
Role of S100A8/A9 in Platelet–Neutrophil Complex Formation during Acute Inflammation

 , , , ,
, , , , 
Abstract
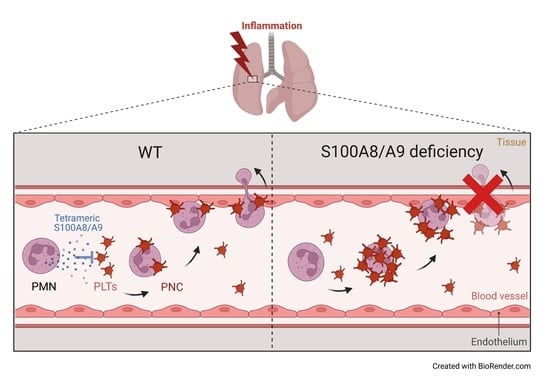
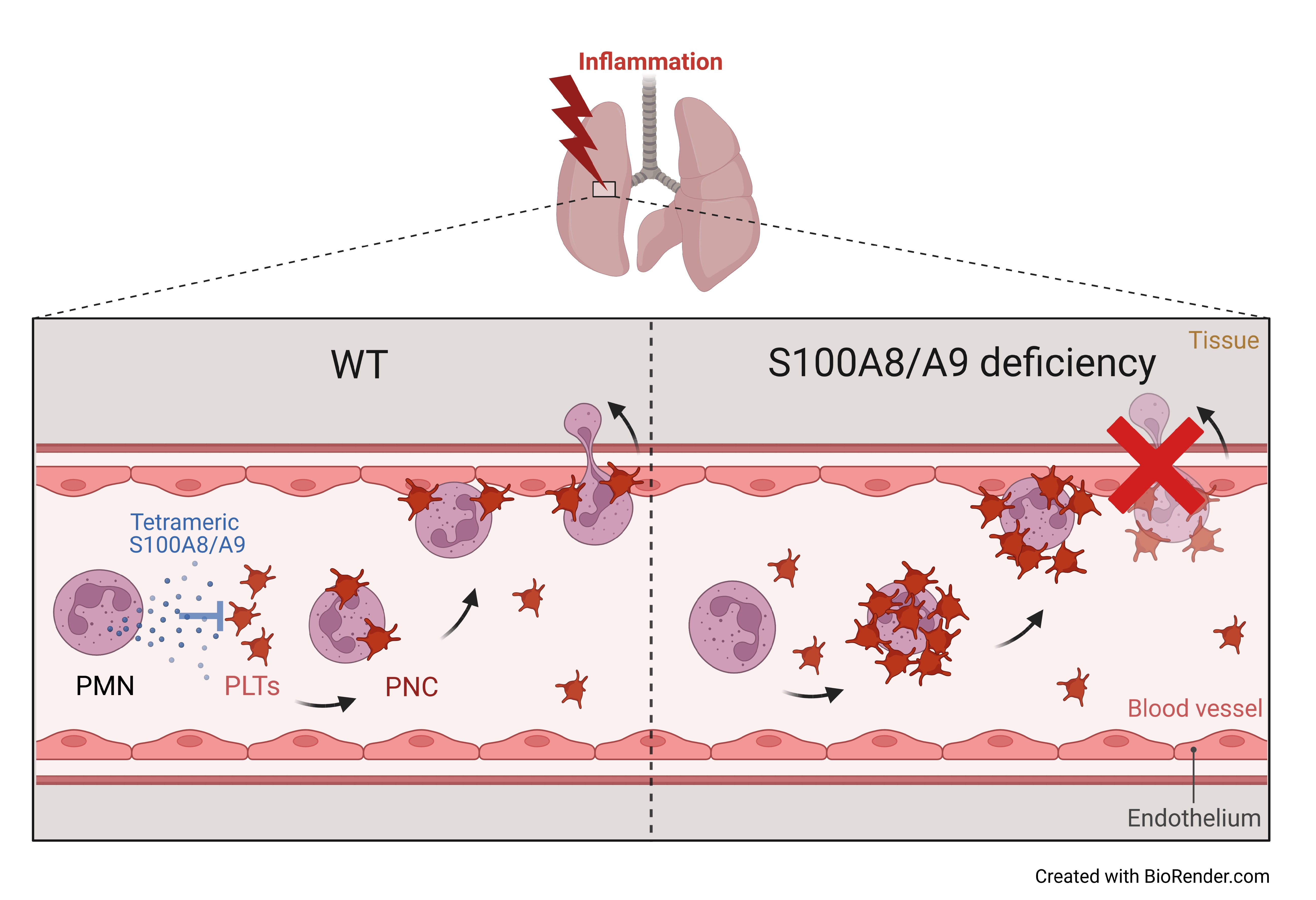{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Reagents
2.2. Klebsiella pneumoniae-Induced Lung Infection
2.3. Luminex Analysis of Inflammatory Mediators
2.4. Isolation of Murine Lung Microvascular Endothelial Cells
2.5. Isolation of Murine Alveolar Epithelial Cells Type II
2.6. Bilayer Transmigration Assay
2.7. ImageStream Analysis
2.8. In Vitro Platelet–Neutrophil Complex Formation
2.9. Intravital Microscopy of the Murine Cremaster Muscle
2.10. Western Blot Analysis
2.11. Flow Cytometry
2.12. Surface Marker Expression
2.13. Fibrinogen ELISA and Blood Cell Counts
2.14. Statistics
3. Results
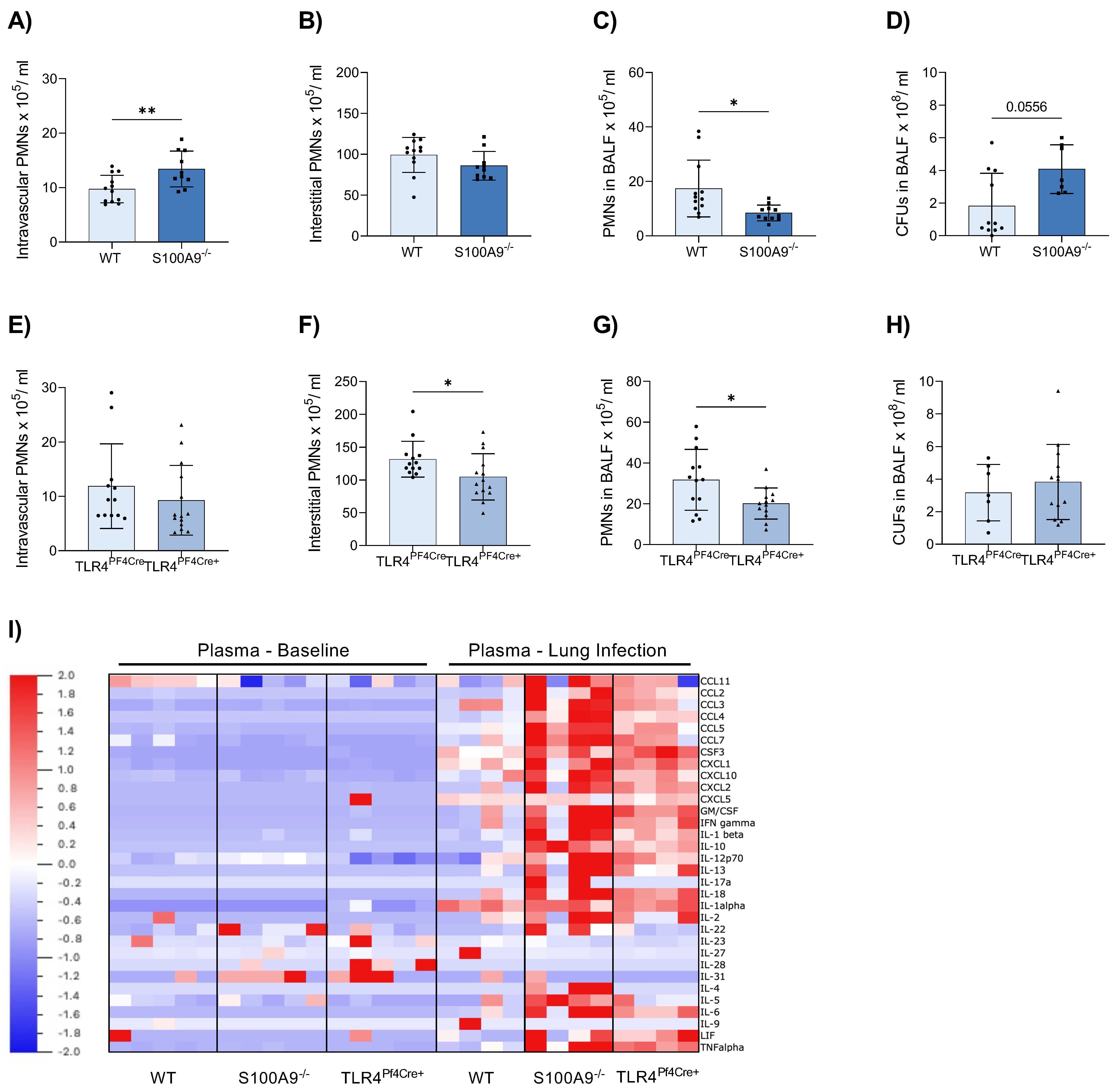
3.1. Neutrophil Response Is Hampered in S100A9−/− and TLR4PF4Cre+ Mice during Pneumonia
3.2. Platelets Do Not Express S100A8/A9
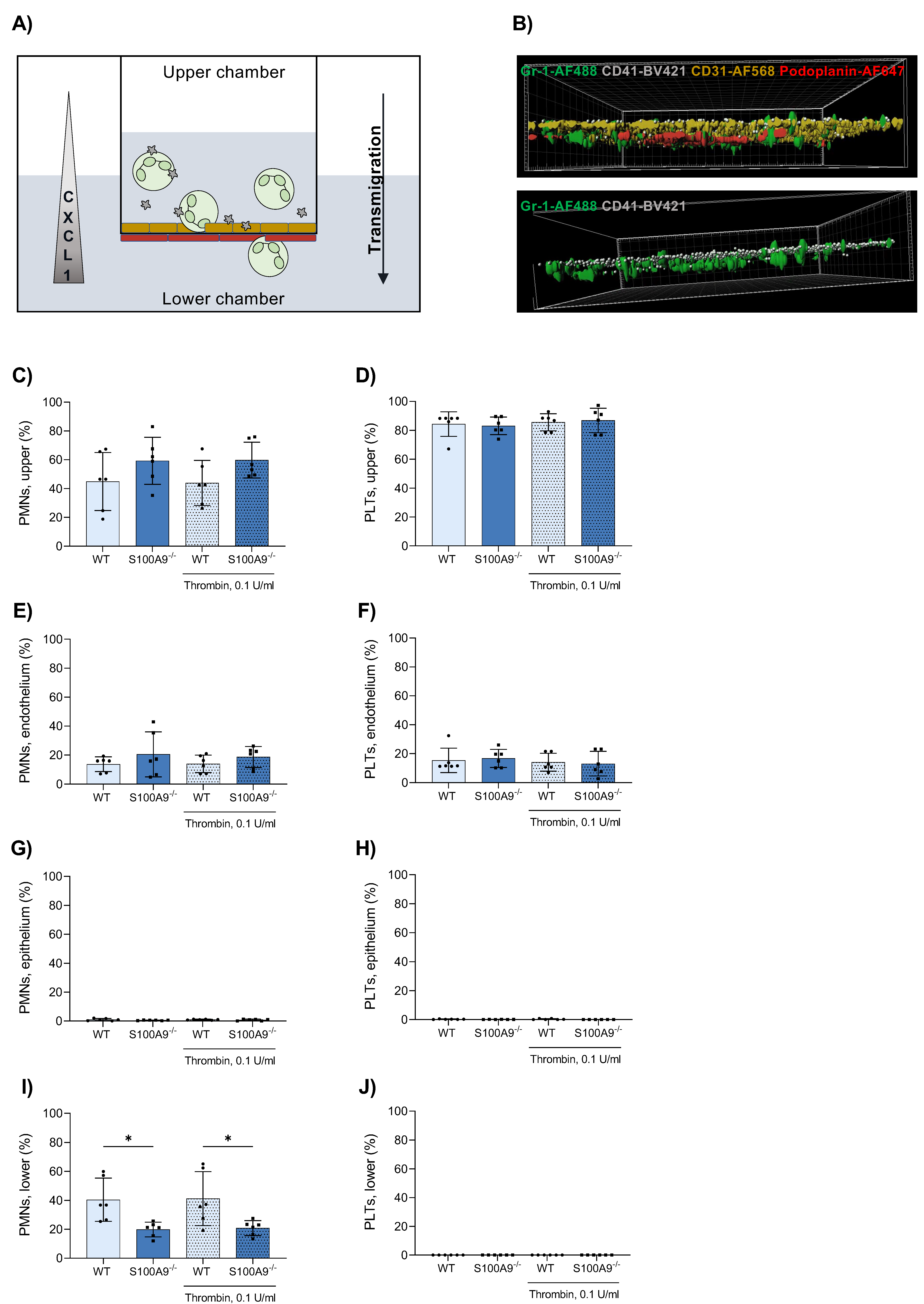
3.3. Platelets Do Not Cross the Endothelial–Epithelial Bilayer during Transmigration in an In Vitro Bilayer Model
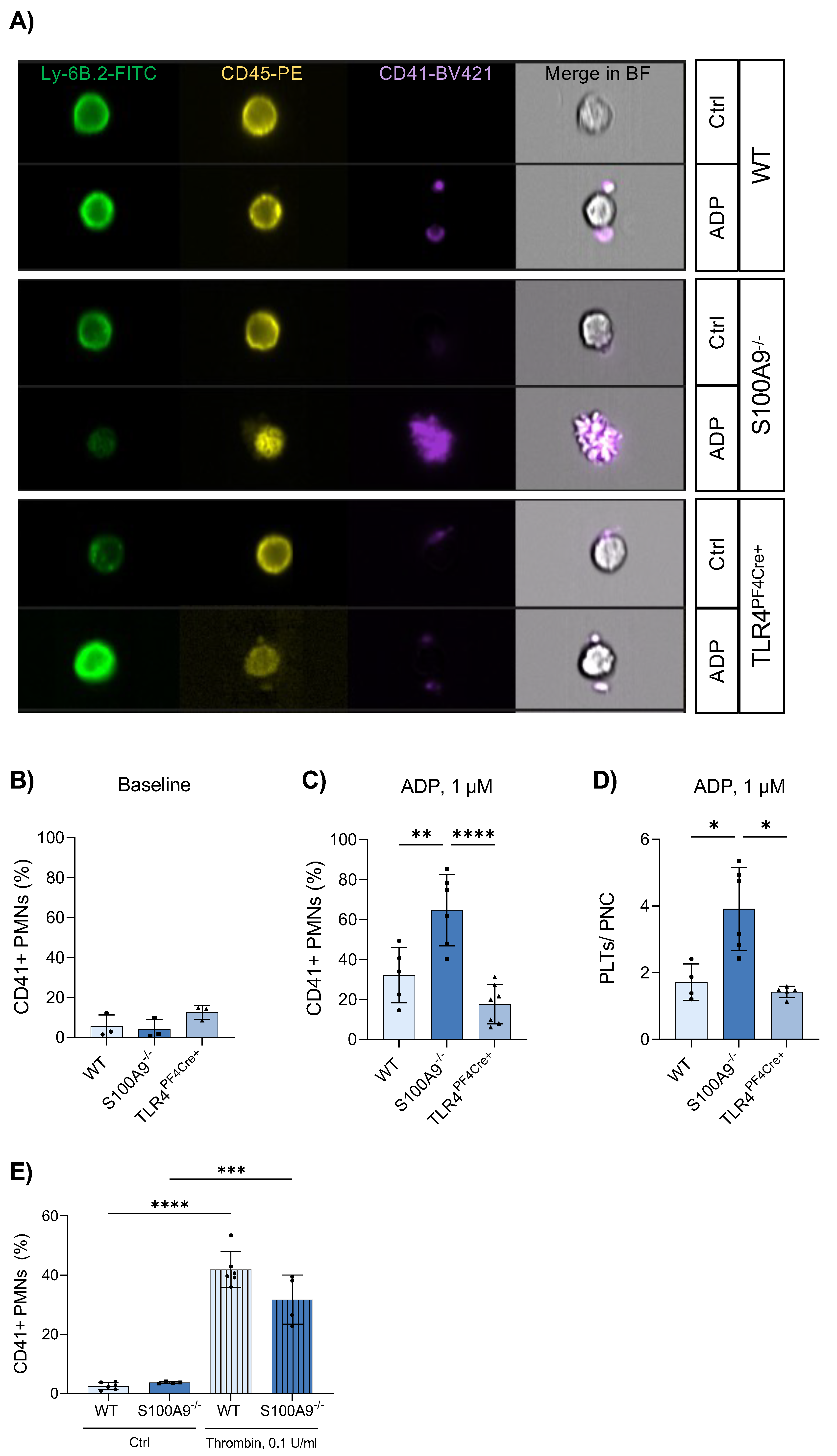
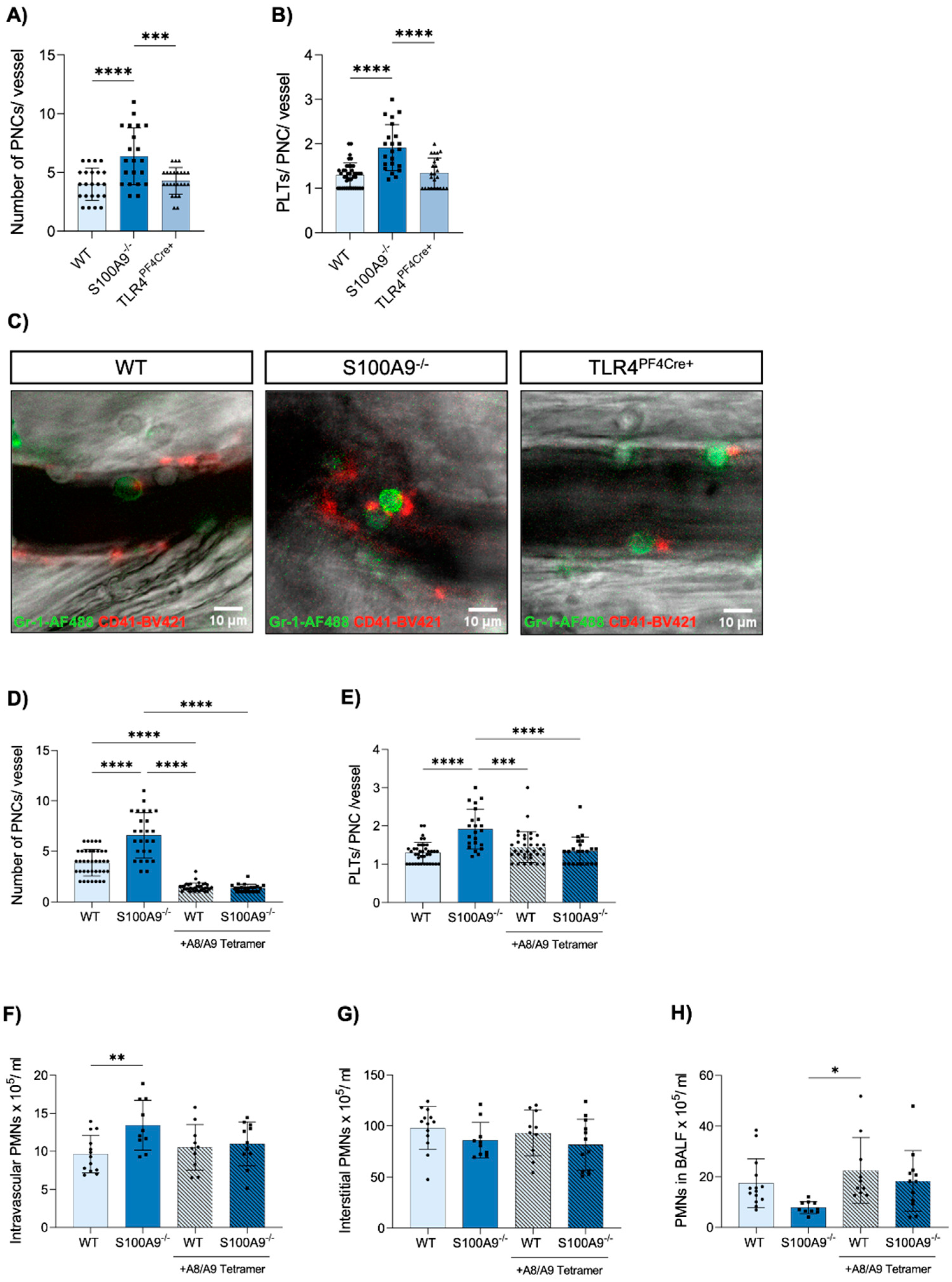
3.4. Platelet–Neutrophil Complexes Are Bigger and More Abundant in S100A9−/− Mice
