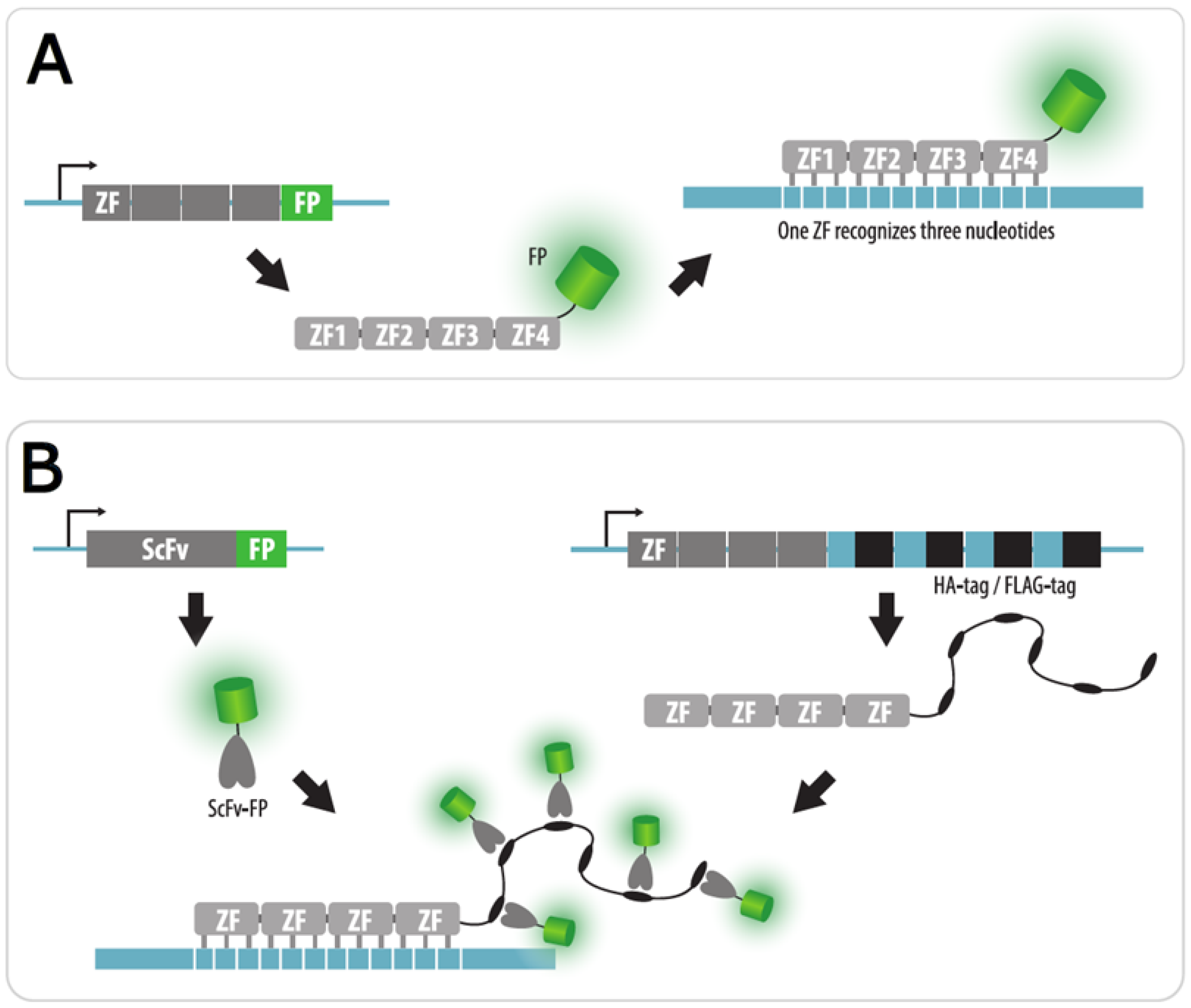
4.1. Zinc Finger Imaging
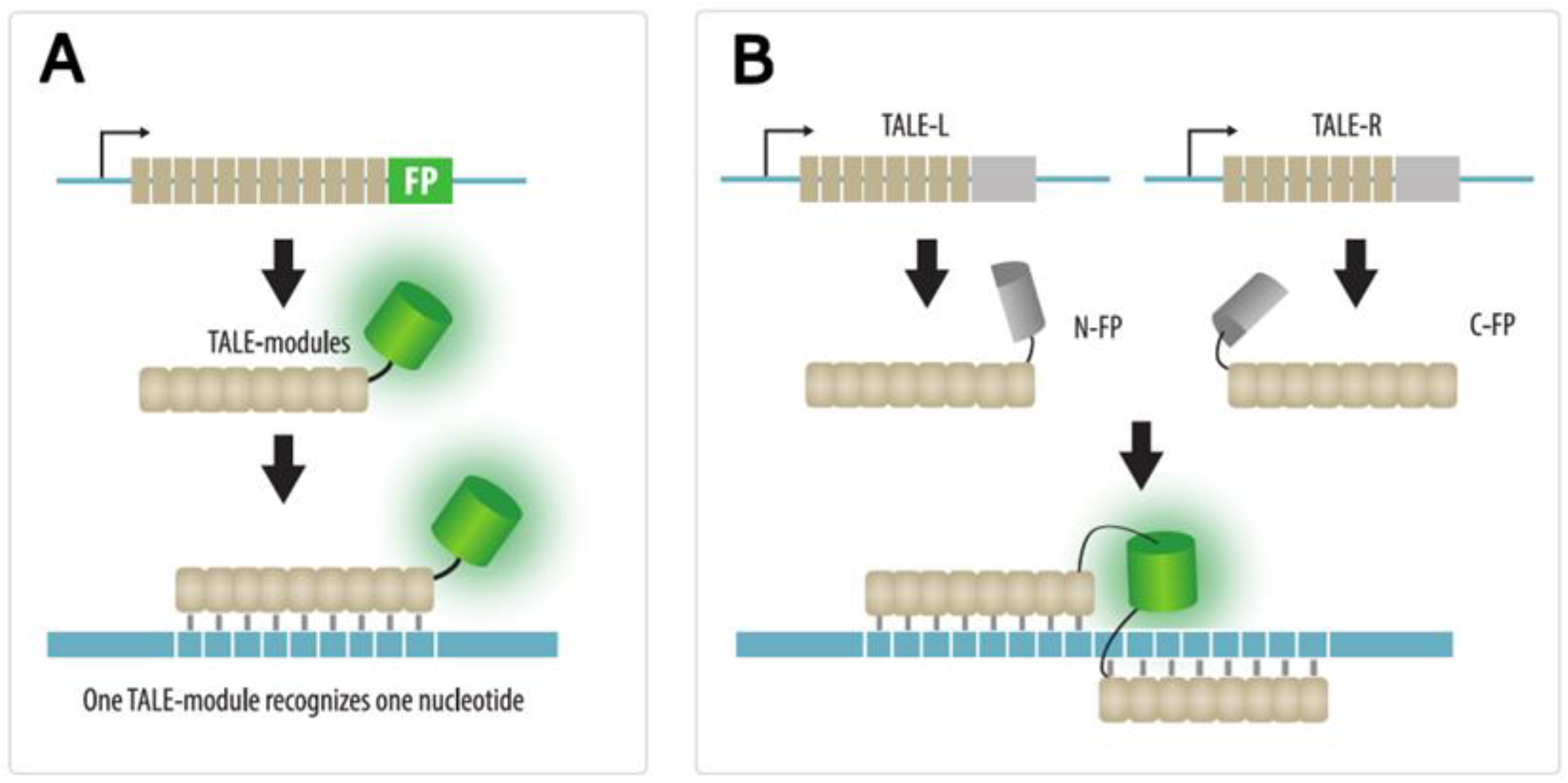
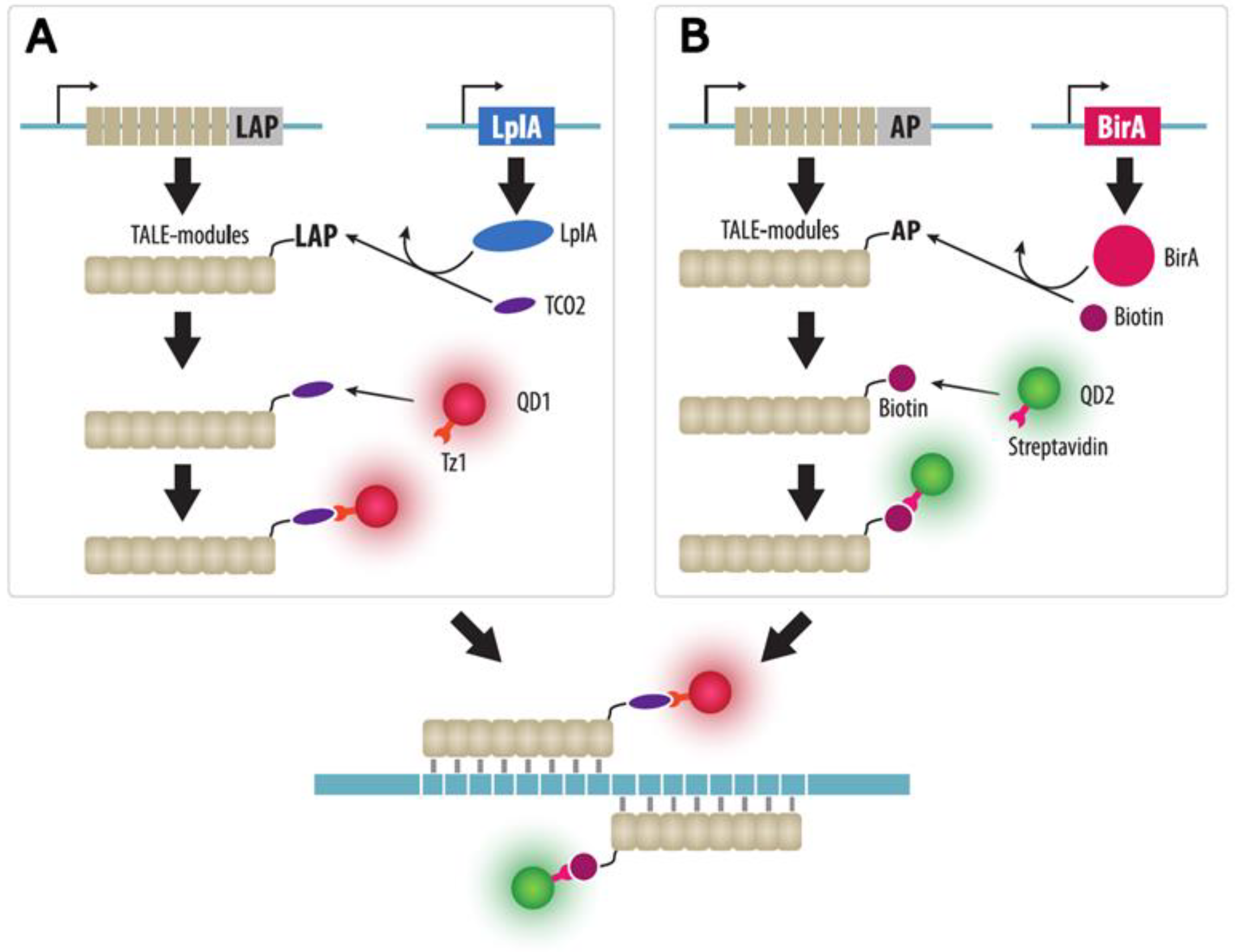
4.2. TALE Imaging
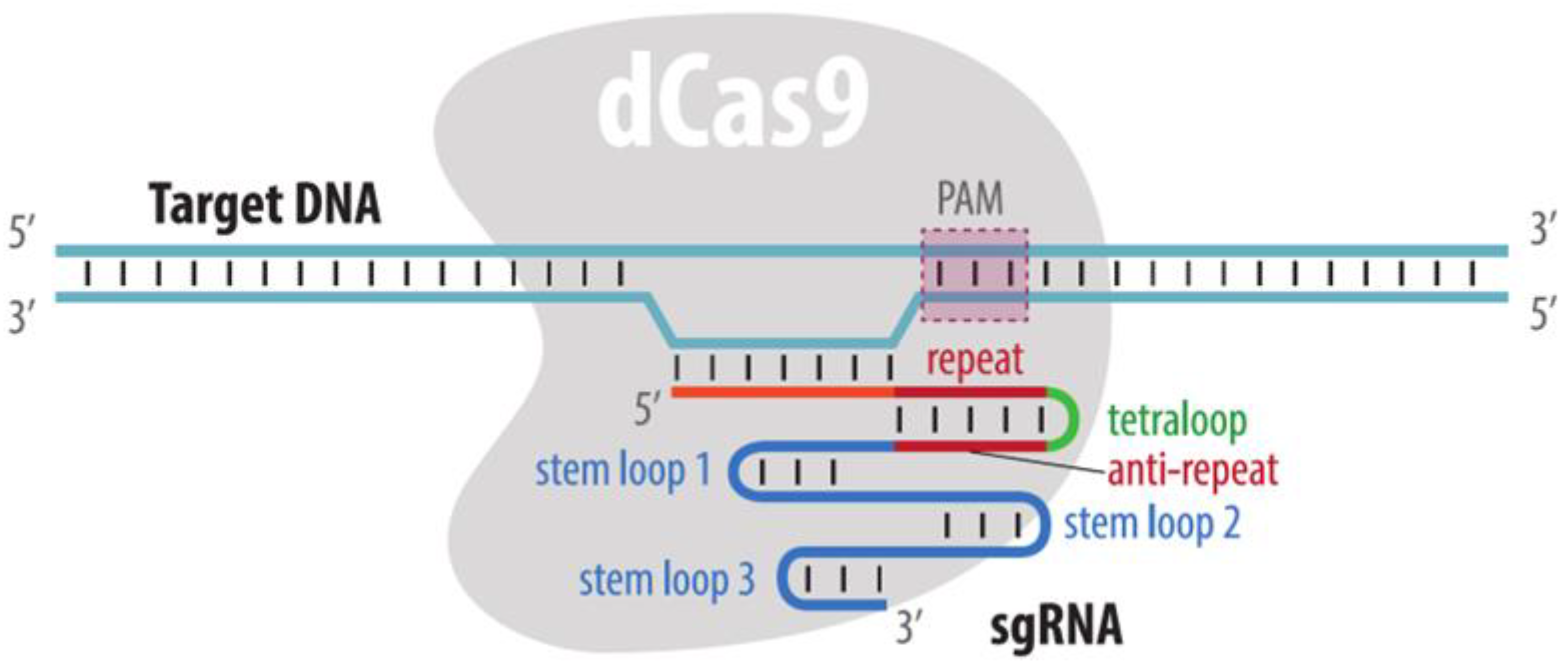
4.3. CRISPR Imaging
4.3.1. Visualization by dCas9 Fused to Fluorescent Protein
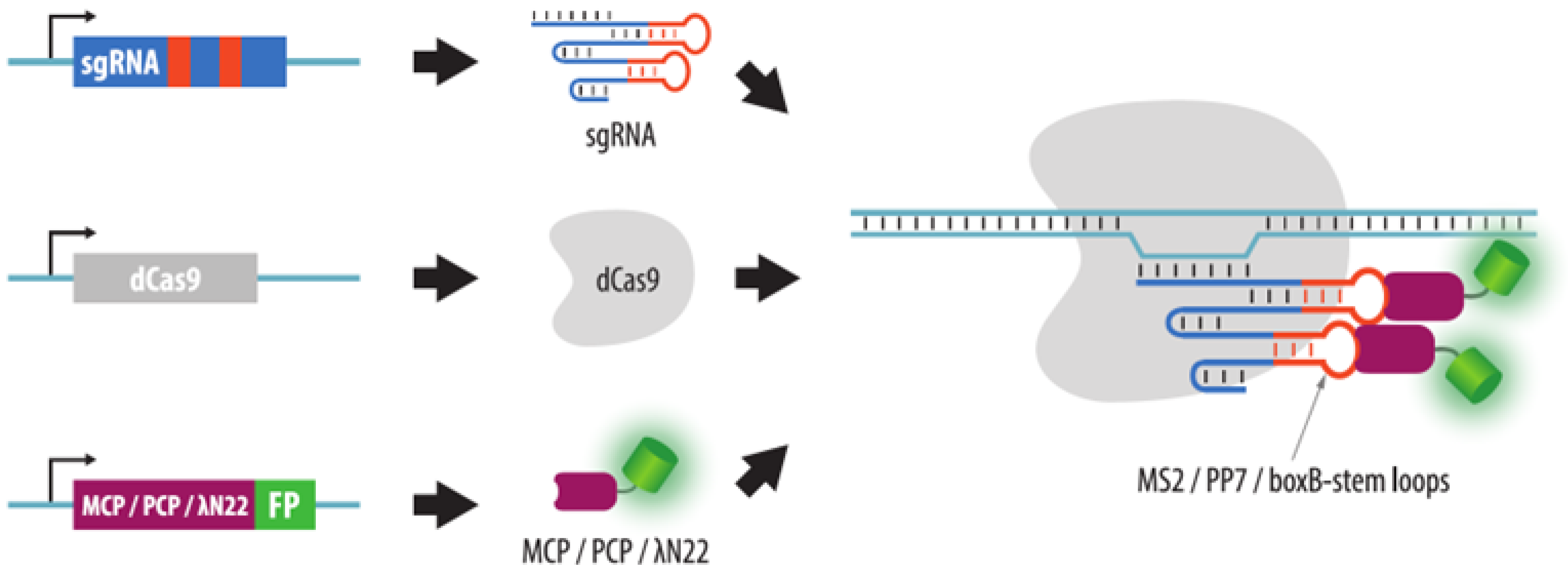
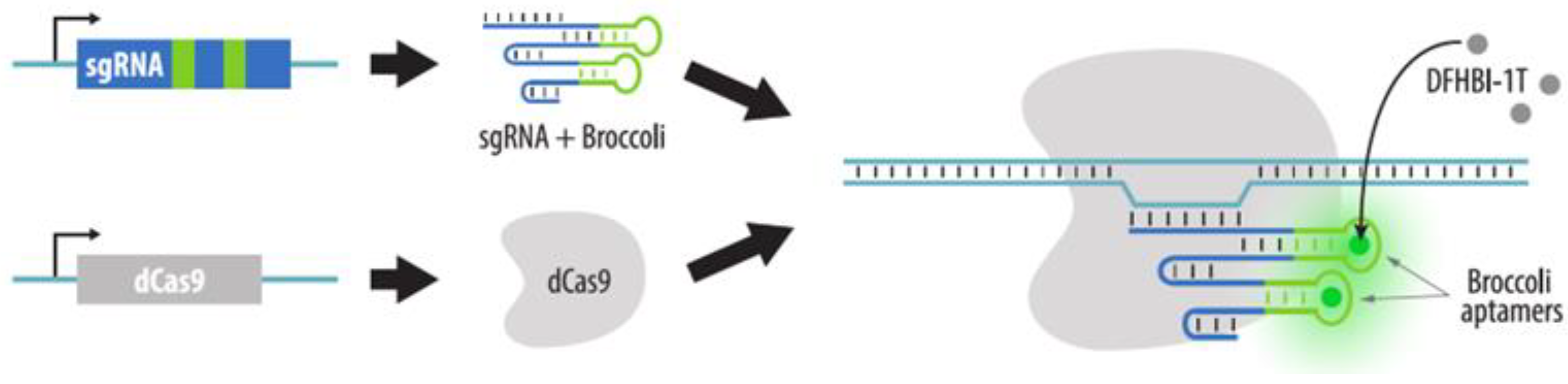
4.3.2. Visualization of Aptamers in sgRNA Structures
4.3.3. Visualization by dCas9 Conjugated to Organic Fluorophores
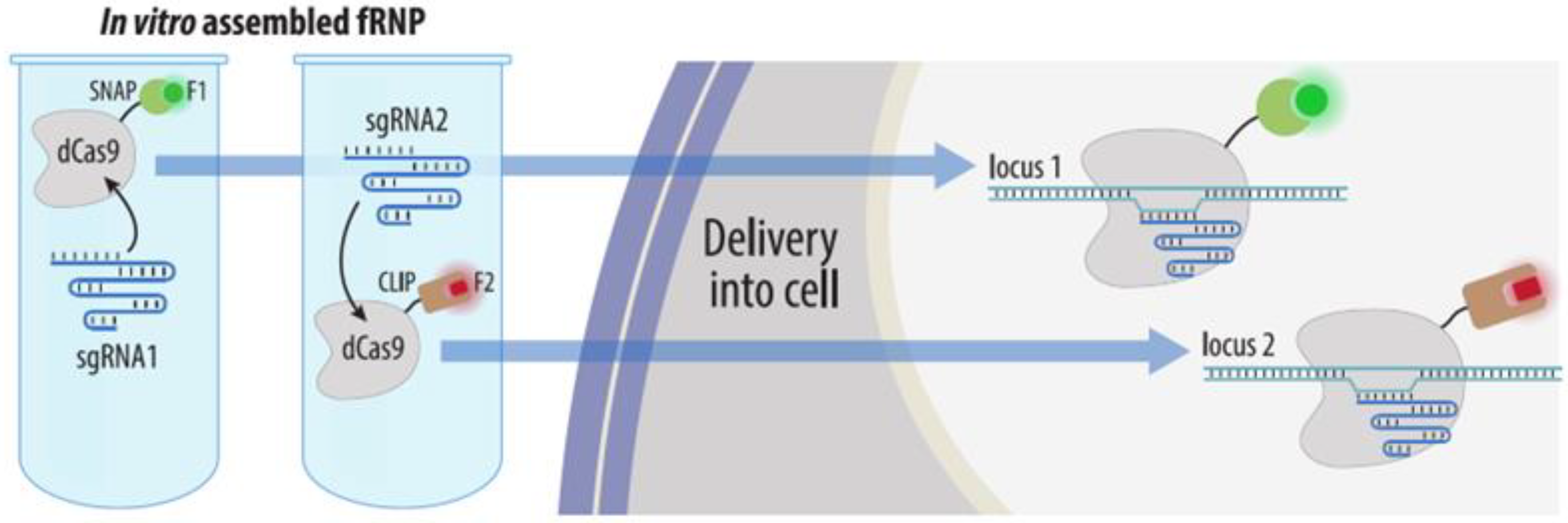
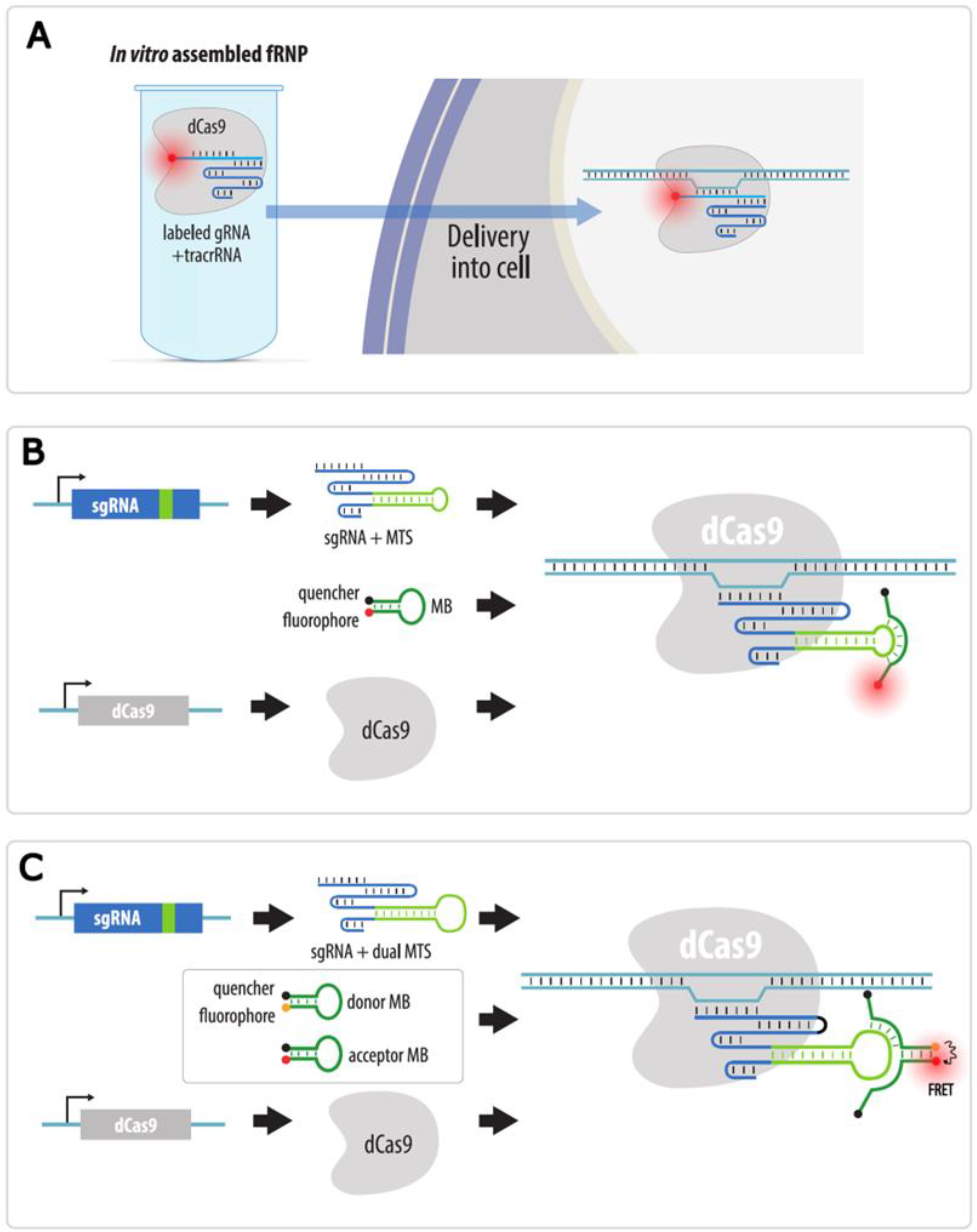
4.3.4. Visualization with gRNAs Conjugated to Organic Fluorophores
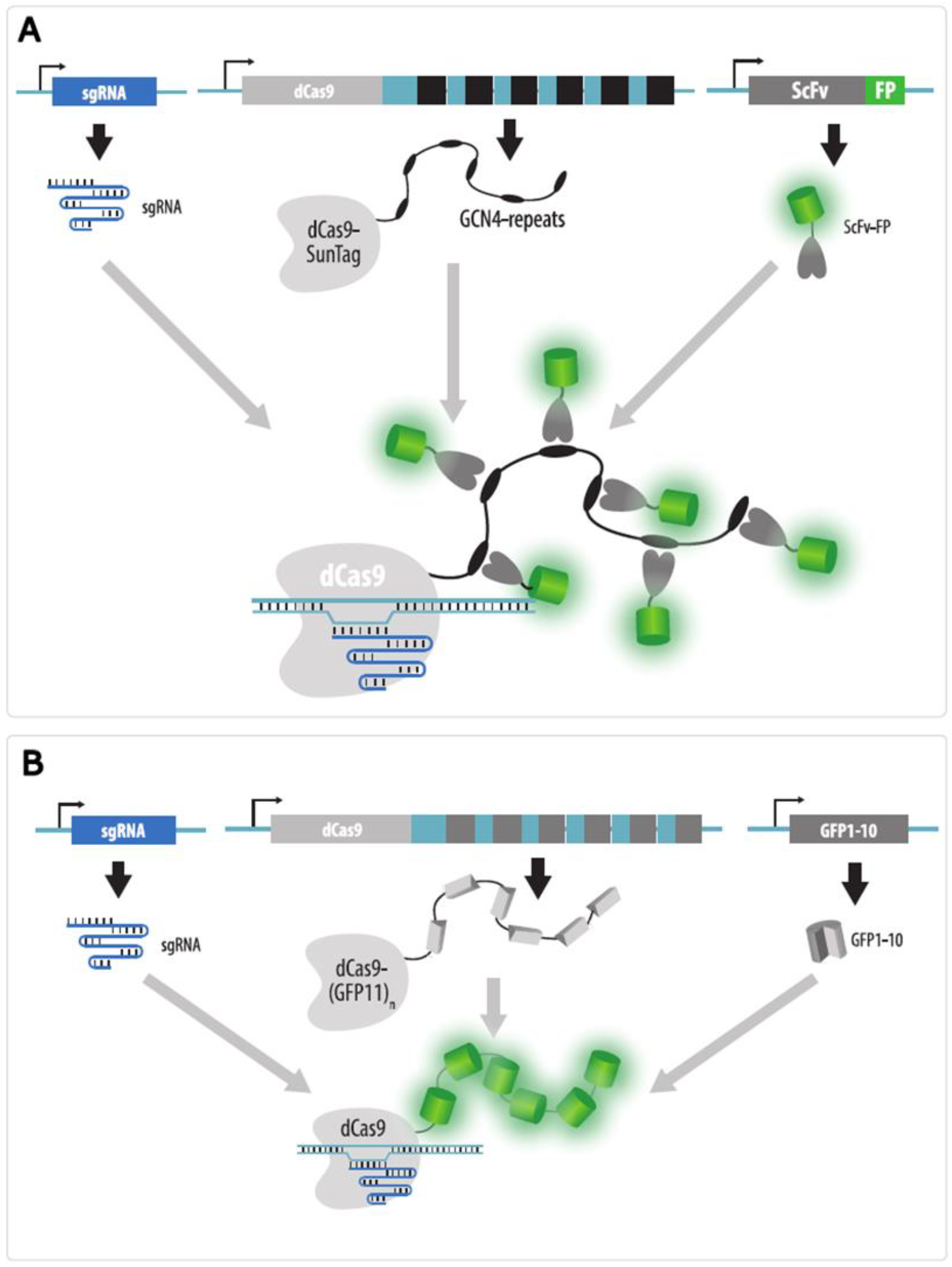
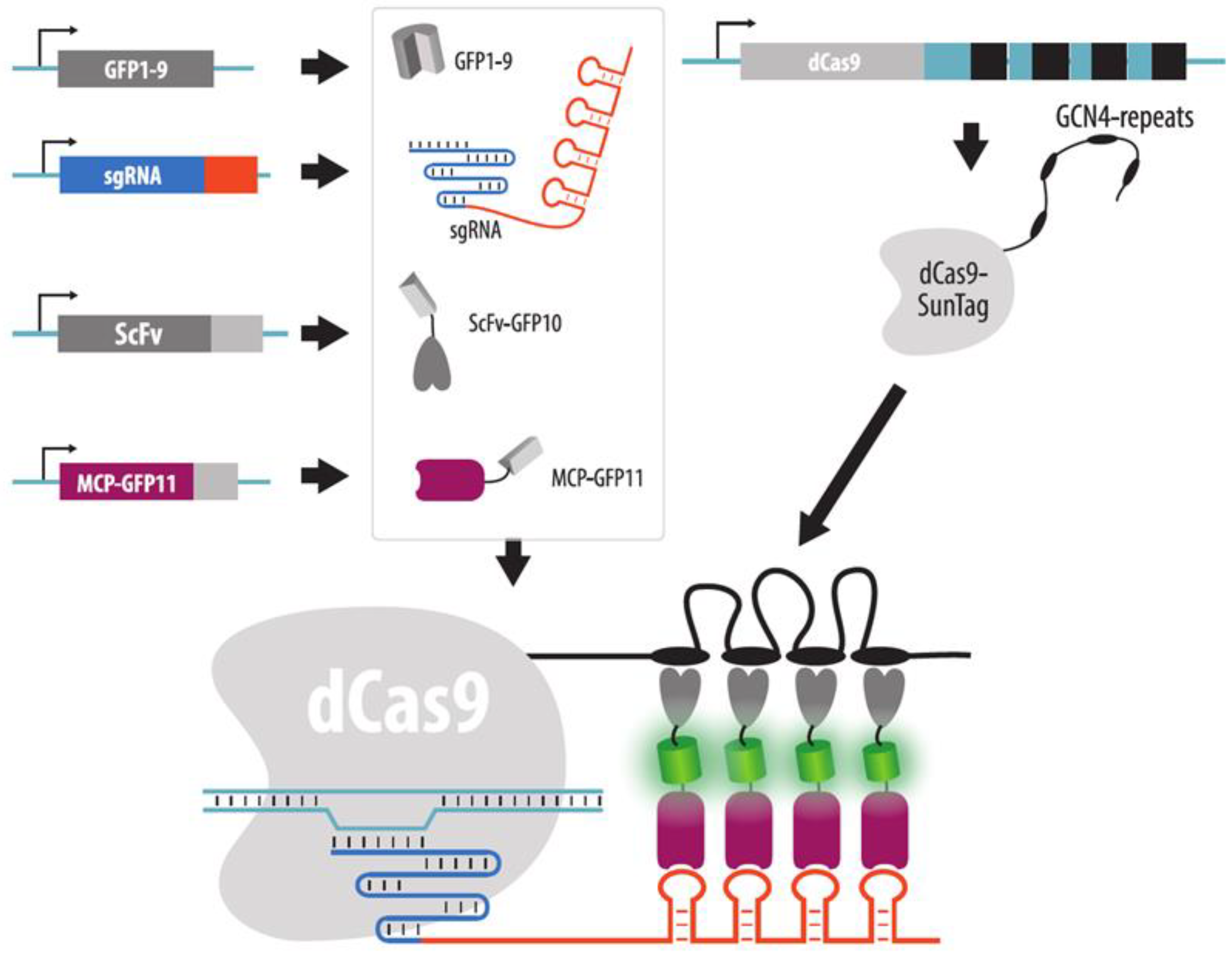
4.3.5. Strategies to Increase Brightness and SNR in CRISPR Imaging
4.3.6. Visualization of Unique Loci without Repeats
4.3.7. CRISPR Imaging in Plant Cells
4.3.8. CRISPR Imaging and Native Dynamics of Chromatin
5. Resolution of Live-Cell Chromatin Imaging
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, Y.; Kireev, I.; Plutz, M.; Ashourian, N.; Belmont, A.S. Large-scale chromatin structure of inducible genes: Transcription on a condensed, linear template. J. Cell Biol. 2009, 185, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhironkina, O.A.; Cherepanynets, V.D.; Strelkova, O.S.; Kireev, I.I.; Belmont, A.S. Cytology of DNA Replication Reveals Dynamic Plasticity of Large-Scale Chromatin Fibers. Curr. Biol. 2016, 26, 2527–2534. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chuang, C.H.; Carpenter, A.E.; Fuchsova, B.; Johnson, T.; de Lanerolle, P.; Belmont, A.S. Long-range directional movement of an interphase chromosome site. Curr. Biol. 2006, 16, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Cabal, G.G.; Genovesio, A.; Rodriguez-Navarro, S.; Zimmer, C.; Gadal, O.; Lesne, A.; Buc, H.; Feuerbach-Fournier, F.; Olivo-Marin, J.C.; Hurt, E.C.; et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature 2006, 441, 770–773. [Google Scholar] [CrossRef]
- Taddei, A.; Van Houwe, G.; Hediger, F.; Kalck, V.; Cubizolles, F.; Schober, H.; Gasser, S.M. Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature 2006, 441, 774–778. [Google Scholar] [CrossRef]
- Dieppois, G.; Iglesias, N.; Stutz, F. Cotranscriptional recruitment to the mRNA export receptor Mex67p contributes to nuclear pore anchoring of activated genes. Mol. Cell Biol. 2006, 26, 7858–7870. [Google Scholar] [CrossRef]
- Brickner, D.G.; Ahmed, S.; Meldi, L.; Thompson, A.; Light, W.; Young, M.; Hickman, T.L.; Chu, F.; Fabre, E.; Brickner, J.H. Transcription factor binding to a DNA zip code controls interchromosomal clustering at the nuclear periphery. Dev. Cell 2012, 22, 1234–1246. [Google Scholar] [CrossRef]
- Brickner, D.G.; Coukos, R.; Brickner, J.H. INO1 transcriptional memory leads to DNA zip code-dependent interchromosomal clustering. Microb. Cell 2015, 2, 481–490. [Google Scholar] [CrossRef]
- Brickner, D.G.; Sood, V.; Tutucci, E.; Coukos, R.; Viets, K.; Singer, R.H.; Brickner, J.H. Subnuclear positioning and interchromosomal clustering of the GAL1-10 locus are controlled by separable, interdependent mechanisms. Mol. Biol. Cell 2016, 27, 2980–2993. [Google Scholar] [CrossRef]
- Dundr, M.; Ospina, J.K.; Sung, M.H.; John, S.; Upender, M.; Ried, T.; Hager, G.L.; Matera, A.G. Actin-dependent intranuclear repositioning of an active gene locus in vivo. J. Cell Biol. 2007, 179, 1095–1103. [Google Scholar] [CrossRef]
- Khanna, N.; Hu, Y.; Belmont, A.S. HSP70 transgene directed motion to nuclear speckles facilitates heat shock activation. Curr. Biol. 2014, 24, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Fudenberg, G.; Abdennur, N.; Imakaev, M.; Goloborodko, A.; Mirny, L.A. Emerging Evidence of Chromosome Folding by Loop Extrusion. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.M. DNA loop extrusion by human cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef]
- Razin, S.V.; Ulianov, S.V.; Gavrilov, A.A. 3D Genomics. Mol. Biol. 2019, 53, 802–812. [Google Scholar] [CrossRef]
