

A Targeted Epigenetic Clock for the Prediction of Biological Age

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cohorts
2.2. EpiTYPER DNAm Analysis
2.3. Predictive Model and Statistical Analyses
3. Results and Discussion
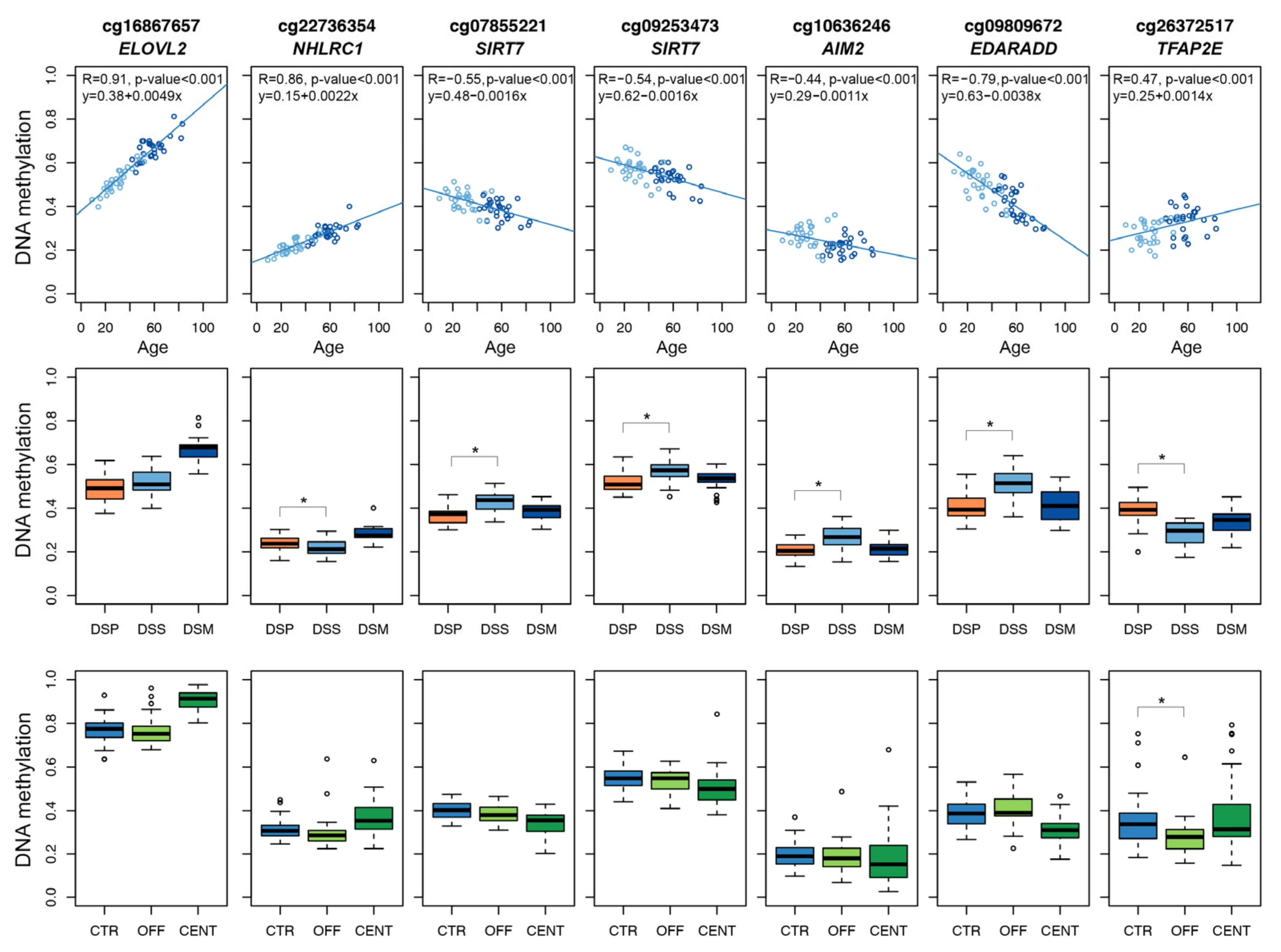
3.1. Rationale for the Selection of Target Genomic Regions
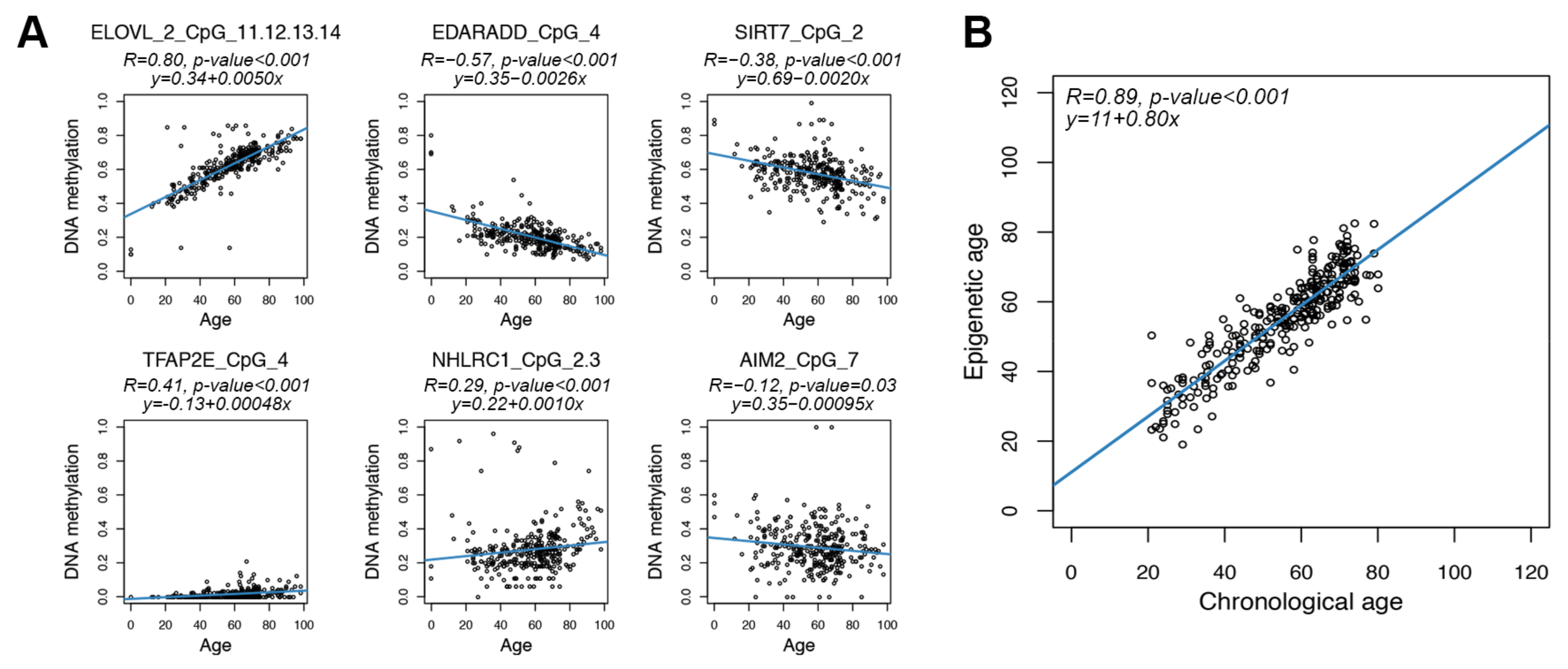
3.2. Design of the Targeted Assay
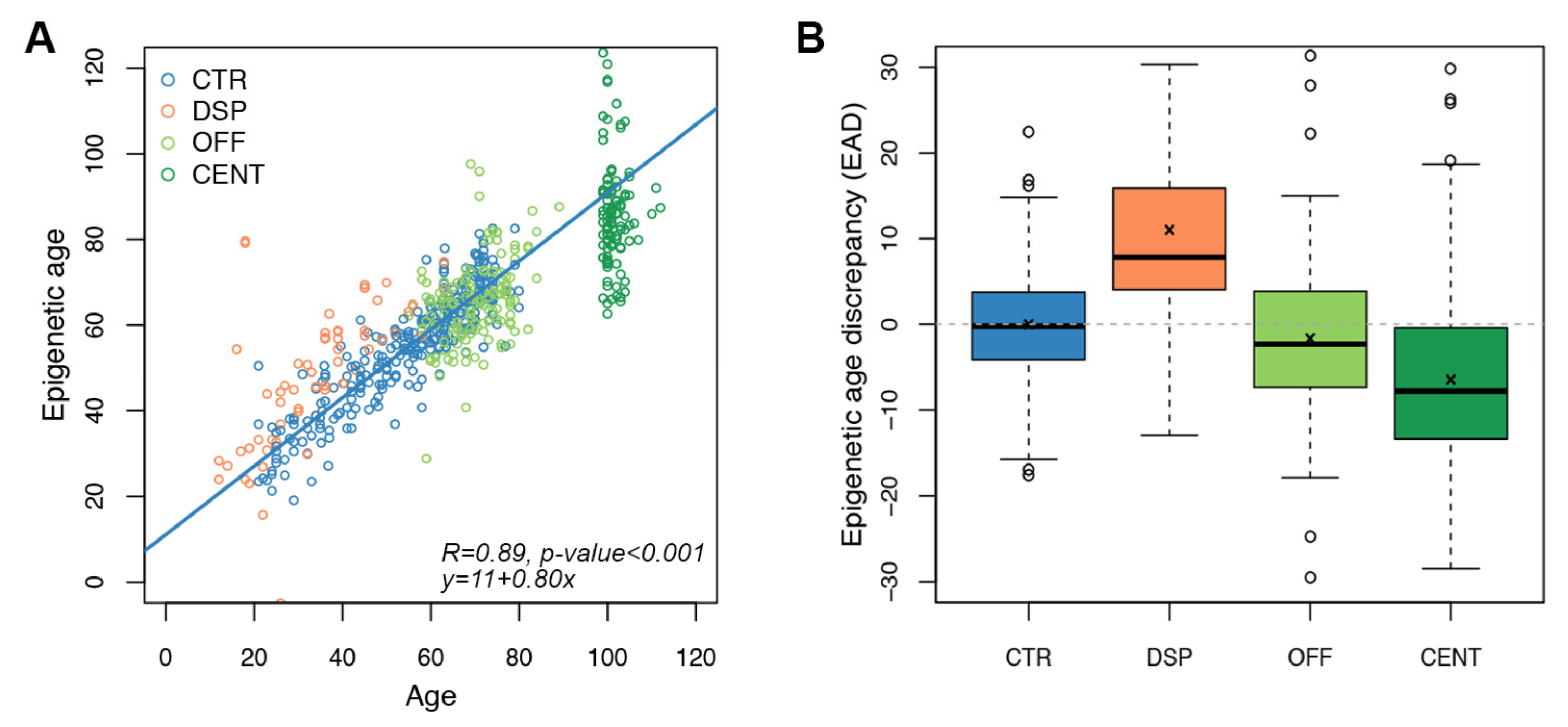
3.3. Age Prediction Using the Targeted Epigenetic Clock
3.4. Application of the Targeted Epigenetic Clock to Models of Increased and Decreased Biological Age
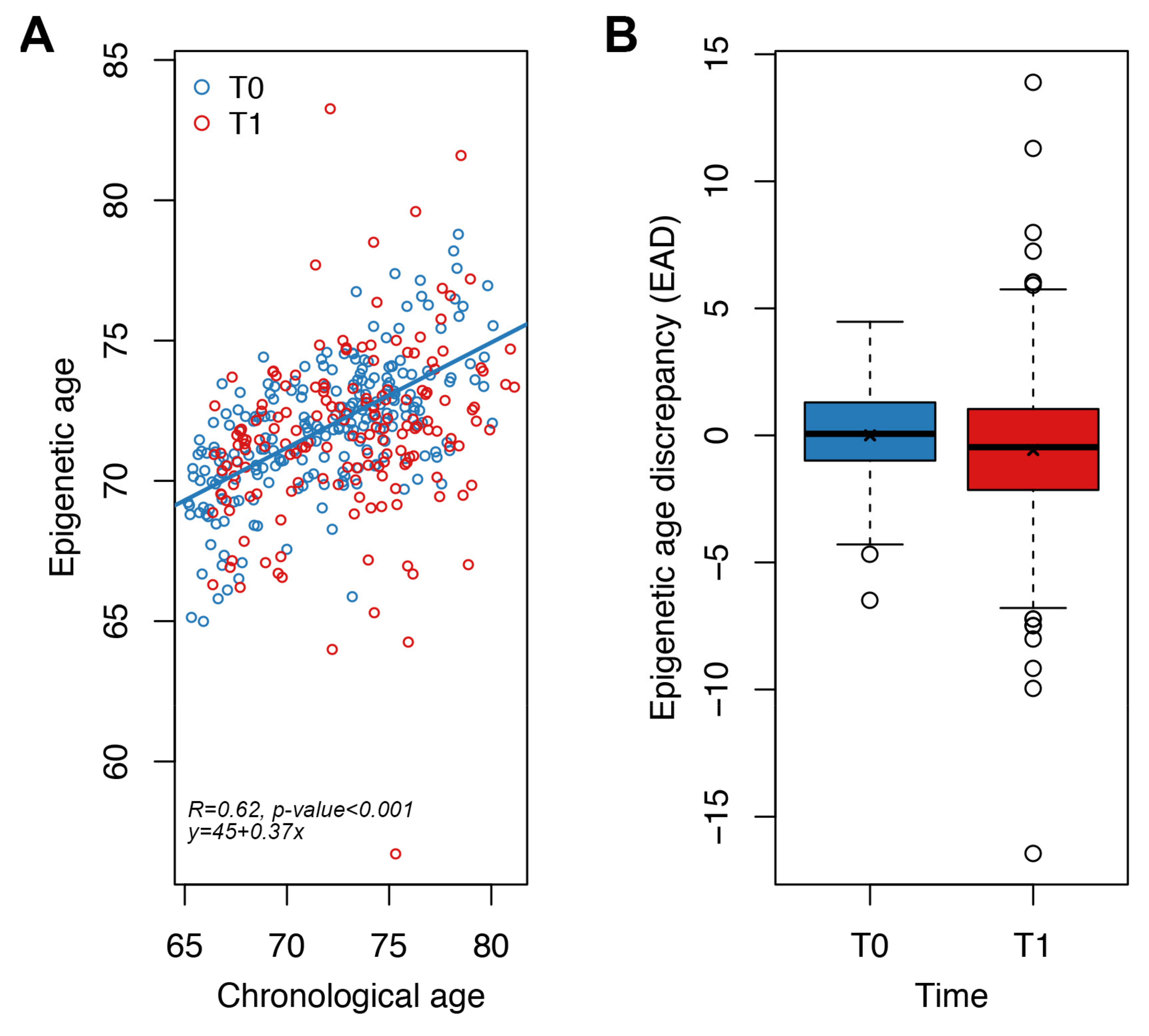
3.5. Application of the Targeted Epigenetic Clock to an Independent Validation Dataset
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological Age Predictors. eBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved Precision of Epigenetic Clock Estimates across Tissues and Its Implication for Biological Ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA Methylation GrimAge Strongly Predicts Lifespan and Healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An Epigenetic Biomarker of Aging for Lifespan and Healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA Methylation Aging Clocks: Challenges and Recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef]
- Belsky, D.W.; Caspi, A.; Arseneault, L.; Baccarelli, A.; Corcoran, D.L.; Gao, X.; Hannon, E.; Harrington, H.L.; Rasmussen, L.J.; Houts, R.; et al. Quantification of the Pace of Biological Aging in Humans through a Blood Test, the DunedinPoAm DNA Methylation Algorithm. eLife 2020, 9, e54870. [Google Scholar] [CrossRef]
- Higgins-Chen, A.T.; Thrush, K.L.; Wang, Y.; Minteer, C.J.; Kuo, P.-L.; Wang, M.; Niimi, P.; Sturm, G.; Lin, J.; Moore, A.Z.; et al. A Computational Solution for Bolstering Reliability of Epigenetic Clocks: Implications for Clinical Trials and Longitudinal Tracking. Nat. Aging 2022, 2, 644–661. [Google Scholar] [CrossRef]
- Maddock, J.; Castillo-Fernandez, J.; Wong, A.; Cooper, R.; Richards, M.; Ong, K.K.; Ploubidis, G.B.; Goodman, A.; Kuh, D.; Bell, J.T.; et al. DNA Methylation Age and Physical and Cognitive Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 504–511. [Google Scholar] [CrossRef]
- McCrory, C.; Fiorito, G.; Hernandez, B.; Polidoro, S.; O’Halloran, A.M.; Hever, A.; Ni Cheallaigh, C.; Lu, A.T.; Horvath, S.; Vineis, P.; et al. GrimAge Outperforms Other Epigenetic Clocks in the Prediction of Age-Related Clinical Phenotypes and All-Cause Mortality. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 76, 741–749. [Google Scholar] [CrossRef]
