
tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions

 , , , , , , , , ,
, , , , , , , , ,  , ,
, ,  , and
, and  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. SHIP Population Study and Cohorts
2.2. CRISPR-Cas9 Experiments
2.3. Cell Culture Studies
2.3.1. Human Monocyte Cultures
2.3.2. THP-1 Monocyte Adhesion to Flow-Primed Human Aortic Endothelial Cells
2.3.3. Tube Formation Angiogenesis Assay
2.4. Reactive Oxygen Species Assay
2.5. Cytokine Measurements
2.6. Cell Proliferation Studies
2.7. Foam Cell Formation and oxLDL Uptake
2.8. Monocyte-Macrophage Transition and Macrophage Polarization Experiments
2.9. FACS Analyses of the Macrophage Clones
2.10. Human Adenovirus and Coxsackievirus B3 Studies
2.11. RNA Sequencing and Data Analysis
2.12. Quantitative RT-PCR
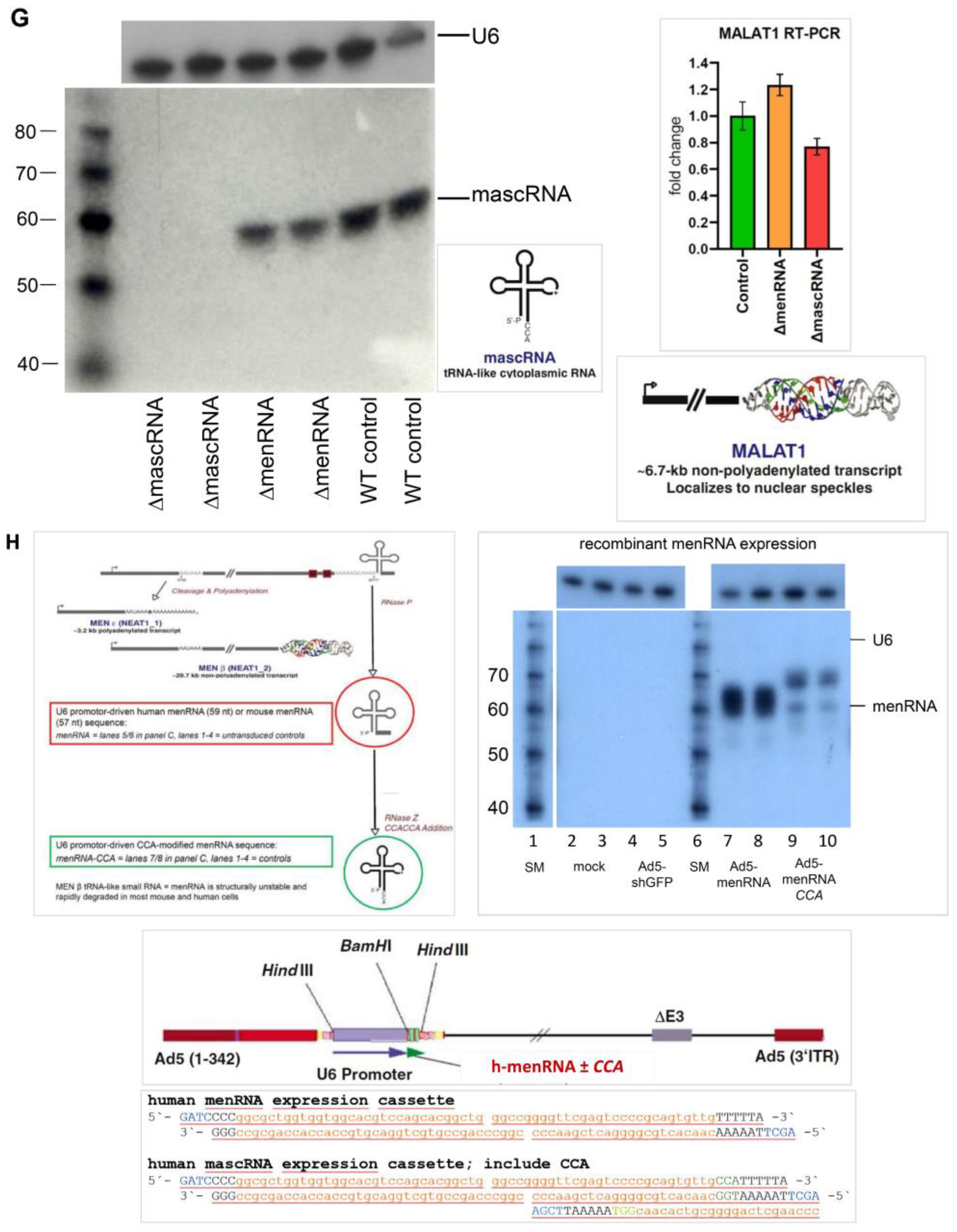
2.13. Cloning and Recombinant Expression of Human menRNA and mascRNA
2.14. Statistical Analyses
3. Results
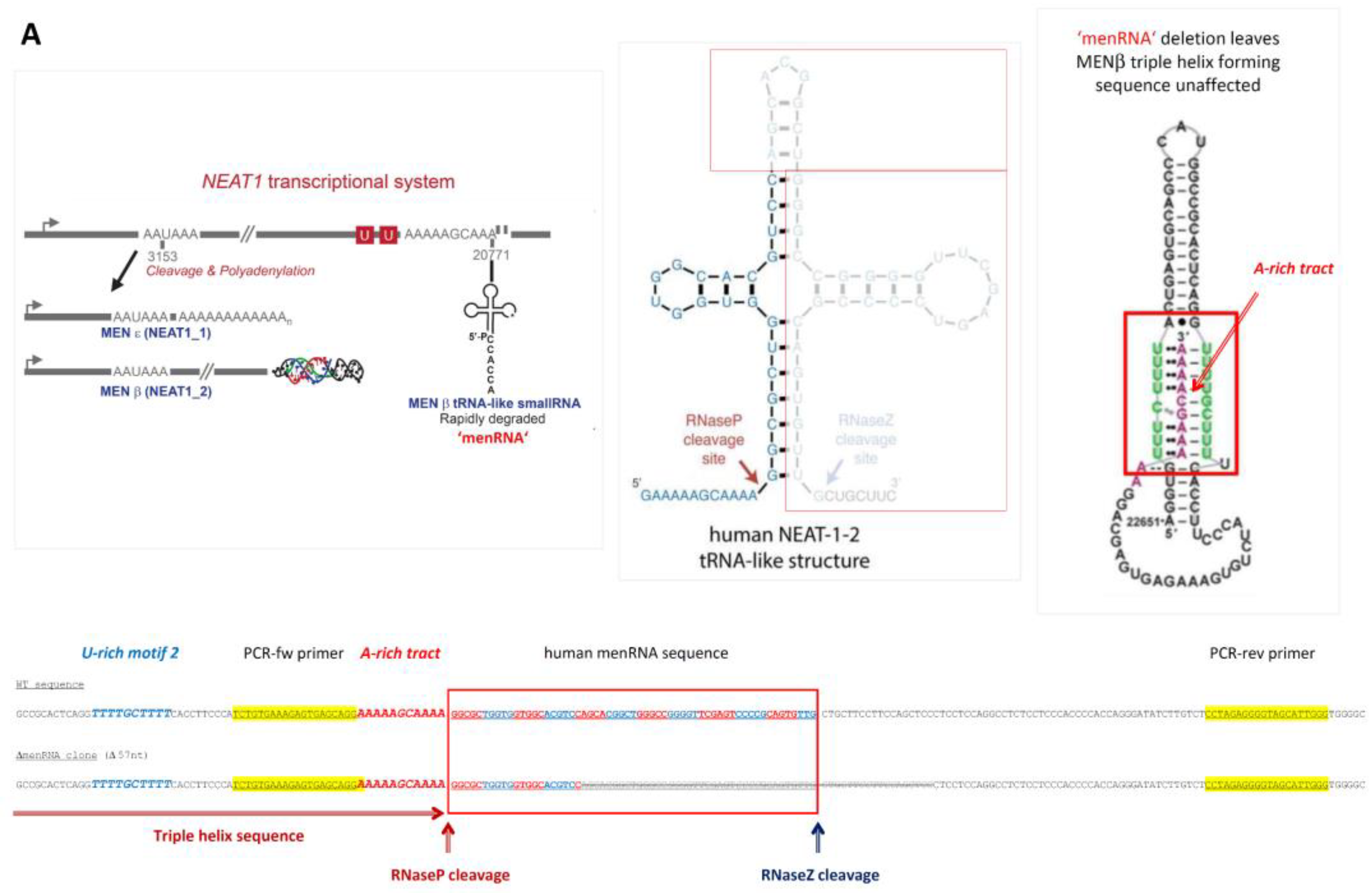
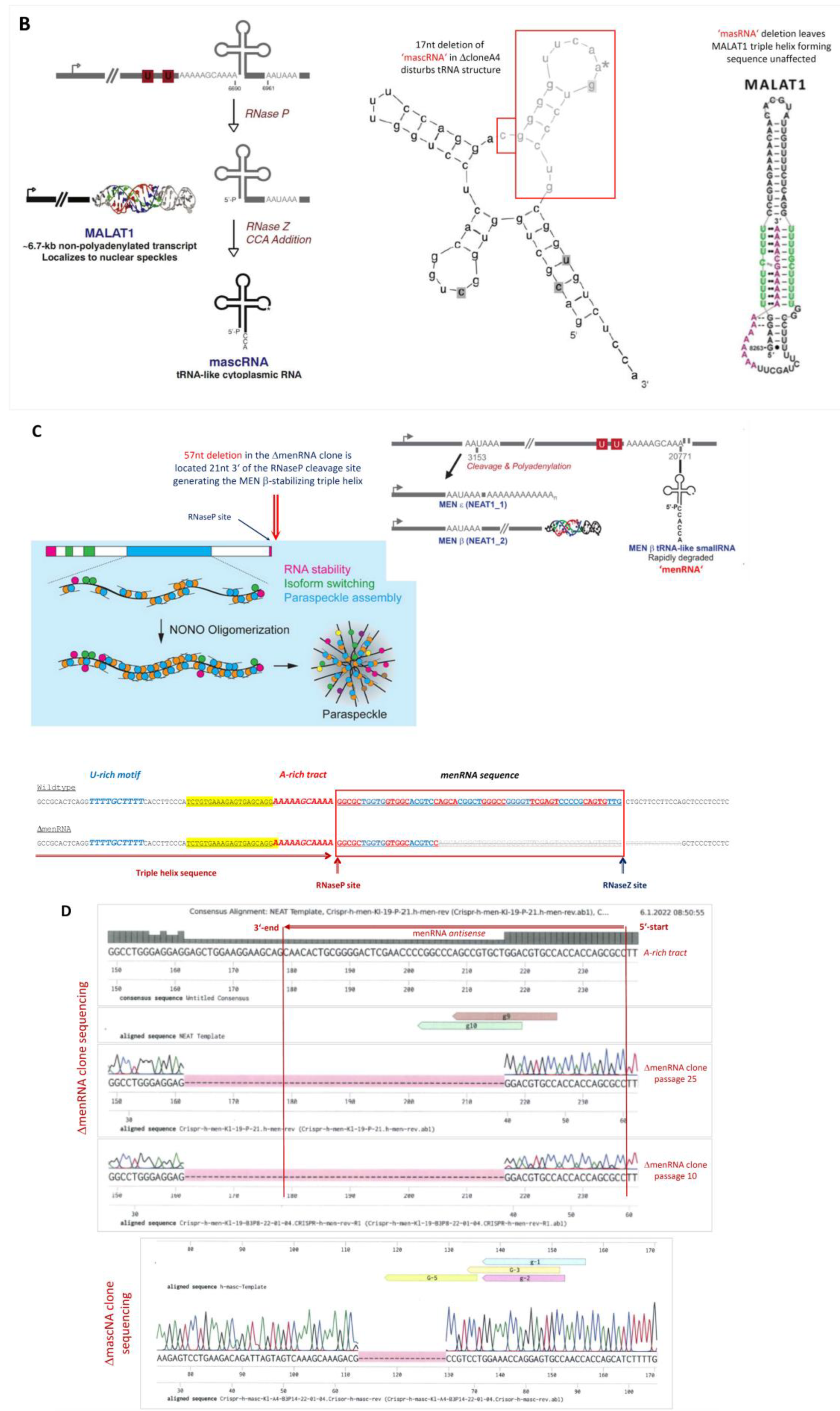
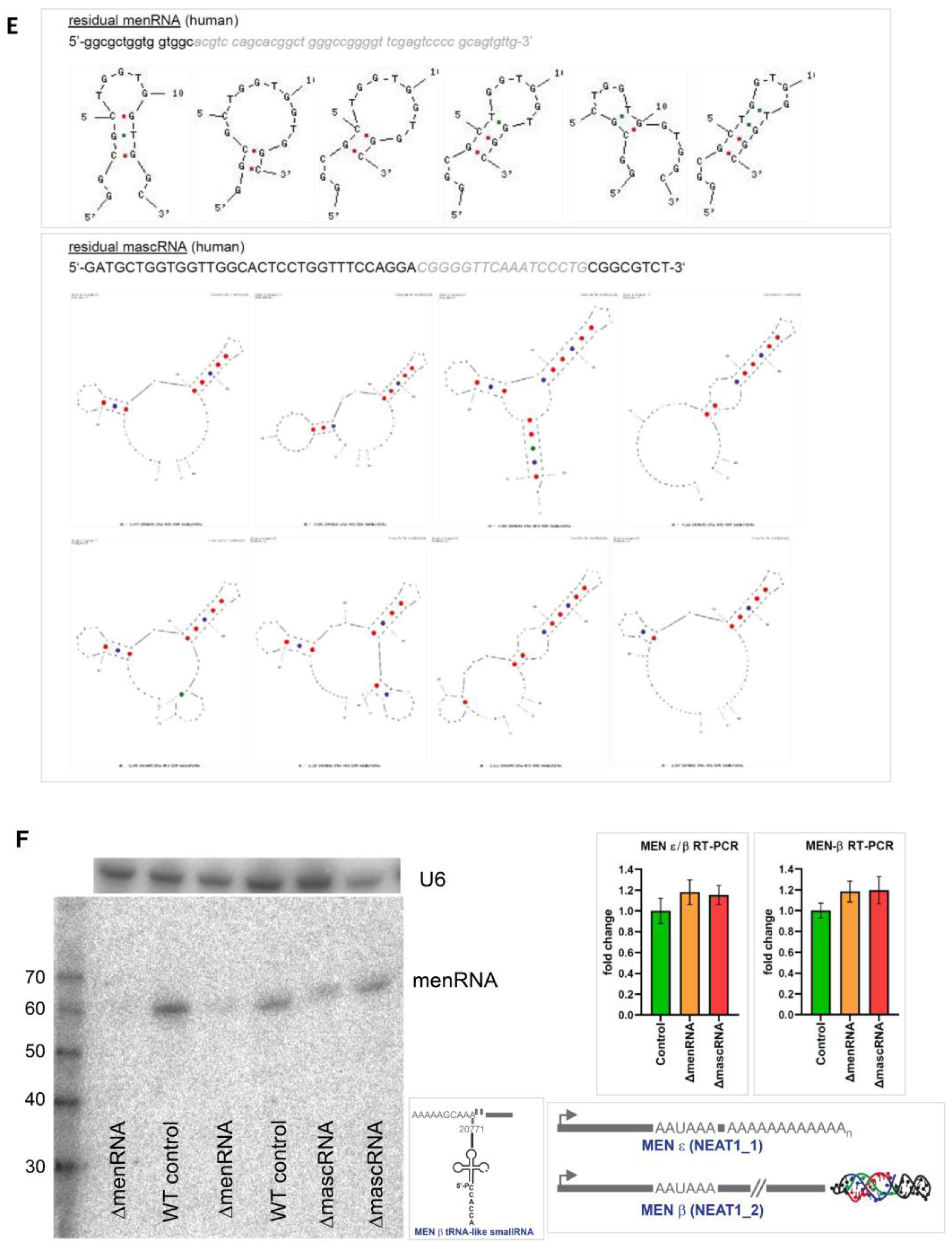
3.1. Targeted Deletion of tRNA-like Transcripts from the NEAT1-MALAT1 Cluster




3.2. Defective Innate Immune Sensing by ΔmenRNA and ΔmascRNA Cells


3.3. Transcription, Translation, and Epigenome Level Anomalies in Defective Monocytes




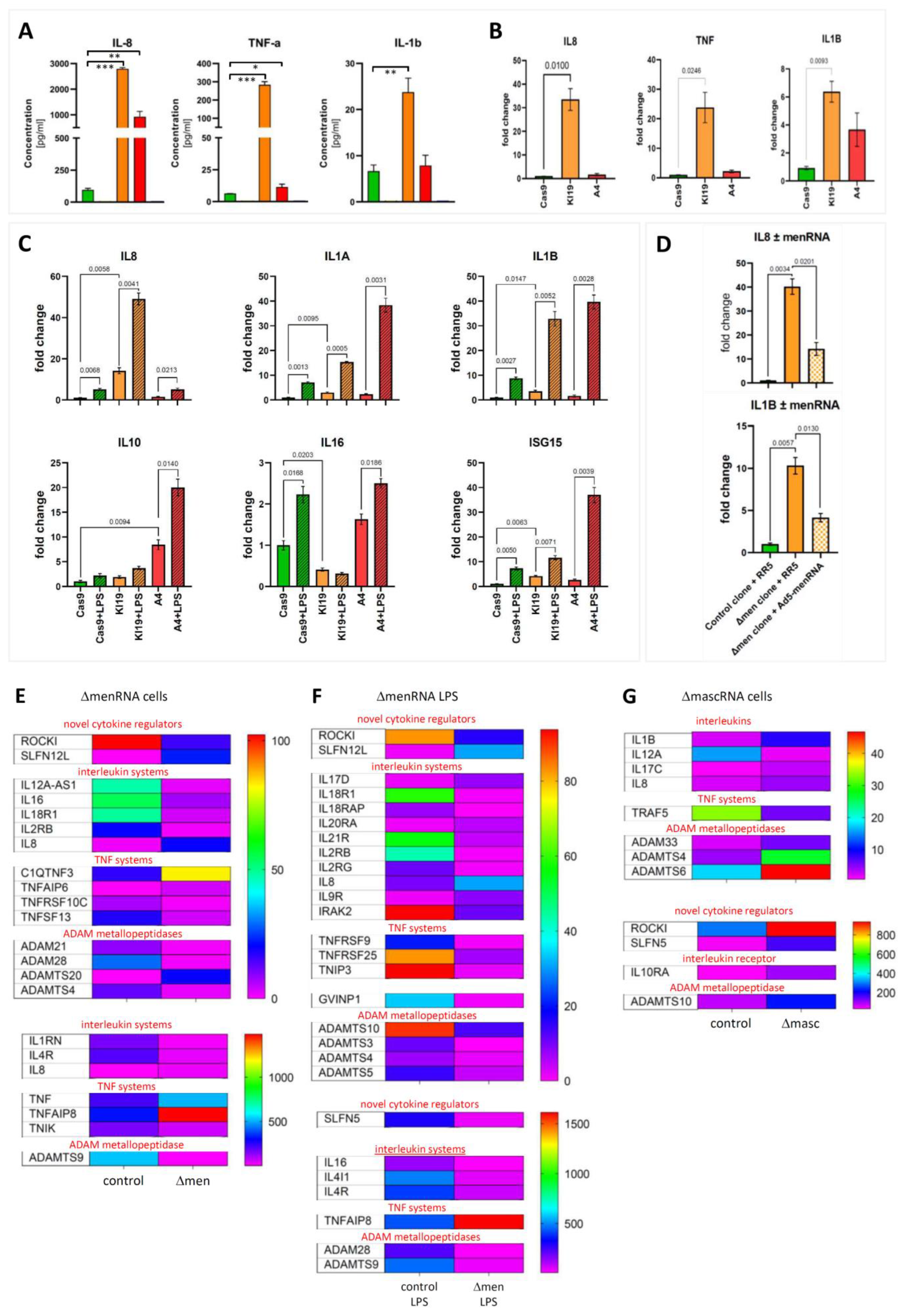
3.4. Excessive Inflammatory Cytokine Production by ΔmenRNA and ΔmascRNA Cells


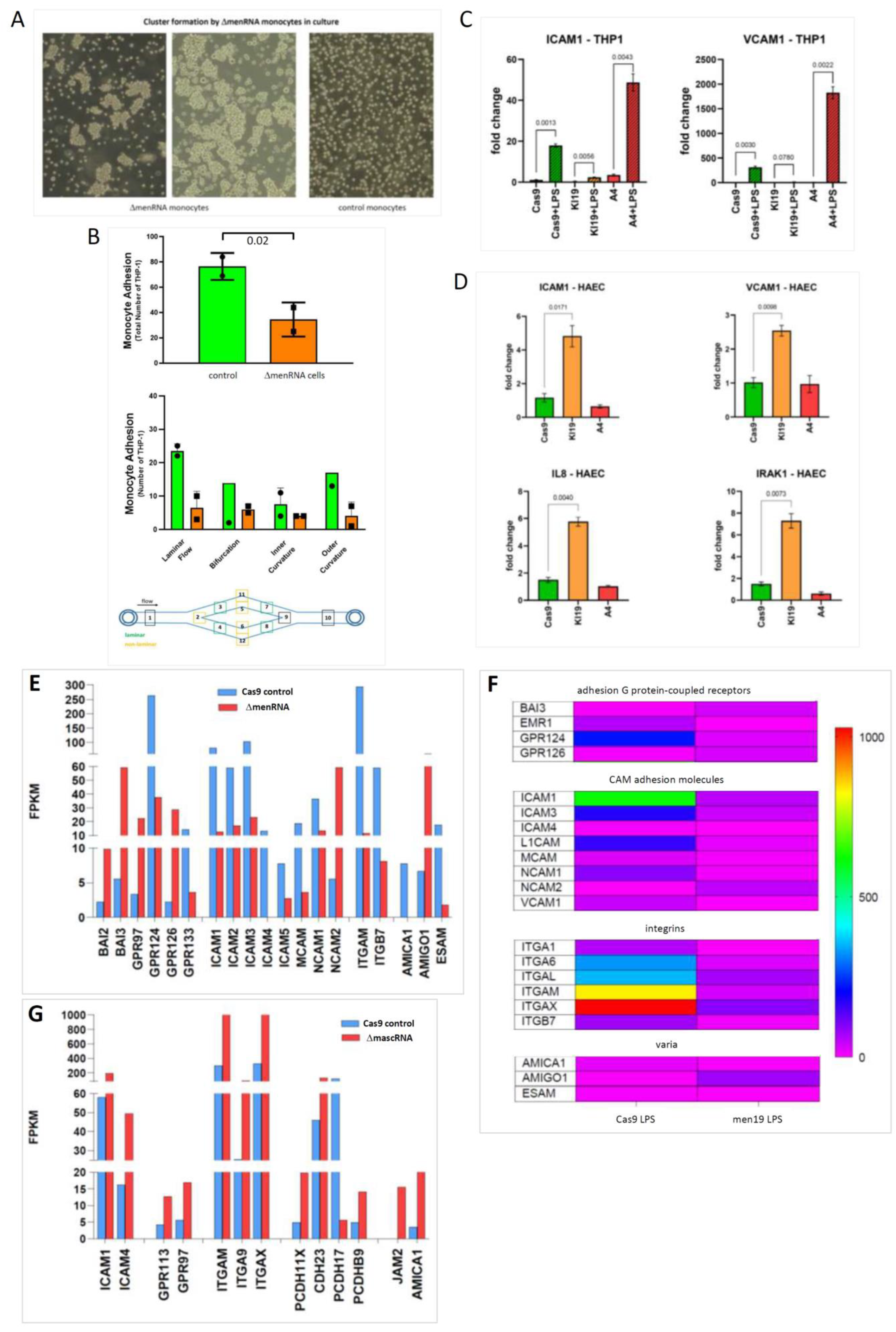
3.5. Disturbed Growth Pattern and Endothelium Interactions of ΔmenRNA Monocytes
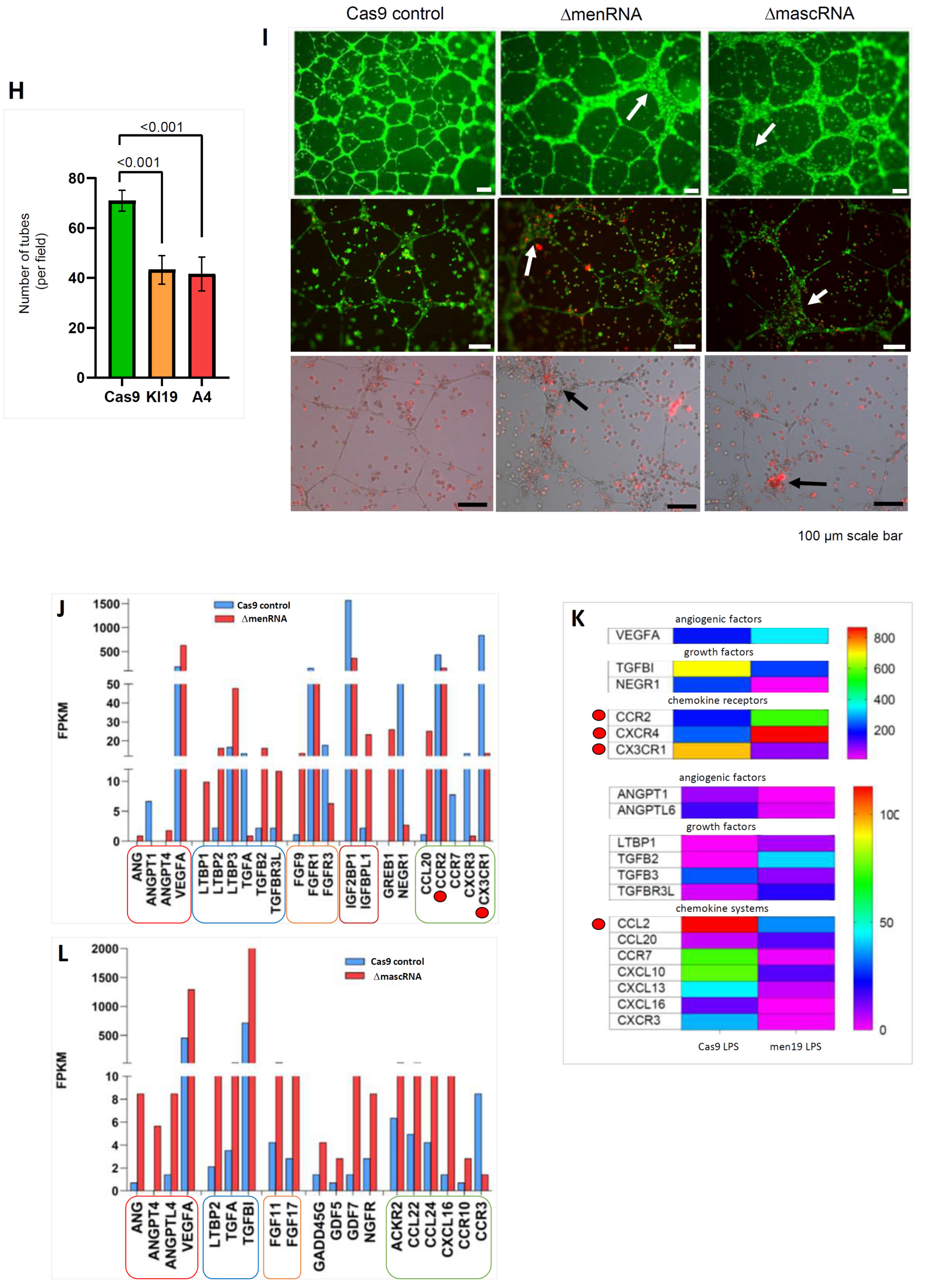
3.6. Impact of ΔmenRNA and ΔmascRNA Monocytes upon Angiogenesis
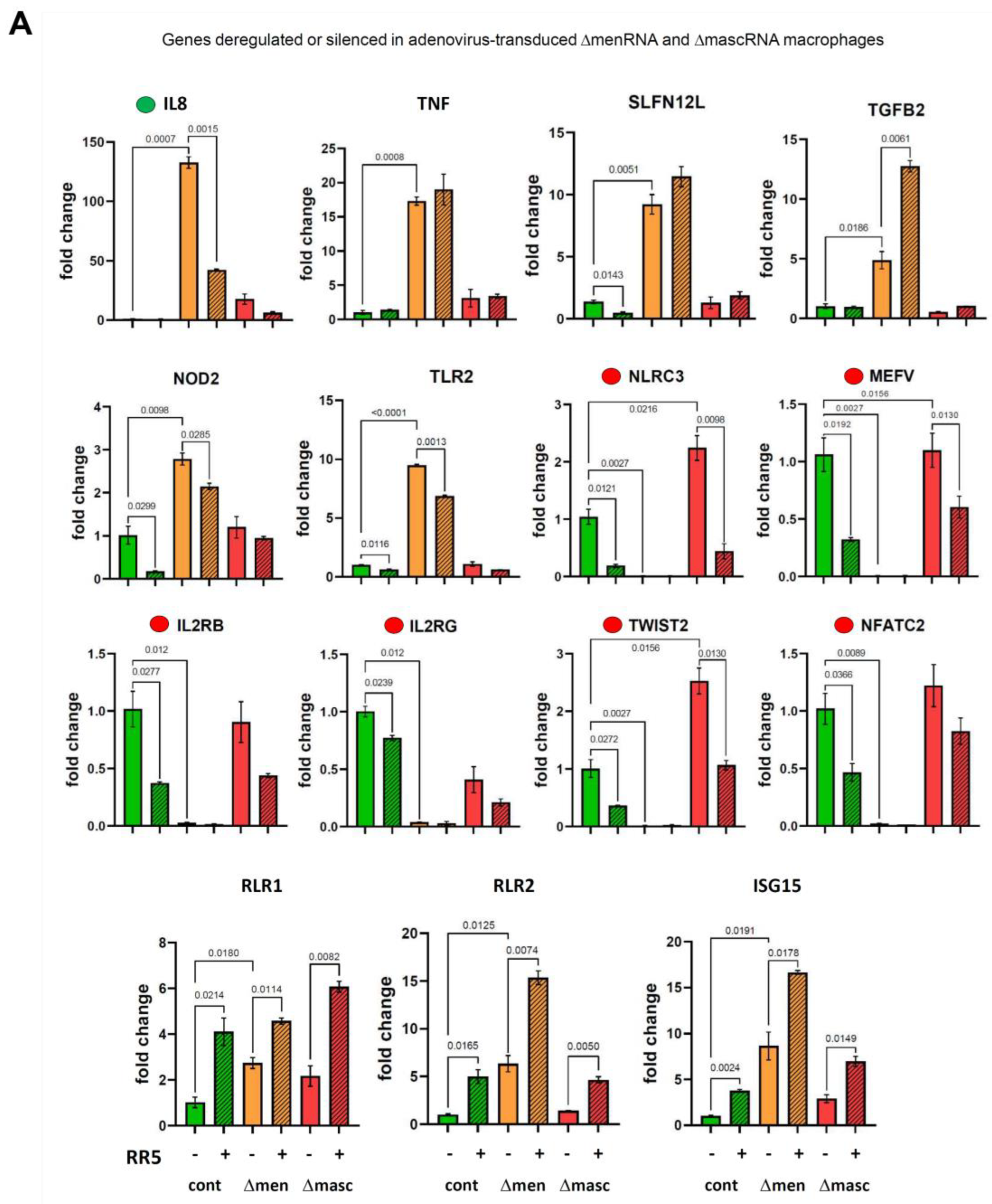
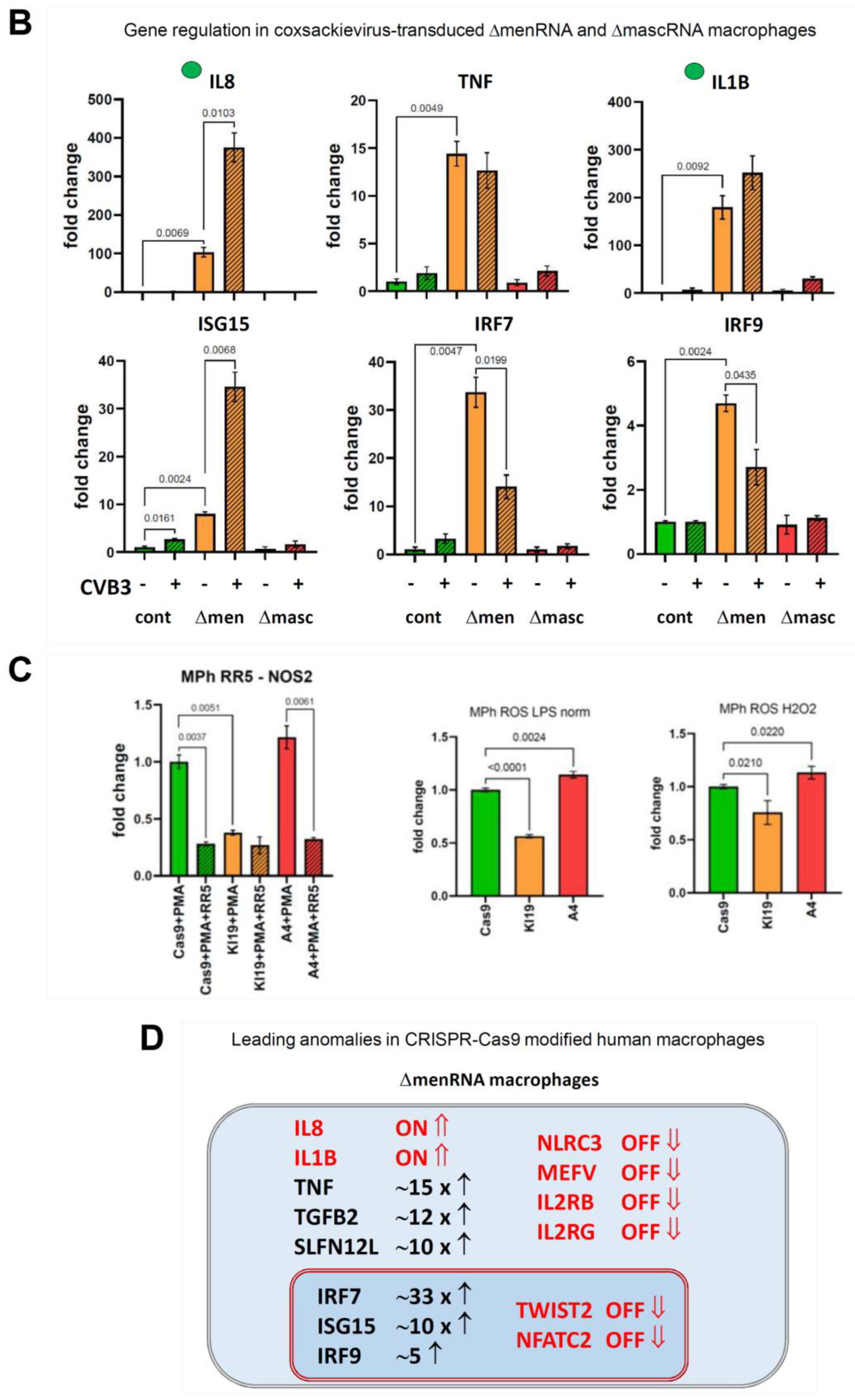
3.7. Response of ΔmenRNA and ΔmascRNA Macrophages to Human-Pathogenic Viruses


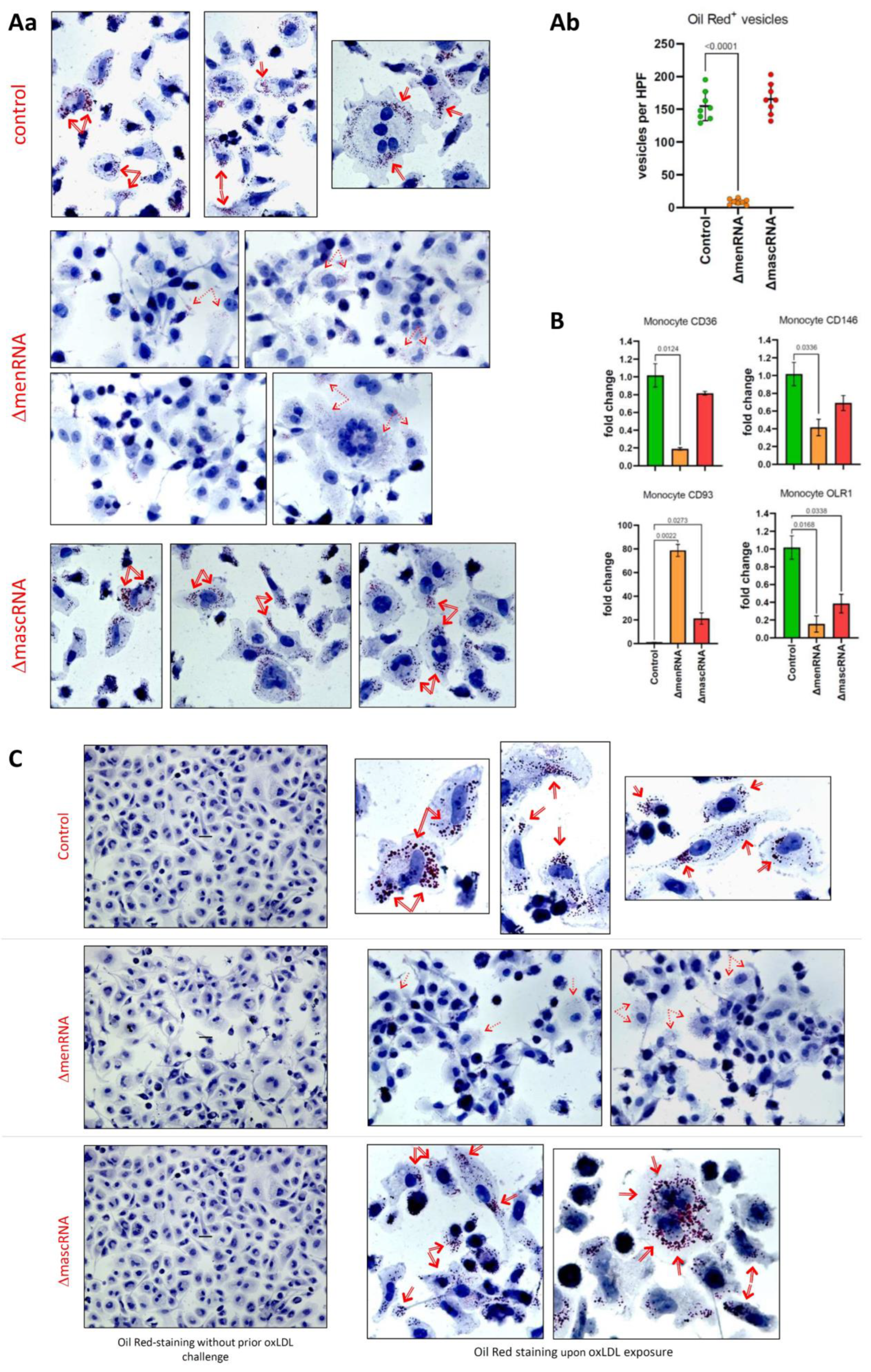
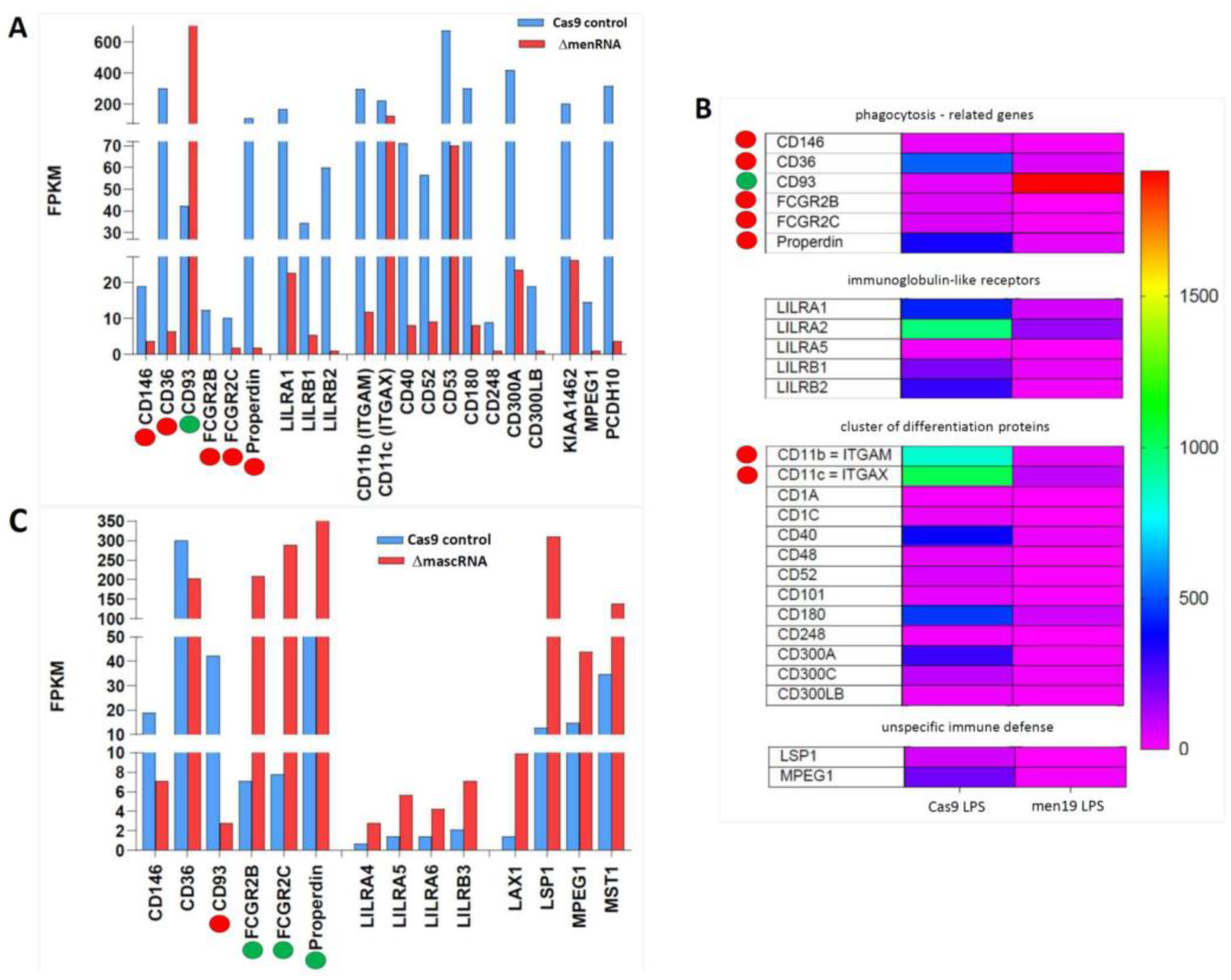
3.8. menRNA Deletion Critically Disturbs Scavenger Receptor Expression and oxLDL Uptake
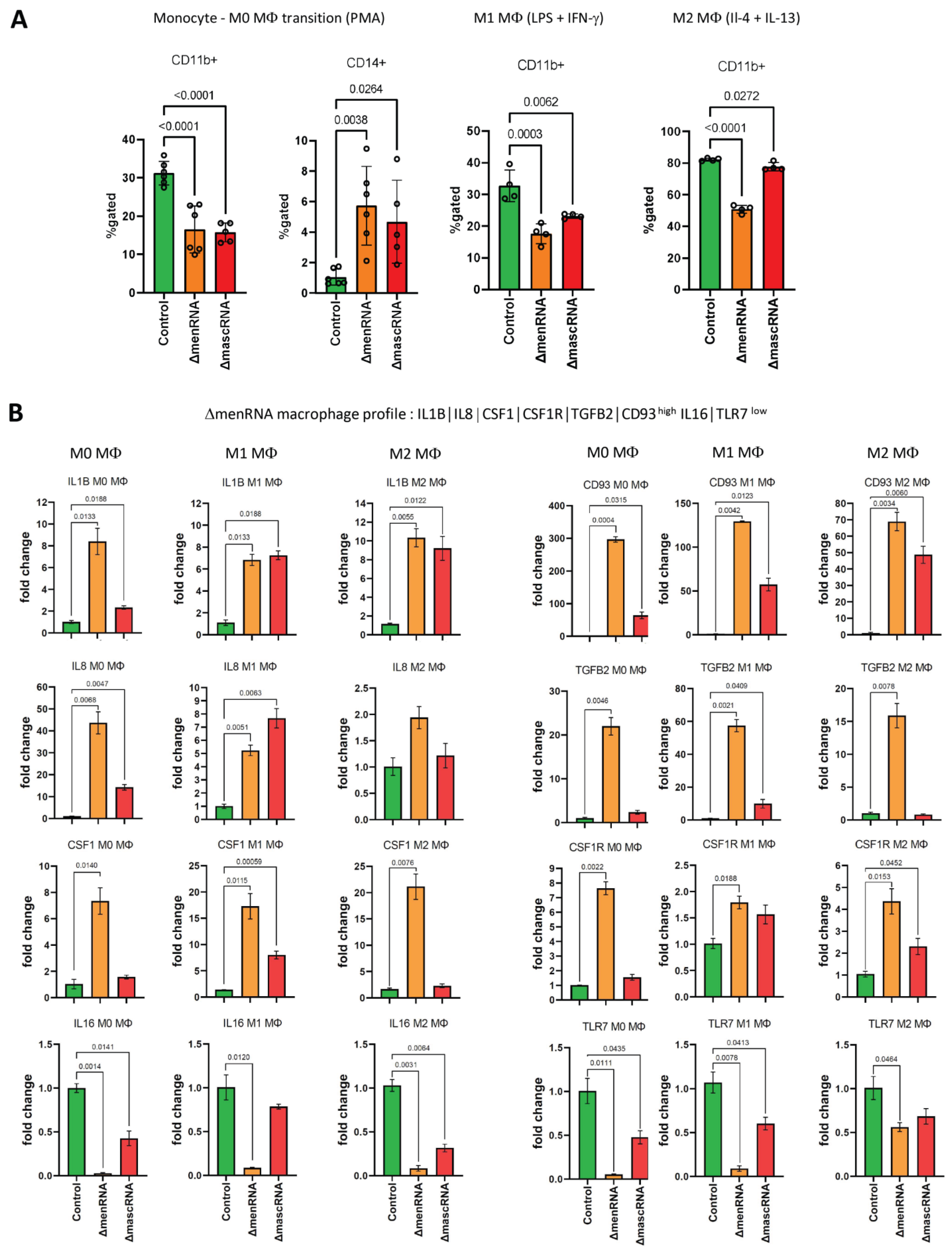
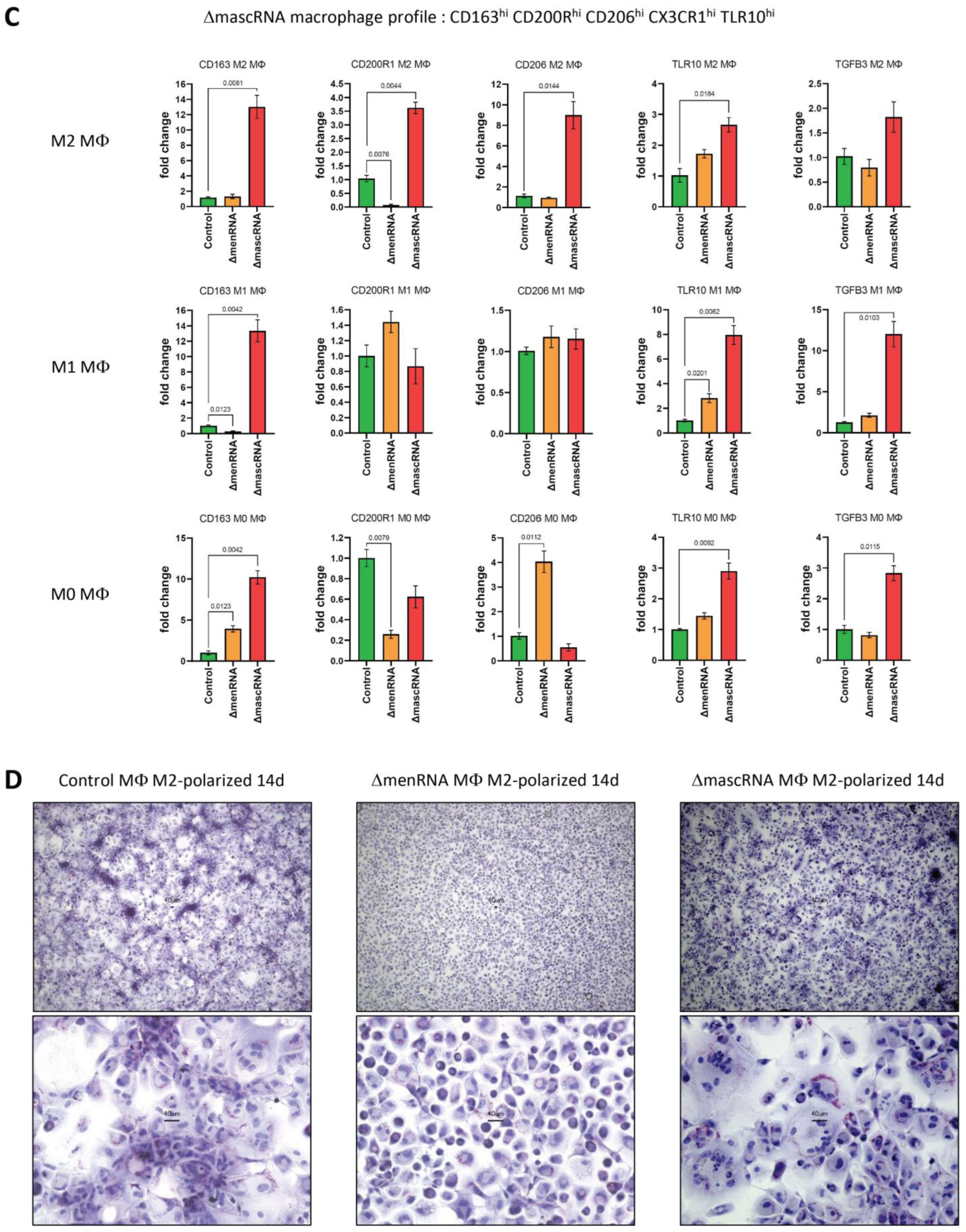
3.9. Defective Monocyte–Macrophage Transition and Polarization of ΔmenRNA and ΔmascRNA Cells




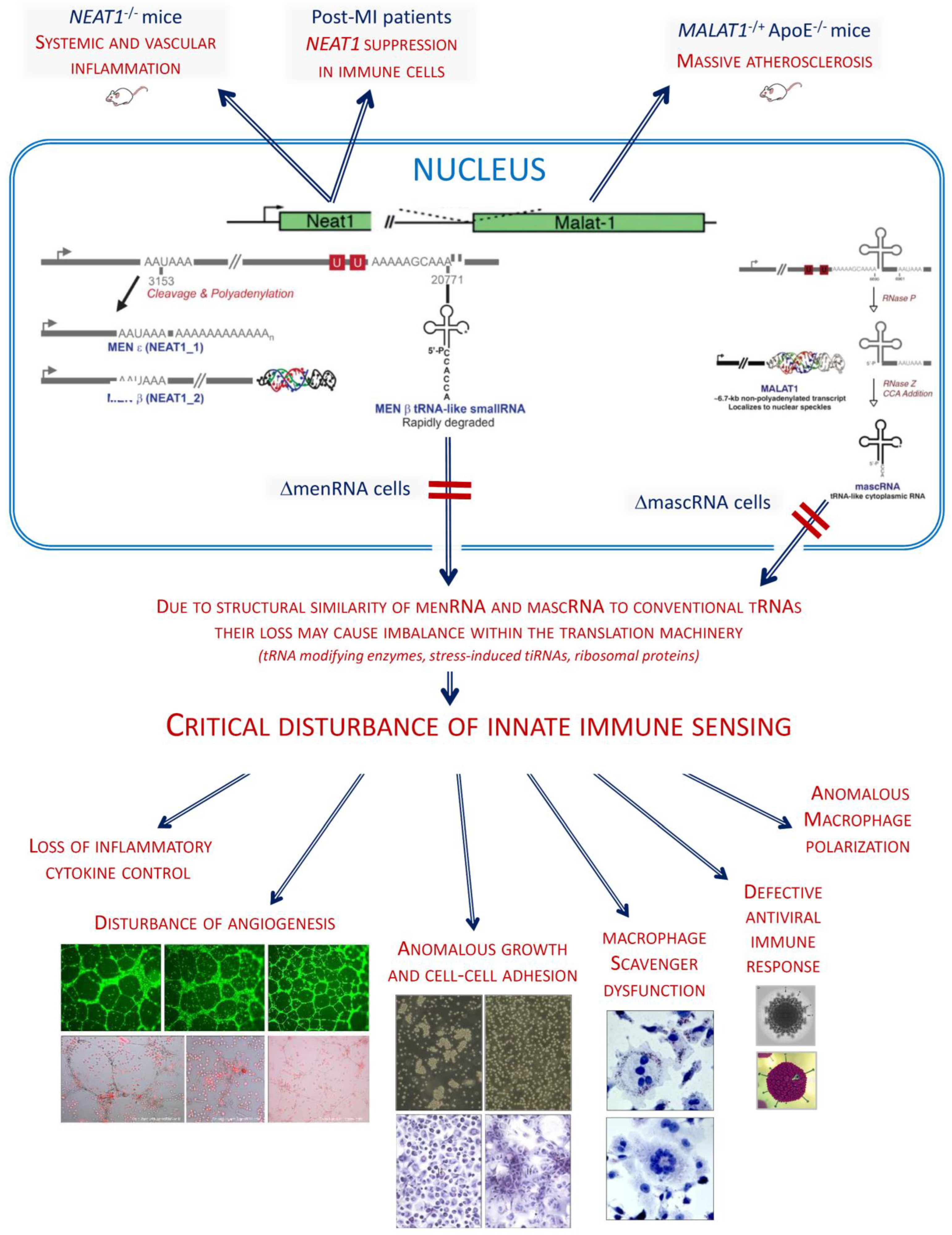
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gast, M.; Rauch, B.H.; Haghikia, A.; Nakagawa, S.; Haas, J.; Stroux, A.; Schmidt, D.; Schumann, P.; Weiss, S.; Jensen, L.; et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc. Res. 2019, 115, 1886–1906. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Bohm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE-/-mice. Cardiovasc. Res. 