- Gabriele, M.; Brandao, H.B.; Grosse-Holz, S.; Jha, A.; Dailey, G.M.; Cattoglio, C.; Hsieh, T.S.; Mirny, L.; Zechner, C.; Hansen, A.S. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022, 376, 496–501. [Google Scholar] [CrossRef]
- Clow, P.A.; Du, M.; Jillette, N.; Taghbalout, A.; Zhu, J.J.; Cheng, A.W. CRISPR-mediated multiplexed live cell imaging of nonrepetitive genomic loci with one guide RNA per locus. Nat. Commun. 2022, 13, 1871. [Google Scholar] [CrossRef]
- Mach, P.; Kos, P.I.; Zhan, Y.; Cramard, J.; Gaudin, S.; Tünnermann, J.; Marchi, E.; Eglinger, J.; Zuin, J.; Kryzhanovska, M.; et al. Live-cell imaging and physical modeling reveal control of chromosome folding dynamics by cohesin and CTCF. bioRxiv 2022. [Google Scholar] [CrossRef]
- Chen, H.; Levo, M.; Barinov, L.; Fujioka, M.; Jaynes, J.B.; Gregor, T. Dynamic interplay between enhancer-promoter topology and gene activity. Nat. Genet. 2018, 50, 1296–1303. [Google Scholar] [CrossRef]
- Alexander, J.M.; Guan, J.; Li, B.; Maliskova, L.; Song, M.; Shen, Y.; Huang, B.; Lomvardas, S.; Weiner, O.D. Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. eLife 2019, 8, e41769. [Google Scholar] [CrossRef]
- Geng, Y.; Pertsinidis, A. Simple and versatile imaging of genomic loci in live mammalian cells and early pre-implantation embryos using CAS-LiveFISH. Sci. Rep. 2021, 11, 12220. [Google Scholar] [CrossRef]
- Iarovaia, O.V.; Rubtsov, M.; Ioudinkova, E.; Tsfasman, T.; Razin, S.V.; Vassetzky, Y.S. Dynamics of double strand breaks and chromosomal translocations. Mol. Cancer 2014, 13, 249. [Google Scholar] [CrossRef]
- Mine-Hattab, J.; Rothstein, R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 2012, 14, 510–517. [Google Scholar] [CrossRef]
- Miné-Hattab, J.; Darzacq, X. Chromatin Dynamics upon DNA Damage. In Chromatin and Epigenetics; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef]
- Krawczyk, P.M.; Borovski, T.; Stap, J.; Cijsouw, T.; ten Cate, R.; Medema, J.P.; Kanaar, R.; Franken, N.A.; Aten, J.A. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 2012, 125, 2127–2133. [Google Scholar] [CrossRef]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef]
- Lomov, N.A.; Viushkov, V.S.; Ulianov, S.V.; Gavrilov, A.A.; Alexeyevsky, D.A.; Artemov, A.V.; Razin, S.V.; Rubtsov, M.A. Recurrent Translocations in Topoisomerase Inhibitor-Related Leukemia Are Determined by the Features of DNA Breaks Rather than by the Proximity of the Translocating Genes. Int. J. Mol. Sci. 2022, 23, 9824. [Google Scholar] [CrossRef]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Muller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef]
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007, 9, 675–682. [Google Scholar] [CrossRef]
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-Scholz, G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177. [Google Scholar] [CrossRef]
- Zhang, Y.; McCord, R.P.; Ho, Y.J.; Lajoie, B.R.; Hildebrand, D.G.; Simon, A.C.; Becker, M.S.; Alt, F.W.; Dekker, J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 2012, 148, 908–921. [Google Scholar] [CrossRef]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial dynamics of chromosome translocations in living cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Germini, D.; Tsfasman, T.; Klibi, M.; El-Amine, R.; Pichugin, A.; Iarovaia, O.V.; Bilhou-Nabera, C.; Subra, F.; Bou Saada, Y.; Sukhanova, A.; et al. HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells. Leukemia 2017, 31, 2515–2522. [Google Scholar] [CrossRef]
- Mariame, B.; Kappler-Gratias, S.; Kappler, M.; Balor, S.; Gallardo, F.; Bystricky, K. Real-Time Visualization and Quantification of Human Cytomegalovirus Replication in Living Cells Using the ANCHOR DNA Labeling Technology. J. Virol. 2018, 92, e00571-18. [Google Scholar] [CrossRef]
- Komatsu, T.; Quentin-Froignant, C.; Carlon-Andres, I.; Lagadec, F.; Rayne, F.; Ragues, J.; Kehlenbach, R.H.; Zhang, W.; Ehrhardt, A.; Bystricky, K.; et al. In Vivo Labelling of Adenovirus DNA Identifies Chromatin Anchoring and Biphasic Genome Replication. J. Virol. 2018, 92, e00795-18. [Google Scholar] [CrossRef]