- Duan, R.; Fu, Q.; Sun, Y.; Li, Q. Epigenetic Clock: A Promising Biomarker and Practical Tool in Aging. Ageing Res. Rev. 2022, 81, 101743. [Google Scholar] [CrossRef] [PubMed]
- Gialluisi, A.; Santoro, A.; Tirozzi, A.; Cerletti, C.; Donati, M.B.; de Gaetano, G.; Franceschi, C.; Iacoviello, L. Epidemiological and Genetic Overlap among Biological Aging Clocks: New Challenges in Biogerontology. Ageing Res. Rev. 2021, 72, 101502. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; English, B.W.; Shokhirev, M.N.; Sinclair, D.A.; Cuellar, T.L. Human Age Reversal: Fact or Fiction? Aging Cell 2022, 21, e13664. [Google Scholar] [CrossRef]
- Han, Y.; Franzen, J.; Stiehl, T.; Gobs, M.; Kuo, C.-C.; Nikolić, M.; Hapala, J.; Koop, B.E.; Strathmann, K.; Ritz-Timme, S.; et al. New Targeted Approaches for Epigenetic Age Predictions. BMC Biol. 2020, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of Blood Can Be Tracked by DNA Methylation Changes at Just Three CpG Sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Ferreira, M.R.P.; Deary, I.J.; Wagner, W. DNA Methylation Levels at Individual Age-Associated CpG Sites Can Be Indicative for Life Expectancy. Aging 2016, 8, 394–401. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, J.; Hou, J.; Fu, X.; Li, L.; Hou, Y. Developing a DNA Methylation Assay for Human Age Prediction in Blood and Bloodstain. Forensic Sci. Int. Genet. 2015, 17, 129–136. [Google Scholar] [CrossRef]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Ż.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a Forensically Useful Age Prediction Method Based on DNA Methylation Analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef]
- Soares Bispo Santos Silva, D.; Antunes, J.; Balamurugan, K.; Duncan, G.; Sampaio Alho, C.; McCord, B. Evaluation of DNA Methylation Markers and Their Potential to Predict Human Aging. Electrophoresis 2015, 36, 1775–1780. [Google Scholar] [CrossRef]
- Spólnicka, M.; Pośpiech, E.; Pepłońska, B.; Zbieć-Piekarska, R.; Makowska, Ż.; Pięta, A.; Karłowska-Pik, J.; Ziemkiewicz, B.; Wężyk, M.; Gasperowicz, P.; et al. DNA Methylation in ELOVL2 and C1orf132 Correctly Predicted Chronological Age of Individuals from Three Disease Groups. Int. J. Leg. Med. 2018, 132, 1–11. [Google Scholar] [CrossRef]