3.5. Administration of Exogenous S100A8/A9 Tetramer Corrects Dysregulated PNC Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Zarbock, A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J. Innate Immun. 2013, 5, 348–357. [Google Scholar] [CrossRef]
- Fan, E.K.Y.; Fan, J. Regulation of alveolar macrophage death in acute lung inflammation. Respir. Res. 2018, 19, 50. [Google Scholar] [CrossRef]
- Chakraborty, D.; Zenker, S.; Rossaint, J.; Hölscher, A.; Pohlen, M.; Zarbock, A.; Roth, J.; Vogl, T. Alarmin S100A8 Activates Alveolar Epithelial Cells in the Context of Acute Lung Injury in a TLR4-Dependent Manner. Front. Immunol. 2017, 8, 1493. [Google Scholar] [CrossRef]
- Menezes, S.L.; Bozza, P.T.; Neto, H.C.; Laranjeira, A.P.; Negri, E.M.; Capelozzi, V.L.; Zin, W.A.; Rocco, P.R. Pulmonary and extrapulmonary acute lung injury: Inflammatory and ultrastructural analyses. J. Appl. Physiol. (1985) 2005, 98, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Wienkamp, A.K.; Erpenbeck, L.; Rossaint, J. Platelets in the NETworks interweaving inflammation and thrombosis. Front. Immunol. 2022, 13, 953129. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef]