- Hinsberger, A.; Graillot, B.; Blachere Lopez, C.; Juliant, S.; Cerutti, M.; King, L.A.; Possee, R.D.; Gallardo, F.; Lopez Ferber, M. Tracing Baculovirus AcMNPV Infection Using a Real-Time Method Based on ANCHOR(TM) DNA Labeling Technology. Viruses 2020, 12, 50. [Google Scholar] [CrossRef]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94, e00135-20. [Google Scholar] [CrossRef]
- Muller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Muller, B.; Krausslich, H.G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. eLife 2021, 10, e64776. [Google Scholar] [CrossRef]
- Szydlowski, N.A.; Go, J.S.; Hu, Y.S. Chromatin imaging and new technologies for imaging the nucleome. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1442. [Google Scholar] [CrossRef]
- Nath, J.; Johnson, K.L. A review of fluorescence in situ hybridization (FISH): Current status and future prospects. Biotech. Histochem. 2000, 75, 54–78. [Google Scholar] [CrossRef]
- Cui, C.; Shu, W.; Li, P. Fluorescence In situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016, 4, 89. [Google Scholar] [CrossRef]
- Zink, D.; Sadoni, N.; Stelzer, E. Visualizing chromatin and chromosomes in living cells. Methods 2003, 29, 42–50. [Google Scholar] [CrossRef]
- Martin, R.M.; Leonhardt, H.; Cardoso, M.C. DNA labeling in living cells. Cytom. A 2005, 67, 45–52. [Google Scholar] [CrossRef]
- Lammle, C.A.; Varady, A.; Muller, T.G.; Sturtzel, C.; Riepl, M.; Mathes, B.; Eichhorst, J.; Sporbert, A.; Lehmann, M.; Krausslich, H.G.; et al. Photocaged Hoechst Enables Subnuclear Visualization and Cell Selective Staining of DNA in vivo. ChemBioChem 2021, 22, 548–556. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.C.; Sun, D.; Hu, J.; Wali, A.; Pass, H.; Fernandez-Madrid, F.; Harbut, M.R.; Tang, N. New insight into the molecular mechanisms of the biological effects of DNA minor groove binders. PLoS ONE 2011, 6, e25822. [Google Scholar] [CrossRef]
- Smith, P.J.; Blunt, N.; Wiltshire, M.; Hoy, T.; Teesdale-Spittle, P.; Craven, M.R.; Watson, J.V.; Amos, W.B.; Errington, R.J.; Patterson, L.H. Characteristics of a novel deep red/infrared fluorescent cell-permeant DNA probe, DRAQ5, in intact human cells analyzed by flow cytometry, confocal and multiphoton microscopy. Cytometry 2000, 40, 280–291. [Google Scholar] [CrossRef]
- Smith, P.J.; Wiltshire, M.; Errington, R.J. DRAQ5 labeling of nuclear DNA in live and fixed cells. Curr. Protoc. Cytom. 2004, 28, 7.25.1–7.25.11. [Google Scholar] [CrossRef]
- Shaban, H.A.; Barth, R.; Recoules, L.; Bystricky, K. Hi-D: Nanoscale mapping of nuclear dynamics in single living cells. Genome Biol. 2020, 21, 95. [Google Scholar] [CrossRef]
- Wojcik, K.; Dobrucki, J.W. Interaction of a DNA intercalator DRAQ5, and a minor groove binder SYTO17, with chromatin in live cells—influence on chromatin organization and histone-DNA interactions. Cytom. A 2008, 73, 555–562. [Google Scholar] [CrossRef]
- Purschke, M.; Rubio, N.; Held, K.D.; Redmond, R.W. Phototoxicity of Hoechst 33342 in time-lapse fluorescence microscopy. Photochem. Photobiol. Sci. 2010, 9, 1634–1639. [Google Scholar] [CrossRef]
- Lukinavicius, G.; Blaukopf, C.; Pershagen, E.; Schena, A.; Reymond, L.; Derivery, E.; Gonzalez-Gaitan, M.; D’Este, E.; Hell, S.W.; Wolfram Gerlich, D.; et al. SiR-Hoechst is a far-red DNA stain for live-cell nanoscopy. Nat. Commun. 2015, 6, 8497. [Google Scholar] [CrossRef]
- Lottersberger, F.; Karssemeijer, R.A.; Dimitrova, N.; de Lange, T. 53BP1 and the LINC Complex Promote Microtubule-Dependent DSB Mobility and DNA Repair. Cell 2015, 163, 880–893. [Google Scholar] [CrossRef]
- Robinett, C.C.; Straight, A.; Li, G.; Willhelm, C.; Sudlow, G.; Murray, A.; Belmont, A.S. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 1996, 135, 1685–1700. [Google Scholar] [CrossRef]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Khanna, N.; Zhang, Y.; Lucas, J.S.; Dudko, O.K.; Murre, C. Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions. Nat. Commun. 2019, 10, 2771. [Google Scholar] [CrossRef]