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Schroen, B.; Voigt, A.; Haas, J.; Kuehl, U.; Lassner, D.; Skurk, C.; Escher, F.; Wang, X.; Kratzer, A.; et al. Long noncoding RNA MALAT1-derived mascRNA is involved in cardiovascular innate immunity. J. Mol. Cell. Biol. 2016, 8, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Cremer, S.; Michalik, K.M.; Fischer, A.; Pfisterer, L.; Jae, N.; Winter, C.; Boon, R.A.; Muhly-Reinholz, M.; John, D.; Uchida, S.; et al. Hematopoietic Deficiency of the Long Noncoding RNA MALAT1 Promotes Atherosclerosis and Plaque Inflammation. Circulation 2019, 139, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, P.; Marechal, P.; Donis, N.; Oury, C. Inflammation, cardiovascular disease, and cancer: A common link with far-reaching implications. Eur. Heart J. 2019, 40, 3910–3912. [Google Scholar] [CrossRef] [PubMed]
- Poller, W.; Dimmeler, S.; Heymans, S.; Zeller, T.; Haas, J.; Karakas, M.; Leistner, D.M.; Jakob, P.; Nakagawa, S.; Blankenberg, S.; et al. Non-coding RNAs in cardiovascular diseases: Diagnostic and therapeutic perspectives. Eur. Heart J. 2018, 39, 2704–2716. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef]
- Kuhn, C.D.; Wilusz, J.E.; Zheng, Y.; Beal, P.A.; Joshua-Tor, L. On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell 2015, 160, 644–658. [Google Scholar] [CrossRef]
- Sun, T.; Wei, C.; Wang, D.; Wang, X.; Wang, J.; Hu, Y.; Mao, X. The small RNA mascRNA differentially regulates TLR-induced proinflammatory and antiviral responses. JCI Insight 2021, 6, e150833. [Google Scholar] [CrossRef]
- Lu, X.; Huang, J.; Wu, S.; Zheng, Q.; Liu, P.; Feng, H.; Su, X.; Fu, H.; Xi, Q.; Wang, G. The tRNA-like small noncoding RNA mascRNA promotes global protein translation. EMBO Rep. 2020, 21, e49684. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Souquere, S.; Chujo, T.; Kobelke, S.; Chong, Y.S.; Fox, A.H.; Bond, C.S.; Nakagawa, S.; Pierron, G.; Hirose, T. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 2018, 70, 1038–1053.e7. [Google Scholar] [CrossRef]
- Suckau, L.; Fechner, H.; Chemaly, E.; Krohn, S.; Hadri, L.; Kockskamper, J.; Westermann, D.; Bisping, E.; Ly, H.; Wang, X.; et al. Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation 2009, 119, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Fechner, H.; Suckau, L.; Kurreck, J.; Sipo, I.; Wang, X.; Pinkert, S.; Loschen, S.; Rekittke, J.; Weger, S.; Dekkers, D.; et al. Highly efficient and specific modulation of cardiac calcium homeostasis by adenovector-derived short hairpin RNA targeting phospholamban. Gene Ther. 2007, 14, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Whipple, J.M.; Phizicky, E.M.; Sharp, P.A. tRNAs marked with CCACCA are targeted for degradation. Science 2011, 334, 817–821. [Google Scholar] [CrossRef]