- Brown, A.J.; Sepuru, K.M.; Sawant, K.V.; Rajarathnam, K. Platelet-Derived Chemokine CXCL7 Dimer Preferentially Exists in the Glycosaminoglycan-Bound Form: Implications for Neutrophil-Platelet Crosstalk. Front. Immunol. 2017, 8, 1248. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Doring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Brandt, E.; Petersen, F.; Ludwig, A.; Ehlert, J.E.; Bock, L.; Flad, H.D. The beta-thromboglobulins and platelet factor 4: Blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J. Leukoc. Biol. 2000, 67, 471–478. [Google Scholar] [CrossRef]
- Abdulnour, R.E.; Gunderson, T.; Barkas, I.; Timmons, J.Y.; Barnig, C.; Gong, M.; Kor, D.J.; Gajic, O.; Talmor, D.; Carter, R.E.; et al. Early Intravascular Events Are Associated with Development of Acute Respiratory Distress Syndrome. A Substudy of the LIPS-A Clinical Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 1575–1585. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e368. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Looney, M.R.; Nguyen, J.X.; Hu, Y.; Van Ziffle, J.A.; Lowell, C.A.; Matthay, M.A. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J. Clin. Investig. 2009, 119, 3450–3461. [Google Scholar] [CrossRef]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef]