- Lassadi, I.; Kamgoue, A.; Goiffon, I.; Tanguy-le-Gac, N.; Bystricky, K. Differential chromosome conformations as hallmarks of cellular identity revealed by mathematical polymer modeling. PLoS Comput. Biol. 2015, 11, e1004306. [Google Scholar] [CrossRef]
- Tasan, I.; Sustackova, G.; Zhang, L.; Kim, J.; Sivaguru, M.; HamediRad, M.; Wang, Y.; Genova, J.; Ma, J.; Belmont, A.S.; et al. CRISPR/Cas9-mediated knock-in of an optimized TetO repeat for live cell imaging of endogenous loci. Nucleic Acids Res. 2018, 46, e100. [Google Scholar] [CrossRef] [PubMed]
- Sofueva, S.; Osman, F.; Lorenz, A.; Steinacher, R.; Castagnetti, S.; Ledesma, J.; Whitby, M.C. Ultrafine anaphase bridges, broken DNA and illegitimate recombination induced by a replication fork barrier. Nucleic Acids Res. 2011, 39, 6568–6584. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, G.W.; Chen, C.; Xie, X.S.; Zhuang, X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science 2011, 333, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Masui, O.; Bonnet, I.; Le Baccon, P.; Brito, I.; Pollex, T.; Murphy, N.; Hupe, P.; Barillot, E.; Belmont, A.S.; Heard, E. Live-cell chromosome dynamics and outcome of X chromosome pairing events during ES cell differentiation. Cell 2011, 145, 447–458. [Google Scholar] [CrossRef]
- Li, G.; Sudlow, G.; Belmont, A.S. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: Precise choreography of condensation/decondensation and nuclear positioning. J. Cell Biol. 1998, 140, 975–989. [Google Scholar] [CrossRef]
- Sinclair, P.; Bian, Q.; Plutz, M.; Heard, E.; Belmont, A.S. Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J. Cell Biol. 2010, 190, 761–776. [Google Scholar] [CrossRef]
- Kato, N.; Lam, E. Detection of chromosomes tagged with green fluorescent protein in live Arabidopsis thaliana plants. Genome Biol. 2001, 2, RESEARCH0045. [Google Scholar] [CrossRef]
- Matzke, A.J.; Huettel, B.; van der Winden, J.; Matzke, M. Use of two-color fluorescence-tagged transgenes to study interphase chromosomes in living plants. Plant Physiol. 2005, 139, 1586–1596. [Google Scholar] [CrossRef]
- Jacome, A.; Fernandez-Capetillo, O. Lac operator repeats generate a traceable fragile site in mammalian cells. EMBO Rep. 2011, 12, 1032–1038. [Google Scholar] [CrossRef]
- Beuzer, P.; Quivy, J.P.; Almouzni, G. Establishment of a replication fork barrier following induction of DNA binding in mammalian cells. Cell Cycle 2014, 13, 1607–1616. [Google Scholar] [CrossRef]
- Dubarry, M.; Loiodice, I.; Chen, C.L.; Thermes, C.; Taddei, A. Tight protein-DNA interactions favor gene silencing. Genes Dev. 2011, 25, 1365–1370. [Google Scholar] [CrossRef]
- Saad, H.; Gallardo, F.; Dalvai, M.; Tanguy-le-Gac, N.; Lane, D.; Bystricky, K. DNA dynamics during early double-strand break processing revealed by non-intrusive imaging of living cells. PLoS Genet. 2014, 10, e1004187. [Google Scholar] [CrossRef]
- Germier, T.; Kocanova, S.; Walther, N.; Bancaud, A.; Shaban, H.A.; Sellou, H.; Politi, A.Z.; Ellenberg, J.; Gallardo, F.; Bystricky, K. Real-Time Imaging of a Single Gene Reveals Transcription-Initiated Local Confinement. Biophys. J. 2017, 113, 1383–1394. [Google Scholar] [CrossRef]
- Germier, T.; Audibert, S.; Kocanova, S.; Lane, D.; Bystricky, K. Real-time imaging of specific genomic loci in eukaryotic cells using the ANCHOR DNA labelling system. Methods 2018, 142, 16–23. [Google Scholar] [CrossRef]
- Meschichi, A.; Ingouff, M.; Picart, C.; Mirouze, M.; Desset, S.; Gallardo, F.; Bystricky, K.; Picault, N.; Rosa, S.; Pontvianne, F. ANCHOR: A Technical Approach to Monitor Single-Copy Locus Localization in Planta. Front. Plant Sci. 2021, 12, 677849. [Google Scholar] [CrossRef]
- Gaj, T.; Sirk, S.J.; Shui, S.L.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef]