- Wang, Z.; Xiang, L.; Shao, J.; Yuan, Z. The 3′ CCACCA sequence of tRNAAla(UGC) is the motif that is important in inducing Th1-like immune response, and this motif can be recognized by Toll-like receptor 3. Clin. Vaccine Immunol. 2006, 13, 733–739. [Google Scholar] [CrossRef]
- Sunwoo, H.; Dinger, M.E.; Wilusz, J.E.; Amaral, P.P.; Mattick, J.S.; Spector, D.L. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009, 19, 347–359. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Ballester, A.; Guijarro, A.; Bravo, B.; Hernandez, J.; Murillas, R.; Gallego, M.I.; Ballester, S. Hedgehog Signalling Modulates Immune Response and Protects against Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2022, 23, 3171. [Google Scholar] [CrossRef]
- Babagana, M.; Oh, K.S.; Chakraborty, S.; Pacholewska, A.; Aqdas, M.; Sung, M.H. Hedgehog dysregulation contributes to tissue-specific inflammaging of resident macrophages. Aging 2021, 13, 19207–19229. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zelkha, S.; Burkatovskaya, M.; Gupte, R.; Leeman, S.E.; Amar, S. Pivotal role of NOD2 in inflammatory processes affecting atherosclerosis and periodontal bone loss. Proc. Natl. Acad. Sci. USA 2013, 110, E5059–E5068. [Google Scholar] [CrossRef]
- Geirsson, A.; Paliwal, I.; Lynch, R.J.; Bothwell, A.L.; Hammond, G.L. Class II transactivator promoter activity is suppressed through regulation by a trophoblast noncoding RNA. Transplantation 2003, 76, 387–394. [Google Scholar] [CrossRef]
- Sundaram, B.; Kanneganti, T.D. Advances in Understanding Activation and Function of the NLRC4 Inflammasome. Int. J. Mol. Sci. 2021, 22, 1048. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Hedl, M.; Abraham, C. Twist1 and Twist2 Induce Human Macrophage Memory upon Chronic Innate Receptor Treatment by HDAC-Mediated Deacetylation of Cytokine Promoters. J. Immunol. 2019, 202, 3297–3308. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef]
- Ignatova, V.V.; Jansen, P.; Baltissen, M.P.; Vermeulen, M.; Schneider, R. The interactome of a family of potential methyltransferases in HeLa cells. Sci. Rep. 2019, 9, 6584. [Google Scholar] [CrossRef]
- Godfrey, A.K.; Naqvi, S.; Chmatal, L.; Chick, J.M.; Mitchell, R.N.; Gygi, S.P.; Skaletsky, H.; Page, D.C. Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 2020, 30, 860–873. [Google Scholar] [CrossRef]