- Alharbi, A.; Thompson, J.P.; Brindle, N.P.; Stover, C.M. Ex vivo modelling of the formation of inflammatory platelet-leucocyte aggregates and their adhesion on endothelial cells, an early event in sepsis. Clin. Exp. Med. 2019, 19, 321–337. [Google Scholar] [CrossRef]
- Caillon, A.; Trimaille, A.; Favre, J.; Jesel, L.; Morel, O.; Kauffenstein, G. Role of neutrophils, platelets, and extracellular vesicles and their interactions in COVID-19-associated thrombopathy. J. Thromb. Haemost. 2022, 20, 17–31. [Google Scholar] [CrossRef]
- Vogl, T.; Ludwig, S.; Goebeler, M.; Strey, A.; Thorey, I.S.; Reichelt, R.; Foell, D.; Gerke, V.; Manitz, M.P.; Nacken, W.; et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 2004, 104, 4260–4268. [Google Scholar] [CrossRef]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef]
- Vogl, T.; Stratis, A.; Wixler, V.; Voller, T.; Thurainayagam, S.; Jorch, S.K.; Zenker, S.; Dreiling, A.; Chakraborty, D.; Frohling, M.; et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J. Clin. Investig. 2018, 128, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Schurmann, H.; Brandt, M.; Scholz, K.; Matos, A.L.L.; Grill, D.; Revenstorff, J.; Rembrink, M.; von Wulffen, M.; Fischer-Riepe, L.; et al. Alarming and Calming: Opposing Roles of S100A8/S100A9 Dimers and Tetramers on Monocytes. Adv. Sci. 2022, e2201505. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Thomas, K.; Mersmann, S.; Skupski, J.; Margraf, A.; Tekath, T.; Jouvene, C.C.; Dalli, J.; Hidalgo, A.; Meuth, S.G.; et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J. Exp. Med. 2021, 218, e20201353. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Boras, M.; Volmering, S.; Bokemeyer, A.; Rossaint, J.; Block, H.; Bardel, B.; Van Marck, V.; Heitplatz, B.; Kliche, S.; Reinhold, A.; et al. Skap2 is required for beta2 integrin-mediated neutrophil recruitment and functions. J. Exp. Med. 2017, 214, 851–874. [Google Scholar] [CrossRef]
- Zarbock, A.; Lowell, C.A.; Ley, K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 2007, 26, 773–783. [Google Scholar] [CrossRef]
- Mueller, H.; Stadtmann, A.; Van Aken, H.; Hirsch, E.; Wang, D.; Ley, K.; Zarbock, A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways. Blood 2010, 115, 3118–3127. [Google Scholar] [CrossRef]
- Colicchia, M.; Schrottmaier, W.C.; Perrella, G.; Reyat, J.S.; Begum, J.; Slater, A.; Price, J.; Clark, J.C.; Zhi, Z.; Simpson, M.; et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbalpha. Blood 2022. [Google Scholar] [CrossRef] [PubMed]
- Manitz, M.P.; Horst, B.; Seeliger, S.; Strey, A.; Skryabin, B.V.; Gunzer, M.; Frings, W.; Schonlau, F.; Roth, J.; Sorg, C.; et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol. Cell. Biol. 2003, 23, 1034–1043. [Google Scholar] [CrossRef]