- Freire-Aradas, A.; Phillips, C.; Mosquera-Miguel, A.; Girón-Santamaría, L.; Gómez-Tato, A.; Casares de Cal, M.; Álvarez-Dios, J.; Ansede-Bermejo, J.; Torres-Español, M.; Schneider, P.M.; et al. Development of a Methylation Marker Set for Forensic Age Estimation Using Analysis of Public Methylation Data and the Agena Bioscience EpiTYPER System. Forensic Sci. Int. Genet. 2016, 24, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Montesanto, A.; D’Aquila, P.; Lagani, V.; Paparazzo, E.; Geracitano, S.; Formentini, L.; Giacconi, R.; Cardelli, M.; Provinciali, M.; Bellizzi, D.; et al. A New Robust Epigenetic Model for Forensic Age Prediction. J. Forensic Sci. 2020, 65, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Freire-Aradas, A.; Girón-Santamaría, L.; Mosquera-Miguel, A.; Ambroa-Conde, A.; Phillips, C.; Casares de Cal, M.; Gómez-Tato, A.; Álvarez-Dios, J.; Pospiech, E.; Aliferi, A.; et al. A Common Epigenetic Clock from Childhood to Old Age. Forensic Sci. Int. Genet. 2022, 60, 102743. [Google Scholar] [CrossRef]
- Horvath, S.; Pirazzini, C.; Bacalini, M.G.; Gentilini, D.; Di Blasio, A.M.; Delledonne, M.; Mari, D.; Arosio, B.; Monti, D.; Passarino, G.; et al. Decreased Epigenetic Age of PBMCs from Italian Semi-Supercentenarians and Their Offspring. Aging 2015, 7, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated Epigenetic Aging in Down Syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Gentilini, D.; Boattini, A.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Scurti, M.; Remondini, D.; Capri, M.; et al. Identification of a DNA Methylation Signature in Blood Cells from Persons with Down Syndrome. Aging 2015, 7, 82–96. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Santoro, A.; Pini, E.; Scurti, M.; Palmas, G.; Berendsen, A.; Brzozowska, A.; Pietruszka, B.; Szczecinska, A.; Cano, N.; Meunier, N.; et al. Combating Inflammaging through a Mediterranean Whole Diet Approach: The NU-AGE Project’s Conceptual Framework and Design. Mech. Ageing Dev. 2014, 136–137, 3–13. [Google Scholar] [CrossRef]
- Berendsen, A.; Santoro, A.; Pini, E.; Cevenini, E.; Ostan, R.; Pietruszka, B.; Rolf, K.; Cano, N.; Caille, A.; Lyon-Belgy, N.; et al. Reprint of: A Parallel Randomized Trial on the Effect of a Healthful Diet on Inflammageing and Its Consequences in European Elderly People: Design of the NU-AGE Dietary Intervention Study. Mech. Ageing Dev. 2014, 136–137, 14–21. [Google Scholar] [CrossRef]
- Berendsen, A.A.M.; Van de Rest, O.; Feskens, E.J.M.; Santoro, A.; Ostan, R.; Pietruszka, B.; Brzozowska, A.; Stelmaszczyk-Kusz, A.; Jennings, A.; Gillings, R.; et al. Changes in Dietary Intake and Adherence to the NU-AGE Diet Following a One-Year Dietary Intervention among European Older Adults—Results of the NU-AGE Randomized Trial. Nutrients 2018, 10, 1905. [Google Scholar] [CrossRef]