- Aguilo, F.; Li, S.; Balasubramaniyan, N.; Sancho, A.; Benko, S.; Zhang, F.; Vashisht, A.; Rengasamy, M.; Andino, B.; Chen, C.H.; et al. Deposition of 5-Methylcytosine on Enhancer RNAs Enables the Coactivator Function of PGC-1alpha. Cell Rep. 2016, 14, 479–492. [Google Scholar] [CrossRef]
- Suzuki, T.; Yashiro, Y.; Kikuchi, I.; Ishigami, Y.; Saito, H.; Matsuzawa, I.; Okada, S.; Mito, M.; Iwasaki, S.; Ma, D.; et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020, 11, 4269. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Lee, S.Y.; McCann, B.J.; Powell, C.A.; Bansal, D.; Vasiliauskaite, L.; Garone, C.; Shin, S.; Kim, J.S.; Frye, M.; et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8720–8733. [Google Scholar] [CrossRef] [PubMed]
- Hunsucker, S.A.; Spychala, J.; Mitchell, B.S. Human cytosolic 5′-nucleotidase I: Characterization and role in nucleoside analog resistance. J. Biol. Chem. 2001, 276, 10498–10504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chao, T.C.; Patil, V.S.; Qin, Y.; Tiwari, S.K.; Chiou, J.; Dobin, A.; Tsai, C.M.; Li, Z.; Dang, J.; et al. The long noncoding RNA ROCKI regulates inflammatory gene expression. EMBO J. 2019, 38, e100041. [Google Scholar] [CrossRef] [PubMed]
- Geirsson, A.; Bothwell, A.L.; Hammond, G.L. Inhibition of alloresponse by a human trophoblast non-coding RNA suppressing class II transactivator promoter III and major histocompatibility class II expression in murine B-lymphocytes. J. Heart Lung Transplant. 2004, 23, 1077–1081. [Google Scholar] [CrossRef]
- Kawase, T.; Ichikawa, H.; Ohta, T.; Nozaki, N.; Tashiro, F.; Ohki, R.; Taya, Y. p53 target gene AEN is a nuclear exonuclease required for p53-dependent apoptosis. Oncogene 2008, 27, 3797–3810. [Google Scholar] [CrossRef]
- Roberts, O.; Paraoan, L. PERP-ing into diverse mechanisms of cancer pathogenesis: Regulation and role of the p53/p63 effector PERP. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188393. [Google Scholar] [CrossRef]
- Cruikshank, W.W.; Kornfeld, H.; Center, D.M. Interleukin-16. J. Leukoc. Biol. 2000, 67, 757–766. [Google Scholar] [CrossRef]
- Sadik, A.; Somarribas Patterson, L.F.; Ozturk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfander, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Zhao, S.; Zhang, H.; Cai, W.; Cai, M.; Ji, X.; Leak, R.K.; Gao, Y.; Chen, J.; et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke 2016, 47, 498–504. [Google Scholar] [CrossRef]