- Germini, D.; Tsfasman, T.; Zakharova, V.V.; Sjakste, N.; Lipinski, M.; Vassetzky, Y. A Comparison of Techniques to Evaluate the Effectiveness of Genome Editing. Trends Biotechnol. 2018, 36, 147–159. [Google Scholar] [CrossRef]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef]
- Lindhout, B.I.; Fransz, P.; Tessadori, F.; Meckel, T.; Hooykaas, P.J.; van der Zaal, B.J. Live cell imaging of repetitive DNA sequences via GFP-tagged polydactyl zinc finger proteins. Nucleic Acids Res. 2007, 35, e107. [Google Scholar] [CrossRef]
- Lindhout, B.I.; Meckel, T.; van der Zaal, B.J. Zinc finger-mediated live cell imaging in Arabidopsis roots. Methods Mol. Biol. 2010, 649, 383–398. [Google Scholar] [CrossRef]
- Casas-Delucchi, C.S.; Becker, A.; Bolius, J.J.; Cardoso, M.C. Targeted manipulation of heterochromatin rescues MeCP2 Rett mutants and re-establishes higher order chromatin organization. Nucleic Acids Res. 2012, 40, e176. [Google Scholar] [CrossRef]
- van Tol, N.; Rolloos, M.; Hooykaas, P.J.J.; van der Zaal, B.J. Two novel strategies to assess in vivo meiotic protein expression in Arabidopsis thaliana. F1000Research 2019, 8, 539. [Google Scholar] [CrossRef]
- Dutta, S.; Madan, S.; Sundar, D. Exploiting the recognition code for elucidating the mechanism of zinc finger protein-DNA interactions. BMC Genom. 2016, 17, 1037. [Google Scholar] [CrossRef][Green Version]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Muller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, N.; Kanemaki, M.T.; Yamamoto, Y.; Sadamura, Y.; Ito, Y.; Tokunaga, M.; Stasevich, T.J.; Kimura, H. Visualizing looping of two endogenous genomic loci using synthetic zinc-finger proteins with anti-FLAG and anti-HA frankenbodies in living cells. Genes Cells 2021, 26, 905–926. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Ma, H.; Reyes-Gutierrez, P.; Pederson, T. Visualization of repetitive DNA sequences in human chromosomes with transcription activator-like effectors. Proc. Natl. Acad. Sci. USA 2013, 110, 21048–21053. [Google Scholar] [CrossRef]
- Miyanari, Y.; Ziegler-Birling, C.; Torres-Padilla, M.E. Live visualization of chromatin dynamics with fluorescent TALEs. Nat. Struct. Mol. Biol. 2013, 20, 1321–1324. [Google Scholar] [CrossRef]
- Thanisch, K.; Schneider, K.; Morbitzer, R.; Solovei, I.; Lahaye, T.; Bultmann, S.; Leonhardt, H. Targeting and tracing of specific DNA sequences with dTALEs in living cells. Nucleic Acids Res. 2014, 42, e38. [Google Scholar] [CrossRef]
- Yuan, K.; Shermoen, A.W.; O’Farrell, P.H. Illuminating DNA replication during Drosophila development using TALE-lights. Curr. Biol. 2014, 24, R144–R145. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.; Sugano, S.S.; Kuwata, K.; Osakabe, K.; Matsunaga, S. Visualization of specific repetitive genomic sequences with fluorescent TALEs in Arabidopsis thaliana. J. Exp. Bot. 2016, 67, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, S.; Schindele, P.; Gladilin, E.; Dunemann, F.; Rutten, T.; Puchta, H.; Houben, A. Application of Aptamers Improves CRISPR-Based Live Imaging of Plant Telomeres. Front. Plant Sci. 2020, 11, 1254. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhang, H.; Wang, S.; Ding, M.; An, H.; Hou, Y.; Yang, X.; Wei, W.; Sun, Y.; Tang, C. Live visualization of genomic loci with BiFC-TALE. Sci. Rep. 2017, 7, 40192. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, M.; Li, W.; Zhang, Z.; Zhang, X.; Tan, T.; Zhang, X.E.; Cui, Z. Live cell imaging of single genomic loci with quantum dot-labeled TALEs. Nat. Commun. 2017, 8, 15318. [Google Scholar] [CrossRef]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Ma, H.; Naseri, A.; Reyes-Gutierrez, P.; Wolfe, S.A.; Zhang, S.; Pederson, T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, 3002–3007. [Google Scholar] [CrossRef]
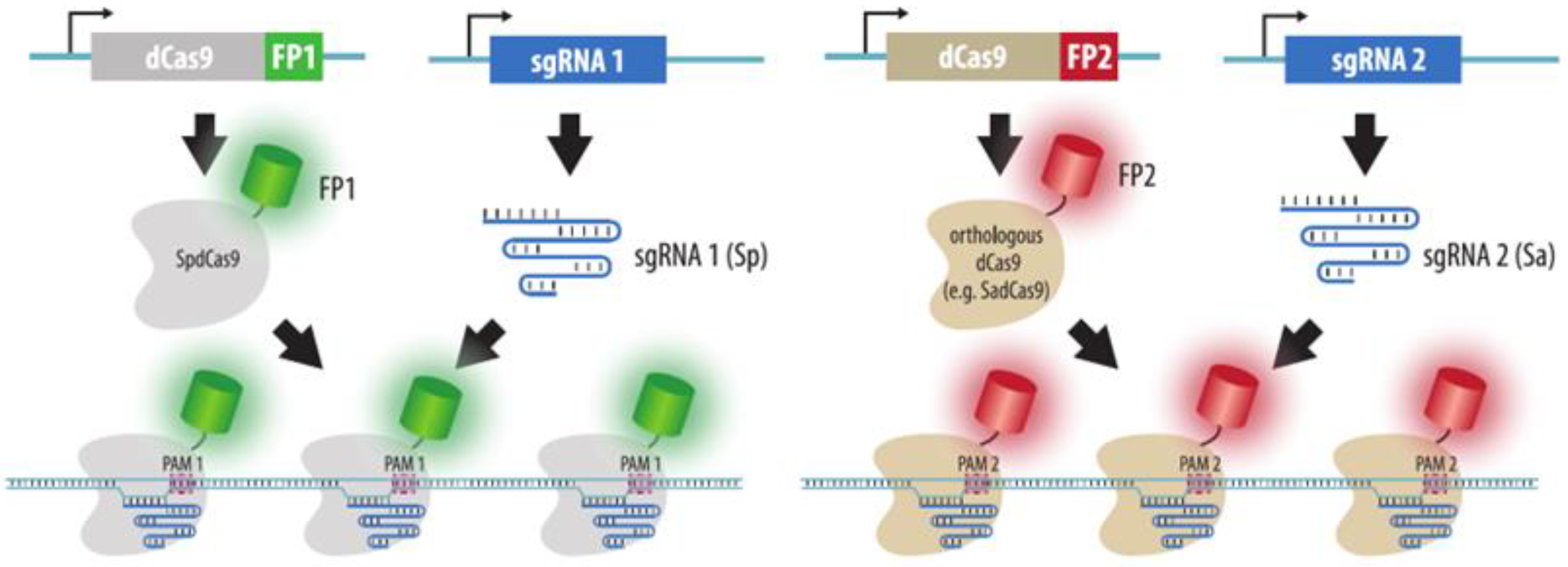
- Chen, B.; Hu, J.; Almeida, R.; Liu, H.; Balakrishnan, S.; Covill-Cooke, C.; Lim, W.A.; Huang, B. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res. 2016, 44, e75. [Google Scholar] [CrossRef]
- Deng, W.; Shi, X.; Tjian, R.; Lionnet, T.; Singer, R.H. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11870–11875. [Google Scholar] [CrossRef]