- Gensous, N.; Garagnani, P.; Santoro, A.; Giuliani, C.; Ostan, R.; Fabbri, C.; Milazzo, M.; Gentilini, D.; di Blasio, A.M.; Pietruszka, B.; et al. One-Year Mediterranean Diet Promotes Epigenetic Rejuvenation with Country- and Sex-Specific Effects: A Pilot Study from the NU-AGE Project. Geroscience 2020, 42, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Van Buuren, S.; Groothuis-Oudshoorn, K. Mice: Multivariate Imputation by Chained Equations in R. J. Stat. Softw. 2011, 45, 1–67. [Google Scholar] [CrossRef]
- Du, P.; Kibbe, W.A.; Lin, S.M. Lumi: A Pipeline for Processing Illumina Microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 Gene as a New Epigenetic Marker of Age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Deelen, J.; Pirazzini, C.; De Cecco, M.; Giuliani, C.; Lanzarini, C.; Ravaioli, F.; Marasco, E.; van Heemst, D.; Suchiman, H.E.D.; et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and In Vitro Evidences of a Cell Replication Process. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1015–1023. [Google Scholar] [CrossRef]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-Related DNA Methylation Changes Are Tissue-Specific with ELOVL2 Promoter Methylation as Exception. Epigenet. Chromatin 2018, 11, 25. [Google Scholar] [CrossRef]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Makowska, Ż.; Spas, A.; Parys-Proszek, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Examination of DNA Methylation Status of the ELOVL2 Marker May Be Useful for Human Age Prediction in Forensic Science. Forensic Sci. Int. Genet. 2015, 14, 161–167. [Google Scholar] [CrossRef]
- Jung, S.-E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.-J.; Lee, H.Y. DNA Methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 Genes for Age Prediction from Blood, Saliva, and Buccal Swab Samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef]
- Han, X.; Xiao, C.; Yi, S.; Li, Y.; Chen, M.; Huang, D. Accurate Age Estimation from Blood Samples of Han Chinese Individuals Using Eight High-Performance Age-Related CpG Sites. Int. J. Leg. Med. 2022, 136, 1655–1665. [Google Scholar] [CrossRef]