- Kim, E.T.; Dybas, J.M.; Kulej, K.; Reyes, E.D.; Price, A.M.; Akhtar, L.N.; Orr, A.; Garcia, B.A.; Boutell, C.; Weitzman, M.D. Comparative proteomics identifies Schlafen 5 (SLFN5) as a herpes simplex virus restriction factor that suppresses viral transcription. Nat. Microbiol. 2021, 6, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Arslan, A.D.; Sassano, A.; Saleiro, D.; Lisowski, P.; Kosciuczuk, E.M.; Fischietti, M.; Eckerdt, F.; Fish, E.N.; Platanias, L.C. Human SLFN5 is a transcriptional co-repressor of STAT1-mediated interferon responses and promotes the malignant phenotype in glioblastoma. Oncogene 2017, 36, 6006–6019. [Google Scholar] [CrossRef] [PubMed]
- Fischietti, M.; Eckerdt, F.; Blyth, G.T.; Arslan, A.D.; Mati, W.M.; Oku, C.V.; Perez, R.E.; Lee-Chang, C.; Kosciuczuk, E.M.; Saleiro, D.; et al. Schlafen 5 as a novel therapeutic target in pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 3273–3286. [Google Scholar] [CrossRef] [PubMed]
- Metzner, F.J.; Huber, E.; Hopfner, K.P.; Lammens, K. Structural and biochemical characterization of human Schlafen 5. Nucleic Acids Res. 2022, 50, 1147–1161. [Google Scholar] [CrossRef]
- Prabhudas, M.; Bowdish, D.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; Means, T.K.; Moestrup, S.K.; et al. Standardizing scavenger receptor nomenclature. J. Immunol. 2014, 192, 1997–2006. [Google Scholar] [CrossRef]
- Ley, K.; Pramod, A.B.; Croft, M.; Ravichandran, K.S.; Ting, J.P. How Mouse Macrophages Sense What Is Going On. Front. Immunol. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Su, H.; Shen, X.; Du, J.; Zhang, X.; Zhao, Y. The immunological function of CD52 and its targeting in organ transplantation. Inflamm. Res. 2017, 66, 571–578. [Google Scholar] [CrossRef]
- Vitalle, J.; Terren, I.; Orrantia, A.; Bilbao, A.; Gamboa, P.M.; Borrego, F.; Zenarruzabeitia, O. The Expression and Function of CD300 Molecules in the Main Players of Allergic Responses: Mast Cells, Basophils and Eosinophils. Int. J. Mol. Sci. 2020, 21, 3173. [Google Scholar] [CrossRef]
- Vitalle, J.; Terren, I.; Orrantia, A.; Zenarruzabeitia, O.; Borrego, F. CD300 receptor family in viral infections. Eur. J. Immunol. 2019, 49, 364–374. [Google Scholar] [CrossRef]
- Lewis Marffy, A.L.; McCarthy, A.J. Leukocyte Immunoglobulin-Like Receptors (LILRs) on Human Neutrophils: Modulators of Infection and Immunity. Front. Immunol. 2020, 11, 857. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.R.; Botto, M.; Morgan, B.P.; Neal, J.W.; Gasque, P. CD93 regulates central nervous system inflammation in two mouse models of autoimmune encephalomyelitis. Immunology 2018, 155, 346–355. [Google Scholar] [CrossRef]