- Shao, S.; Zhang, W.; Hu, H.; Xue, B.; Qin, J.; Sun, C.; Sun, Y.; Wei, W.; Sun, Y. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res. 2016, 44, e86. [Google Scholar] [CrossRef]
- Wang, S.; Su, J.H.; Zhang, F.; Zhuang, X. An RNA-aptamer-based two-color CRISPR labeling system. Sci. Rep. 2016, 6, 26857. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016, 34, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Parlak, M.; Kuscu, C.; Bandaria, J.; Mir, M.; Szlachta, K.; Singh, R.; Darzacq, X.; Yildiz, A.; Adli, M. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017, 8, 14725. [Google Scholar] [CrossRef]
- Adamala, K.P.; Martin-Alarcon, D.A.; Boyden, E.S. Programmable RNA-binding protein composed of repeats of a single modular unit. Proc. Natl. Acad. Sci. USA 2016, 113, E2579–E2588. [Google Scholar] [CrossRef]
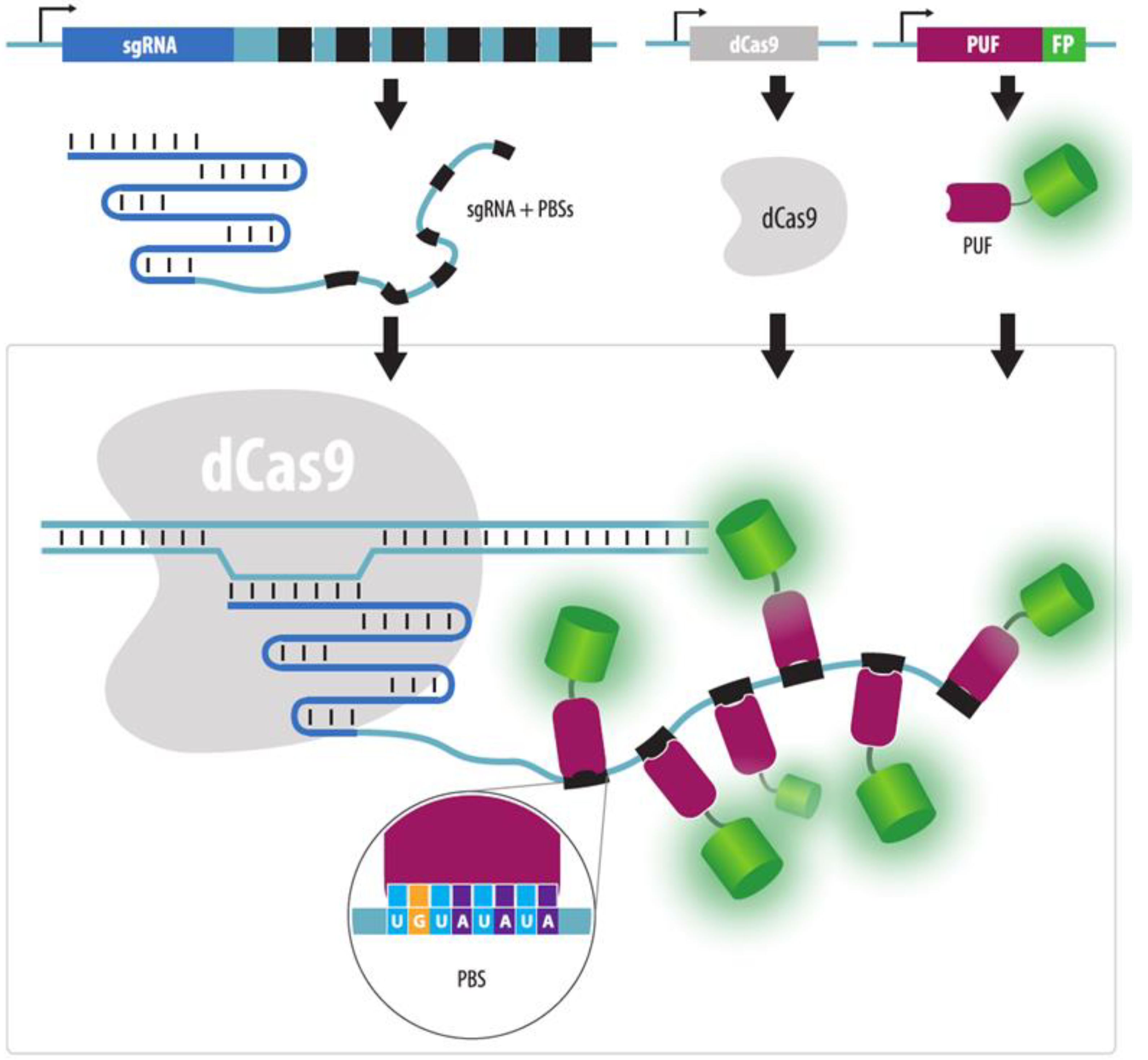
- Cheng, A.W.; Jillette, N.; Lee, P.; Plaskon, D.; Fujiwara, Y.; Wang, W.; Taghbalout, A.; Wang, H. Casilio: A versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016, 26, 254–257. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016, 214, 529–537. [Google Scholar] [CrossRef]
- Maass, P.G.; Barutcu, A.R.; Shechner, D.M.; Weiner, C.L.; Mele, M.; Rinn, J.L. Spatiotemporal allele organization by allele-specific CRISPR live-cell imaging (SNP-CLING). Nat. Struct. Mol. Biol. 2018, 25, 176–184. [Google Scholar] [CrossRef]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Wang, S.; Hao, Y.; Zhang, L.; Wang, F.; Li, J.; Wang, L.; Fan, C. Multiplexed Superresolution CRISPR Imaging of Chromatin in Living Cells. CCS Chem. 2019, 1, 278–285. [Google Scholar] [CrossRef]
- Wu, X.; Mao, S.; Yang, Y.; Rushdi, M.N.; Krueger, C.J.; Chen, A.K. A CRISPR/molecular beacon hybrid system for live-cell genomic imaging. Nucleic Acids Res. 2018, 46, e80. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Ying, Y.; Wu, X.; Krueger, C.J.; Chen, A.K. CRISPR/dual-FRET molecular beacon for sensitive live-cell imaging of non-repetitive genomic loci. Nucleic Acids Res. 2019, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Rong, Z.; Lin, Y. Live cell imaging of genomic loci using dCas9-SunTag system and a bright fluorescent protein. Protein Cell 2017, 8, 853–855. [Google Scholar] [CrossRef]
- Boersma, S.; Khuperkar, D.; Verhagen, B.M.P.; Sonneveld, S.; Grimm, J.B.; Lavis, L.D.; Tanenbaum, M.E. Multi-Color Single-Molecule Imaging Uncovers Extensive Heterogeneity in mRNA Decoding. Cell 2019, 178, 458–472 e419. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zou, W.; Xu, H.; Liang, Y.; Huang, B. Efficient labeling and imaging of protein-coding genes in living cells using CRISPR-Tag. Nat. Commun. 2018, 9, 5065. [Google Scholar] [CrossRef]
- Xu, H.; Wang, J.; Liang, Y.; Fu, Y.; Li, S.; Huang, J.; Xu, H.; Zou, W.; Chen, B. TriTag: An integrative tool to correlate chromatin dynamics and gene expression in living cells. Nucleic Acids Res. 2020, 48, 13013–13014. [Google Scholar] [CrossRef]
- Hong, Y.; Lu, G.; Duan, J.; Liu, W.; Zhang, Y. Comparison and optimization of CRISPR/dCas9/gRNA genome-labeling systems for live cell imaging. Genome Biol. 2018, 19, 39. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.C.; Chung, Y.C.; Naseri, A.; Grunwald, D.; Zhang, S.; Pederson, T. Cell cycle- and genomic distance-dependent dynamics of a discrete chromosomal region. J. Cell Biol. 2019, 218, 1467–1477. [Google Scholar] [CrossRef]
- Ma, H.; Tu, L.C.; Naseri, A.; Chung, Y.C.; Grunwald, D.; Zhang, S.; Pederson, T. CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging. Nat. Methods 2018, 15, 928–931. [Google Scholar] [CrossRef]