- Correia Dias, H.; Manco, L.; Corte Real, F.; Cunha, E. A Blood-Bone-Tooth Model for Age Prediction in Forensic Contexts. Biology 2021, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Durso, D.F.; Bacalini, M.G.; Sala, C.; Pirazzini, C.; Marasco, E.; Bonafé, M.; do Valle, Í.F.; Gentilini, D.; Castellani, G.; Faria, A.M.C.; et al. Acceleration of Leukocytes’ Epigenetic Age as an Early Tumor and Sex-Specific Marker of Breast and Colorectal Cancer. Oncotarget 2017, 8, 23237–23245. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Zigman, W.B. Atypical Aging in Down Syndrome. Dev. Disabil. Res. Rev. 2013, 18, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M. Centenarians as a Model for Healthy Aging. Biochem. Soc. Trans. 2003, 31, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Bucci, L.; Ostan, R.; Cevenini, E.; Pini, E.; Scurti, M.; Vitale, G.; Mari, D.; Caruso, C.; Sansoni, P.; Fanelli, F.; et al. Centenarians’ Offspring as a Model of Healthy Aging: A Reappraisal of the Data on Italian Subjects and a Comprehensive Overview. Aging 2016, 8, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Gensous, N.; Bacalini, M.G.; Franceschi, C.; Garagnani, P. Down Syndrome, Accelerated Aging and Immunosenescence. Semin. Immunopathol. 2020, 42, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Henneman, P.; Bouman, A.; Mul, A.; Knegt, L.; van der Kevie-Kersemaekers, A.-M.; Zwaveling-Soonawala, N.; Meijers-Heijboer, H.E.J.; van Trotsenburg, A.S.P.; Mannens, M.M. Widespread Domain-like Perturbations of DNA Methylation in Whole Blood of Down Syndrome Neonates. PLoS ONE 2018, 13, e0194938. [Google Scholar] [CrossRef]
- Muskens, I.S.; Li, S.; Jackson, T.; Elliot, N.; Hansen, H.M.; Myint, S.S.; Pandey, P.; Schraw, J.M.; Roy, R.; Anguiano, J.; et al. The Genome-Wide Impact of Trisomy 21 on DNA Methylation and Its Implications for Hematopoiesis. Nat. Commun. 2021, 12, 821. [Google Scholar] [CrossRef]
- Xu, K.; Li, S.; Muskens, I.S.; Elliott, N.; Myint, S.S.; Pandey, P.; Hansen, H.M.; Morimoto, L.M.; Kang, A.Y.; Ma, X.; et al. Accelerated Epigenetic Aging in Newborns with Down Syndrome. Aging Cell 2022, 21, e13652. [Google Scholar] [CrossRef]
- Naumova, O.Y.; Lipschutz, R.; Rychkov, S.Y.; Zhukova, O.V.; Grigorenko, E.L. DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome. Genes 2021, 12, 1115. [Google Scholar] [CrossRef] [PubMed]
- Clas, G.S.; Fernández, E.A.; Vázquez, J.C.G.; Pertierra, L.; Guimet, N.M.; Tapajoz, F.; Helou, B.; Itzcovich, T.; Kennedy, M.B.; Martinetto, H.; et al. Accelerated Epigenetic Aging in Adults with Down Syndrome in the Argentine Population. Alzheimers Dement. 2021, 17, e058593. [Google Scholar] [CrossRef]