- McCormack, R.; Podack, E.R. Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. J. Leukoc. Biol. 2015, 98, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Mikulska, M.; Lanini, S.; Gudiol, C.; Drgona, L.; Ippolito, G.; Fernandez-Ruiz, M.; Salzberger, B. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: An infectious diseases perspective (Agents targeting lymphoid cells surface antigens [I]: CD19, CD20 and CD52). Clin. Microbiol. Infect. 2018, 24 (Suppl. 2), S71–S82. [Google Scholar] [CrossRef] [PubMed]
- Bassilana, F.; Nash, M.; Ludwig, M.G. Adhesion G protein-coupled receptors: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2019, 18, 869–884. [Google Scholar] [CrossRef]
- Pereira, M.; Ribeiro, D.R.; Pinheiro, M.M.; Ferreira, M.; Kellner, S.; Soares, A.R. m(5)U54 tRNA Hypomodification by Lack of TRMT2A Drives the Generation of tRNA-Derived Small RNAs. Int. J. Mol. Sci. 2021, 22, 2941. [Google Scholar] [CrossRef]
- Yue, T.; Zhan, X.; Zhang, D.; Jain, R.; Wang, K.W.; Choi, J.H.; Misawa, T.; Su, L.; Quan, J.; Hildebrand, S.; et al. SLFN2 protection of tRNAs from stress-induced cleavage is essential for T cell-mediated immunity. Science 2021, 372, eaba4220. [Google Scholar] [CrossRef]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Takai, T.; Ono, M.; Hikida, M.; Ohmori, H.; Ravetch, J.V. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996, 379, 346–349. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, W.; Torphy, R.J.; Yao, S.; Zhu, G.; Lin, R.; Lugano, R.; Miller, E.N.; Fujiwara, Y.; Bian, L.; et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci. Transl. Med. 2021, 13, eabc8922. [Google Scholar] [CrossRef]
- Kielar, M.; Dumnicka, P.; Ignacak, E.; Bedkowska-Prokop, A.; Gala-Bladzinska, A.; Maziarz, B.; Ceranowicz, P.; Kusnierz-Cabala, B. Soluble Complement Component 1q Receptor 1 (sCD93) Is Associated with Graft Function in Kidney Transplant Recipients. Biomolecules 2021, 11, 1623. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Caldi, E.; Parolini, B.; Toti, P.; Neri, G.; Nardi, F.; Traversi, C.; Cevenini, G.; Marigliani, D.; Nuti, E.; et al. CD93 as a Potential Target in Neovascular Age-Related Macular Degeneration. J. Cell. Physiol. 2017, 232, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Oosting, M.; Cheng, S.C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; van Eenennaam, H.; Sturm, P.; et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Sjaarda, J.; Gerstein, H.; Chong, M.; Yusuf, S.; Meyre, D.; Anand, S.S.; Hess, S.; Pare, G. Blood CSF1 and CXCL12 as Causal Mediators of Coronary Artery Disease. J. Am. Coll. Cardiol 2018, 72, 300–310. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, Q.; Zhang, N.; Du, X.; Xu, G.; Yan, X. CD146, from a melanoma cell adhesion molecule to a signaling receptor. Signal. Transduct. Target. Ther. 2020, 5, 148. [Google Scholar] [CrossRef]