- Achouiti, A.; Vogl, T.; Van der Meer, A.J.; Stroo, I.; Florquin, S.; de Boer, O.J.; Roth, J.; Zeerleder, S.; van ‘t Veer, C.; de Vos, A.F.; et al. Myeloid-related protein-14 deficiency promotes inflammation in staphylococcal pneumonia. Eur. Respir. J. 2015, 46, 464–473. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, C.; Gao, H.; Bilodeau, M.L.; Zhang, Z.; Croce, K.; Liu, S.; Morooka, T.; Sakuma, M.; Nakajima, K.; et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J. Clin. Investig. 2014, 124, 2160–2171. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Chen, C.; Li, S.; Li, M.; Zeng, Z.; Zhang, Y.; Xia, L.; Li, X.; Zheng, D.; Lin, Q.; et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat. Cardiovasc. Res. 2022, 1, 732–747. [Google Scholar] [CrossRef]
- Edgeworth, J.; Gorman, M.; Bennett, R.; Freemont, P.; Hogg, N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J. Biol. Chem. 1991, 266, 7706–7713. [Google Scholar] [CrossRef]
- Lam, F.W.; Burns, A.R.; Smith, C.W.; Rumbaut, R.E. Platelets enhance neutrophil transendothelial migration via P-selectin glycoprotein ligand-1. Am. J. Physiology. Heart Circ. Physiol. 2011, 300, H468–H475. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Hessenauer, M.E.; Kurz, A.R.; Rehberg, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Spatiotemporal expression dynamics of selectins govern the sequential extravasation of neutrophils and monocytes in the acute inflammatory response. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hirata, T.; Croce, K.; Merrill-Skoloff, G.; Tchernychev, B.; Williams, E.; Flaumenhaft, R.; Furie, B.C.; Furie, B. Targeted gene disruption demonstrates that P-selectin glycoprotein ligand 1 (PSGL-1) is required for P-selectin-mediated but not E-selectin-mediated neutrophil rolling and migration. J. Exp. Med. 1999, 190, 1769–1782. [Google Scholar] [CrossRef]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef]
- Weber, C.; Springer, T.A. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef]
- Gavioli, R.; Risso, A.; Smilovich, D.; Baldissarro, I.; Capra, M.C.; Bargellesi, A.; Cosulich, M.E. CD69 molecule in human neutrophils: Its expression and role in signal-transducing mechanisms. Cell. Immunol. 1992, 142, 186–196. [Google Scholar] [CrossRef]
- Testi, R.; Pulcinelli, F.; Frati, L.; Gazzaniga, P.P.; Santoni, A. CD69 is expressed on platelets and mediates platelet activation and aggregation. J. Exp. Med. 1990, 172, 701–707. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revenstorff, J.; Ludwig, N.; Hilger, A.; Mersmann, S.; Lehmann, M.; Grenzheuser, J.C.; Kardell, M.; Bone, J.; Kötting, N.M.; Marx, N.C.; et al. Role of S100A8/A9 in Platelet–Neutrophil Complex Formation during Acute Inflammation. Cells 2022, 11, 3944. https://doi.org/10.3390/cells11233944
Revenstorff J, Ludwig N, Hilger A, Mersmann S, Lehmann M, Grenzheuser JC, Kardell M, Bone J, Kötting NM, Marx NC, et al. Role of S100A8/A9 in Platelet–Neutrophil Complex Formation during Acute Inflammation. Cells. 2022; 11(23):3944. https://doi.org/10.3390/cells11233944
Chicago/Turabian StyleRevenstorff, Julian, Nadine Ludwig, Annika Hilger, Sina Mersmann, Martin Lehmann, Julia Chiara Grenzheuser, Marina Kardell, Julia Bone, Niklas Martin Kötting, Nina Christine Marx, and et al. 2022. "Role of S100A8/A9 in Platelet–Neutrophil Complex Formation during Acute Inflammation" Cells 11, no. 23: 3944. https://doi.org/10.3390/cells11233944
APA StyleRevenstorff, J., Ludwig, N., Hilger, A., Mersmann, S., Lehmann, M., Grenzheuser, J. C., Kardell, M., Bone, J., Kötting, N. M., Marx, N. C., Roth, J., Vogl, T., & Rossaint, J. (2022). Role of S100A8/A9 in Platelet–Neutrophil Complex Formation during Acute Inflammation. Cells, 11(23), 3944. https://doi.org/10.3390/cells11233944