- Giuliani, C.; Pirazzini, C.; Delledonne, M.; Xumerle, L.; Descombes, P.; Marquis, J.; Mengozzi, G.; Monti, D.; Bellizzi, D.; Passarino, G.; et al. Centenarians as Extreme Phenotypes: An Ecological Perspective to Get Insight into the Relationship between the Genetics of Longevity and Age-Associated Diseases. Mech. Ageing Dev. 2017, 165, 195–201. [Google Scholar] [CrossRef]
- Franceschi, C.; Ostan, R.; Santoro, A. Nutrition and Inflammation: Are Centenarians Similar to Individuals on Calorie-Restricted Diets? Annu. Rev. Nutr. 2018, 38, 329–356. [Google Scholar] [CrossRef] [PubMed]
- Gutman, D.; Rivkin, E.; Fadida, A.; Sharvit, L.; Hermush, V.; Rubin, E.; Kirshner, D.; Sabin, I.; Dwolatzky, T.; Atzmon, G. Exceptionally Long-Lived Individuals (ELLI) Demonstrate Slower Aging Rate Calculated by DNA Methylation Clocks as Possible Modulators for Healthy Longevity. Int. J. Mol. Sci. 2020, 21, 615. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic Predictor of Age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef]
- Correia Dias, H.; Cordeiro, C.; Corte Real, F.; Cunha, E.; Manco, L. Age Estimation Based on DNA Methylation Using Blood Samples From Deceased Individuals. J. Forensic Sci. 2019, 65, 465–470. [Google Scholar] [CrossRef]
- Vidaki, A.; Ballard, D.; Aliferi, A.; Miller, T.H.; Barron, L.P.; Syndercombe Court, D. DNA Methylation-Based Forensic Age Prediction Using Artificial Neural Networks and next Generation Sequencing. Forensic Sci. Int. Genet. 2017, 28, 225–236. [Google Scholar] [CrossRef]
- Longo, V.D.; Kennedy, B.K. Sirtuins in Aging and Age-Related Disease. Cell 2006, 126, 257–268. [Google Scholar] [CrossRef]
- Paredes, S.; Angulo-Ibanez, M.; Tasselli, L.; Carlson, S.M.; Zheng, W.; Li, T.-M.; Chua, K.F. The Epigenetic Regulator SIRT7 Guards against Mammalian Cellular Senescence Induced by Ribosomal DNA Instability. J. Biol. Chem. 2018, 293, 11242–11250. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. SIRT7 in the Aging Process. Cell Mol. Life Sci. 2022, 79, 297. [Google Scholar] [CrossRef] [PubMed]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, M.J.; Beyer, A.; et al. Dietary Restriction Protects from Age-Associated DNA Methylation and Induces Epigenetic Reprogramming of Lipid Metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Ligthart, S.; Marzi, C.; Aslibekyan, S.; Mendelson, M.M.; Conneely, K.N.; Tanaka, T.; Colicino, E.; Waite, L.L.; Joehanes, R.; Guan, W.; et al. DNA Methylation Signatures of Chronic Low-Grade Inflammation Are Associated with Complex Diseases. Genome Biol. 2016, 17, 255. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA Methylation Patterns Associate with Genetic and Gene Expression Variation in HapMap Cell Lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef]