- Kraler, S.; Wenzl, F.A.; Georgiopoulos, G.; Obeid, S.; Liberale, L.; von Eckardstein, A.; Muller, O.; Mach, F.; Raber, L.; Losdat, S.; et al. Soluble lectin-like oxidized low-density lipoprotein receptor-1 predicts premature death in acute coronary syndromes. Eur. Heart J. 2022, 43, 1849–1860. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, P.; Li, W.J.; Zhang, J.; Wang, G.P.; Jiang, D.F.; Chen, F.P. LncRNA NEAT1 sponges miR-214 to regulate M2 macrophage polarization by regulation of B7-H3 in multiple myeloma. Mol. Immunol. 2020, 117, 20–28. [Google Scholar] [CrossRef]
- Li, X.; Ye, S.; Lu, Y. Long non-coding RNA NEAT1 overexpression associates with increased exacerbation risk, severity, and inflammation, as well as decreased lung function through the interaction with microRNA-124 in asthma. J. Clin. Lab. Anal. 2020, 34, e23023. [Google Scholar] [CrossRef]
- Boros, F.A.; Maszlag-Torok, R.; Vecsei, L.; Klivenyi, P. Increased level of NEAT1 long non-coding RNA is detectable in peripheral blood cells of patients with Parkinson’s disease. Brain Res. 2020, 1730, 146672. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, E.; Lellahi, S.M.; Aure, M.R.; Nord, S.; Fismen, S.; Larsen, K.B.; Gabriel, M.T.; Hedberg, A.; Bjorklund, S.S.; Oslo Breast Cancer Research, C.; et al. The expression of the long NEAT1_2 isoform is associated with human epidermal growth factor receptor 2-positive breast cancers. Sci. Rep. 2020, 10, 1277. [Google Scholar] [CrossRef]
- Apostolakis, S.; Vogiatzi, K.; Amanatidou, V.; Spandidos, D.A. Interleukin 8 and cardiovascular disease. Cardiovasc. Res. 2009, 84, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, I.M.; Frumento, P.; Johansson, K.; Berglund, A.; de Faire, U.; Leander, K.; Gigante, B. Association of interleukin 8 with myocardial infarction: Results from the Stockholm Heart Epidemiology Program. Int. J. Cardiol. 2014, 172, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Szomjak, E.; Der, H.; Kerekes, G.; Veres, K.; Csiba, L.; Toth, J.; Peter, M.; Soltesz, P.; Szodoray, P. Immunological parameters, including CXCL8 (IL-8) characterize cerebro- and cardiovascular events in patients with peripheral artery diseases. Scand. J. Immunol. 2010, 71, 283–291. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-kappaB signaling in macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef] [PubMed]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Tomioka, Y.; Abe, T.; Anderson, P.; Ivanov, P. In lysate RNA digestion provides insights into the angiogenin’s specificity towards transfer RNAs. RNA Biol. 2021, 18, 2546–2555. [Google Scholar] [CrossRef]