- Chaudhary, N.; Nho, S.H.; Cho, H.; Gantumur, N.; Ra, J.S.; Myung, K.; Kim, H. Background-suppressed live visualization of genomic loci with an improved CRISPR system based on a split fluorophore. Genome Res. 2020, 30, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.; Matsunaga, S. Visualization of Chromatin Loci with Transiently Expressed CRISPR/Cas9 in Plants. Cytologia 2017, 82, 559–562. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, P.; Tian, F.; Gao, G.; Huang, L.; Wei, W.; Xie, X.S. Painting a specific chromosome with CRISPR/Cas9 for live-cell imaging. Cell Res. 2017, 27, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Swigut, T.; Spencley, A.; Bauer, M.R.; Chung, M.; Meyer, T.; Wysocka, J. Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 2018, 359, 1050–1055. [Google Scholar] [CrossRef]
- Ochiai, H.; Sugawara, T.; Yamamoto, T. Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res. 2015, 43, e127. [Google Scholar] [CrossRef]
- Dreissig, S.; Schiml, S.; Schindele, P.; Weiss, O.; Rutten, T.; Schubert, V.; Gladilin, E.; Mette, M.F.; Puchta, H.; Houben, A. Live-cell CRISPR imaging in plants reveals dynamic telomere movements. Plant J. 2017, 91, 565–573. [Google Scholar] [CrossRef]
- Anton, T.; Bultmann, S.; Leonhardt, H.; Markaki, Y. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus 2014, 5, 163–172. [Google Scholar] [CrossRef]
- Duan, J.; Lu, G.; Hong, Y.; Hu, Q.; Mai, X.; Guo, J.; Si, X.; Wang, F.; Zhang, Y. Live imaging and tracking of genome regions in CRISPR/dCas9 knock-in mice. Genome Biol. 2018, 19, 192. [Google Scholar] [CrossRef]
- Athmane, N.; Williamson, I.; Boyle, S.; Biddie, S.C.; Bickmore, W.A. MUC4 is not expressed in cell lines used for live cell imaging. Wellcome Open Res. 2021, 6, 265. [Google Scholar] [CrossRef]
- Gao, X.; Tsang, J.C.; Gaba, F.; Wu, D.; Lu, L.; Liu, P. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 2014, 42, e155. [Google Scholar] [CrossRef]
- Shariati, S.A.; Dominguez, A.; Xie, S.; Wernig, M.; Qi, L.S.; Skotheim, J.M. Reversible Disruption of Specific Transcription Factor-DNA Interactions Using CRISPR/Cas9. Mol. Cell 2019, 74, 622–633 e624. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.; Murphy, S. Advances in Chromatin Imaging at Kilobase-Scale Resolution. Trends Genet. 2020, 36, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.J.; Murphy, S.E.; Hafner, A.; Cinquini, I.S.; Walker, C.A.; Boettiger, A.N. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 2019, 568, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Imai, R.; Tanbo, M.; Nagashima, R.; Tamura, S.; Tani, T.; Joti, Y.; Tomita, M.; Hibino, K.; Kanemaki, M.T.; et al. Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging. Mol. Cell 2017, 67, 282–293.e287. [Google Scholar] [CrossRef] [PubMed]
- Mehra, D.; Adhikari, S.; Banerjee, C.; Puchner, E.M. Characterizing locus specific chromatin structure and dynamics with correlative conventional and super-resolution imaging in living cells. Nucleic Acids Res. 2022, 50, e78. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
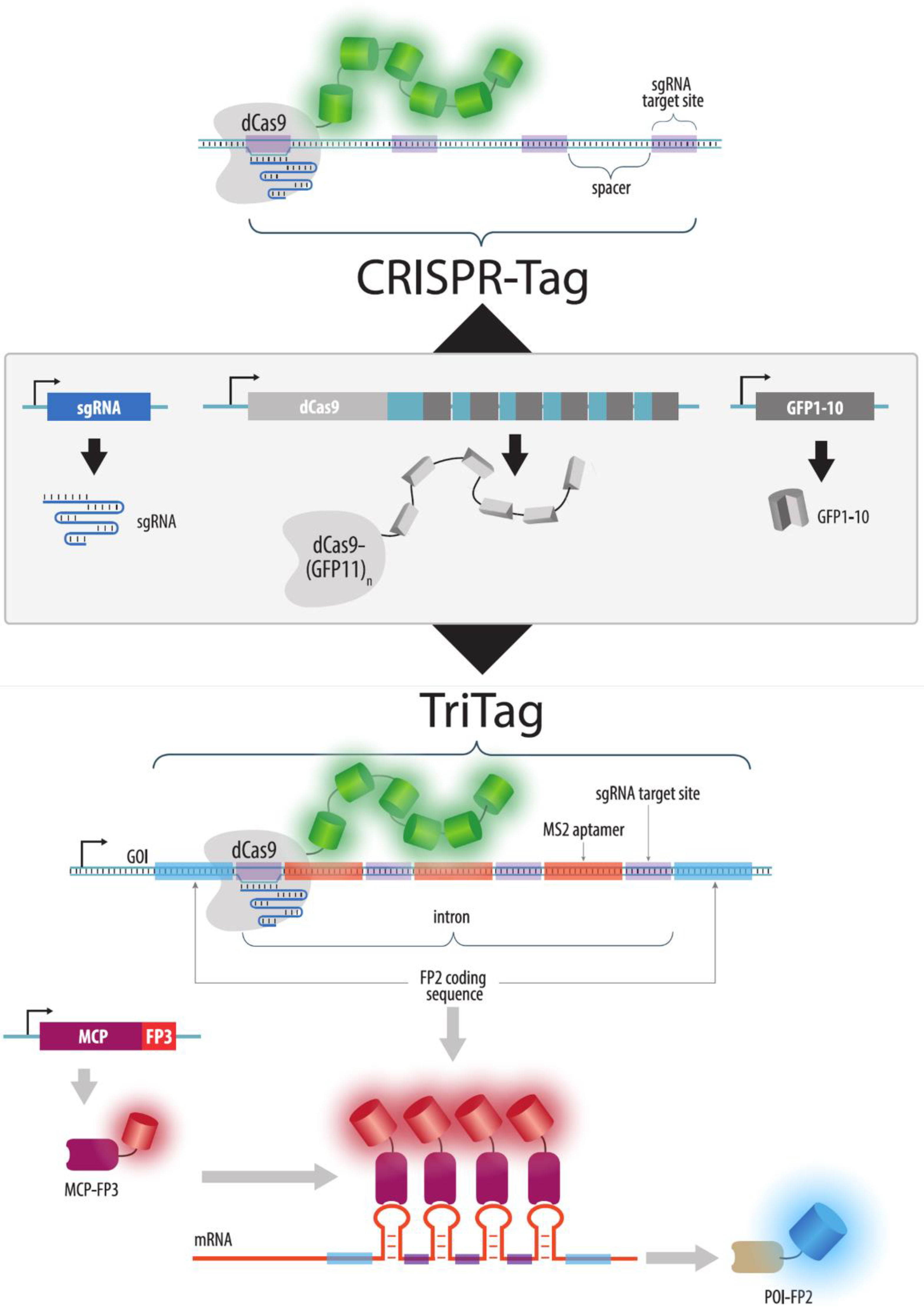{kind=link}
| Method | Visualization Principle | Locus Specific Imaging | Genome Editing Required | Visualization of Unique Loci without Endogenous Repeats | Advantages | Disadvantages | References to Corresponding Sections |
|---|---|---|---|---|---|---|---|
| Crude chromatin imaging with DNA-binding dyes, fluorescent histones or fluorescent nucleotides | Chromatin is stained by DNA binding dyes, by expression of fluorescent histones or by addition of fluorescent nucleotides | No | No | No | Reliable and easy to implement (compared to the other techniques) | 1. Not intended to visualize specific loci | Section 2 |
| 2. DNA-binding dyes may be toxic to cells | |||||||
| FROS | Array of multiple copies of an operator sequence is integrated into the target genomic locus. Bacterial repressor protein fused to a FP is expressed in cells and binds operator sequences | Yes | Yes: insertion of an array of operators | Yes | Reliable | 1. Strong binding of the repressor to the operator may cause replication block and transcription silencing | Section 3.1 |
| 2. The need to integrate the construct into a specific locus | |||||||
| 3. Difficulty in cloning repetitive constructs | |||||||
| 4. A cell line must be generated for every studied locus | |||||||