- Marioni, R.E.; Suderman, M.; Chen, B.H.; Horvath, S.; Bandinelli, S.; Morris, T.; Beck, S.; Ferrucci, L.; Pedersen, N.L.; Relton, C.L.; et al. Tracking the Epigenetic Clock Across the Human Life Course: A Meta-Analysis of Longitudinal Cohort Data. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 57–61. [Google Scholar] [CrossRef]
- Simpson, D.J.; Chandra, T. Epigenetic Age Prediction. Aging Cell 2021, 20, e13452. [Google Scholar] [CrossRef]
- Adalsteinsson, B.T.; Gudnason, H.; Aspelund, T.; Harris, T.B.; Launer, L.J.; Eiriksdottir, G.; Smith, A.V.; Gudnason, V. Heterogeneity in White Blood Cells Has Potential to Confound DNA Methylation Measurements. PLoS ONE 2012, 7, e46705. [Google Scholar] [CrossRef]
- Tan, Q.; Heijmans, B.T.; Hjelmborg, J.V.; Soerensen, M.; Christensen, K.; Christiansen, L. Handling Blood Cell Composition in Epigenetic Studies on Ageing. Int. J. Epidemiol. 2017, 46, 1717–1718. [Google Scholar] [CrossRef]
- Sontag, S.; Bocova, L.; Hubens, W.H.G.; Nüchtern, S.; Schnitker, M.; Look, T.; Schröder, K.M.; Plümäkers, B.; Tharmapalan, V.; Wessiepe, M.; et al. Toward Clinical Application of Leukocyte Counts Based on Targeted DNA Methylation Analysis. Clin. Chem. 2022, 68, 646–656. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | N (F: Females, M: Males | Overlap with Infinium Microarray Data [26] | Age Range (Years) (Mean ± SD 1) | Epigenetic Age (Years) (Mean ± SD 1) | EAD 2 (Years) (Mean ± SD 1) | p-Value 3 |
|---|---|---|---|---|---|---|
| Controls (entire cohort) | 315 (132 F, 180 M, 3 NA) | 32 subjects | 0–98 years (57.32 ± 18.71) | - | - | - |
| Controls (age range 20–80 years) | 278 (117 F, 161 M) | 32 subjects | 21–80 years (54.96 ± 15.03) | 54.96 ± 13.43 | 0 ± 6.04 | - |
| Persons with Down syndrome | 62 (27 F, 35 M) | 11 subjects | 12–66 years (33.97 ± 13.46) | 49.23 ± 34.94 | +11.02 ± 33.33 | <0.001 |
| Centenarians | 106 (82 F, 24 M) | 11 subjects | 100–112 years (101.5 ± 2.44) | 85.66 ± 12.23 | −6.45 ± 12.43 | <0.001 |
| Centenarians’ offspring | 143 (81 F, 62 M) | 19 subjects | 55–89 years (70.06 ± 6.69) | 65.35 ± 9.75 | −1.65 ± 8.96 | 0.015 |
| All | Italy | Poland | |
|---|---|---|---|
| Subjects (N) | 233 | 124 | 109 |
| Overlap with Infinium microarray data [31] | 120 | 60 | 60 |
| Males/Females (N) | 105/128 | 60/64 | 45/64 |