- Betat, H.; Morl, M. The CCA-adding enzyme: A central scrutinizer in tRNA quality control. Bioessays 2015, 37, 975–982. [Google Scholar] [CrossRef]
- Fujishima, K.; Kanai, A. tRNA gene diversity in the three domains of life. Front. Genet. 2014, 5, 142. [Google Scholar] [CrossRef]
- Giege, R. Toward a more complete view of tRNA biology. Nat. Struct. Mol. Biol. 2008, 15, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. Controlling translation via modulation of tRNA levels. Wiley Interdiscip. Rev. RNA 2015, 6, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Puck, A.; Aigner, R.; Modak, M.; Cejka, P.; Blaas, D.; Stockl, J. Expression and regulation of Schlafen (SLFN) family members in primary human monocytes, monocyte-derived dendritic cells and T cells. Results Immunol. 2015, 5, 23–32. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G1_human_mascRNA | taatacgactcactataGGTTGGCACTCCTGGTTTCCgttttagagctagaaatagc |
| G5_human_mascRNA | taatacgactcactataGGACGGGGTTCAAATCCCTGgttttagagctagaaatagc |
| G9_human_menRNA | taatacgactcactataGGGGCACGTCCAGCACGGCTgttttagagctagaaatagc |
| G10_human_menRNA | taatacgactcactataGGTCCAGCACGGCTGGGCCGgttttagagctagaaatagc |
| universal reverse | AGCACCGACTCGGTGCCACT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gast, M.; Nageswaran, V.; Kuss, A.W.; Tzvetkova, A.; Wang, X.; Mochmann, L.H.; Rad, P.R.; Weiss, S.; Simm, S.; Zeller, T.; et al. tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions. Cells 2022, 11, 3970. https://doi.org/10.3390/cells11243970
Gast M, Nageswaran V, Kuss AW, Tzvetkova A, Wang X, Mochmann LH, Rad PR, Weiss S, Simm S, Zeller T, et al. tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions. Cells. 2022; 11(24):3970. https://doi.org/10.3390/cells11243970
Chicago/Turabian StyleGast, Martina, Vanasa Nageswaran, Andreas W. Kuss, Ana Tzvetkova, Xiaomin Wang, Liliana H. Mochmann, Pegah Ramezani Rad, Stefan Weiss, Stefan Simm, Tanja Zeller, and et al. 2022. "tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions" Cells 11, no. 24: 3970. https://doi.org/10.3390/cells11243970
APA StyleGast, M., Nageswaran, V., Kuss, A. W., Tzvetkova, A., Wang, X., Mochmann, L. H., Rad, P. R., Weiss, S., Simm, S., Zeller, T., Voelzke, H., Hoffmann, W., Völker, U., Felix, S. B., Dörr, M., Beling, A., Skurk, C., Leistner, D.-M., Rauch, B. H., ... Poller, W. (2022). tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions. Cells, 11(24), 3970. https://doi.org/10.3390/cells11243970