| ParB-INT | Bacterial INT sequence is integrated into the target genomic locus. ParB-FP is expressed in cells, binds operator sequences and oligomerizes on it | Yes | Yes: insertion of INT-sequence | Yes | A small sequence inserted into target locus. More native than FROS | 1. The need to integrate the construct into a specific locus | Section 3.2 |
| 2. A cell line must be generated for every studied locus | |||||||
| ZFP | Zinc finger protein fused to a FP are expressed in cells | Yes | No | Possible by signal amplification with scFv | No need to insert a tag in the locus of interest | 1. The technique is mainly applicable for highly repeated sequences | Section 4.1 |
| 2. Complex engineering of ZFP | |||||||
| TALE | TALE protein fused to a FP are expressed in cells | Yes | No | Possible by quantum dot-labeled TALE | No need to insert a tag in the locus of interest | 1. The technique is mainly applicable for highly repeated sequences | Section 4.2 |
| 2. Complex engineering of TALEs | |||||||
| CRISPR: dCas9-FP | dCas9-FP and sgRNAs are expressed in cells | Yes | No * | Requires multiple sgRNA. Alternatively, CRISPR-Tag may be integrated into a locus of interest | 1. No need to insert a tag in the locus of interest | 1. dCas9 binding may change the expression of a target locus | Section 4.3.1 |
| 2. Target change is easy | 2. Multicolor imaging requires different dCas9 proteins, their cognate gRNAs and PAM sequences | ||||||
| 3. Difficult visualization of non-repeated loci | |||||||
| CRISPR: sgRNA + aptamers | dCas9, sgRNAs with aptameric modules and fluorescent proteins that bind aptamers are expressed in cells | Yes | No * | Possible with sgRNA containing multiple aptamers | 1. No need to insert a tag in the locus of interest | 1. Difficult visualization of non-repeated loci | Section 4.3.2 |
| 2. Target change is easy | |||||||
| 3. Easy multicolor imaging | 2. Aptamer-binding proteins may accumulate in the nucleoli | ||||||
| 4. High versatility and customization potential | |||||||
| CRISPR: dCas9-organic fluorophore | Recombinant dCas9 conjugated to a fluorophore complexed to sgRNA are transfected into cells | Yes | No * | Difficult, requires multiple sgRNA synthesis | 1. No laborious cloning | 1. Difficult visualization of non-repeated loci | Section 4.3.3 |
| 2. No need to insert a tag in the locus of interest | 2. Need for transfection of fluorescent RNP prior to each experiment | ||||||
| 3. Target change is easy | |||||||
| 4. Organic fluorophores are bright | |||||||
| CRISPR: sgRNA-organic fluorophore | Fluorescent gRNA or molecular beacon probes to gRNA are transfected into cells expressing dCas9; or fRNP of fluorescent gRNA, dCas9 and tracrRNA is pre-assembled and then transfected into cells | Yes | No * | Possible with several sgRNAs and dual-FRET molecular beacon system | 1. No laborious cloning | 1. Difficult visualization of non-repeated loci | Section 4.3.4 |
| 2. No need to insert a tag in the locus of interest | |||||||
| 3. Target change is easy | 2. Need for transfection of fluorescent RNP, sgRNA or molecular beacons prior to each experiment | ||||||
| 4. Organic fluorophores are bright |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viushkov, V.S.; Lomov, N.A.; Rubtsov, M.A.; Vassetzky, Y.S. Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging. Cells 2022, 11, 4086. https://doi.org/10.3390/cells11244086
Viushkov VS, Lomov NA, Rubtsov MA, Vassetzky YS. Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging. Cells. 2022; 11(24):4086. https://doi.org/10.3390/cells11244086
Chicago/Turabian StyleViushkov, Vladimir S., Nikolai A. Lomov, Mikhail A. Rubtsov, and Yegor S. Vassetzky. 2022. "Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging" Cells 11, no. 24: 4086. https://doi.org/10.3390/cells11244086
APA StyleViushkov, V. S., Lomov, N. A., Rubtsov, M. A., & Vassetzky, Y. S. (2022). Visualizing the Genome: Experimental Approaches for Live-Cell Chromatin Imaging. Cells, 11(24), 4086. https://doi.org/10.3390/cells11244086