| Chronological age at T0 (years) mean ± SD 1 | 71.89 ± 3.91 | 72.16 ± 3.79 | 71.58 ± 4.03 |
| Epigenetic age at T0 (years) mean ± SD 1 | 71.89 ± 2.34 | 71.84 ± 2.19 | 71.94 ± 2.51 |
| EAD 2 at T0 (years) mean ± SD 1 | 0.00 ± 1.82 | −0.14 ± 1.69 | 0.17 ± 1.96 |
| Chronological age at T1 (years) mean ± SD 1 | 72.93 ± 3.91 | 73.22 ± 3.78 | 72.59 ± 4.03 |
| Epigenetic age at T1 (years) mean ± SD 1 | 71.70 ± 3.11 | 71.51 ± 2.97 | 71.92 ± 3.26 |
| EAD 2 at T1 (years) mean ± SD 1 | −0.58 ± 3.16 | −0.87 ± 3.03 | −0.23 ± 3.28 |
| CpG Probe | Location | Associated Gene | Region Assessed in the Targeted Assay | Assessable CpG Units |
|---|---|---|---|---|
| cg16867657 | chr6:11,044,877 | ELOVL2 | chr6:11,044,680–11,045,053 | 15 |
| cg22736354 | chr6:18,122,719 | NHLRC1 | chr6:18,122,552–18,123,149 | 21 |
| cg07855221 | chr17:79,877,314 | SIRT7/MAFG | chr17:79,877,158–79,877,497 | 6 |
| cg09253473 | chr17:79,877,390 | SIRT7/MAFG | chr17:79,877,158–79,877,497 | 6 |
| cg10636246 | chr1:159,046,973 | AIM2 | chr1:159,046,884–159,047,270 | 7 |
| cg09809672 | chr1:236,557,683 | EDARADD | chr1:236,557,384–236,557,805 | 5 |
| cg26372517 | chr1:36,039,159 | TFAP2E | chr1:36,038,876–36,039,325 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gensous, N.; Sala, C.; Pirazzini, C.; Ravaioli, F.; Milazzo, M.; Kwiatkowska, K.M.; Marasco, E.; De Fanti, S.; Giuliani, C.; Pellegrini, C.; et al. A Targeted Epigenetic Clock for the Prediction of Biological Age. Cells 2022, 11, 4044. https://doi.org/10.3390/cells11244044
Gensous N, Sala C, Pirazzini C, Ravaioli F, Milazzo M, Kwiatkowska KM, Marasco E, De Fanti S, Giuliani C, Pellegrini C, et al. A Targeted Epigenetic Clock for the Prediction of Biological Age. Cells. 2022; 11(24):4044. https://doi.org/10.3390/cells11244044
Chicago/Turabian StyleGensous, Noémie, Claudia Sala, Chiara Pirazzini, Francesco Ravaioli, Maddalena Milazzo, Katarzyna Malgorzata Kwiatkowska, Elena Marasco, Sara De Fanti, Cristina Giuliani, Camilla Pellegrini, and et al. 2022. "A Targeted Epigenetic Clock for the Prediction of Biological Age" Cells 11, no. 24: 4044. https://doi.org/10.3390/cells11244044
APA StyleGensous, N., Sala, C., Pirazzini, C., Ravaioli, F., Milazzo, M., Kwiatkowska, K. M., Marasco, E., De Fanti, S., Giuliani, C., Pellegrini, C., Santoro, A., Capri, M., Salvioli, S., Monti, D., Castellani, G., Franceschi, C., Bacalini, M. G., & Garagnani, P. (2022). A Targeted Epigenetic Clock for the Prediction of Biological Age. Cells, 11(24), 4044. https://doi.org/10.3390/cells11244044