
Mushroom Natural Products in Neurodegenerative Disease Drug Discovery


Abstract
1. Ageing & Neurodegenerative Diseases
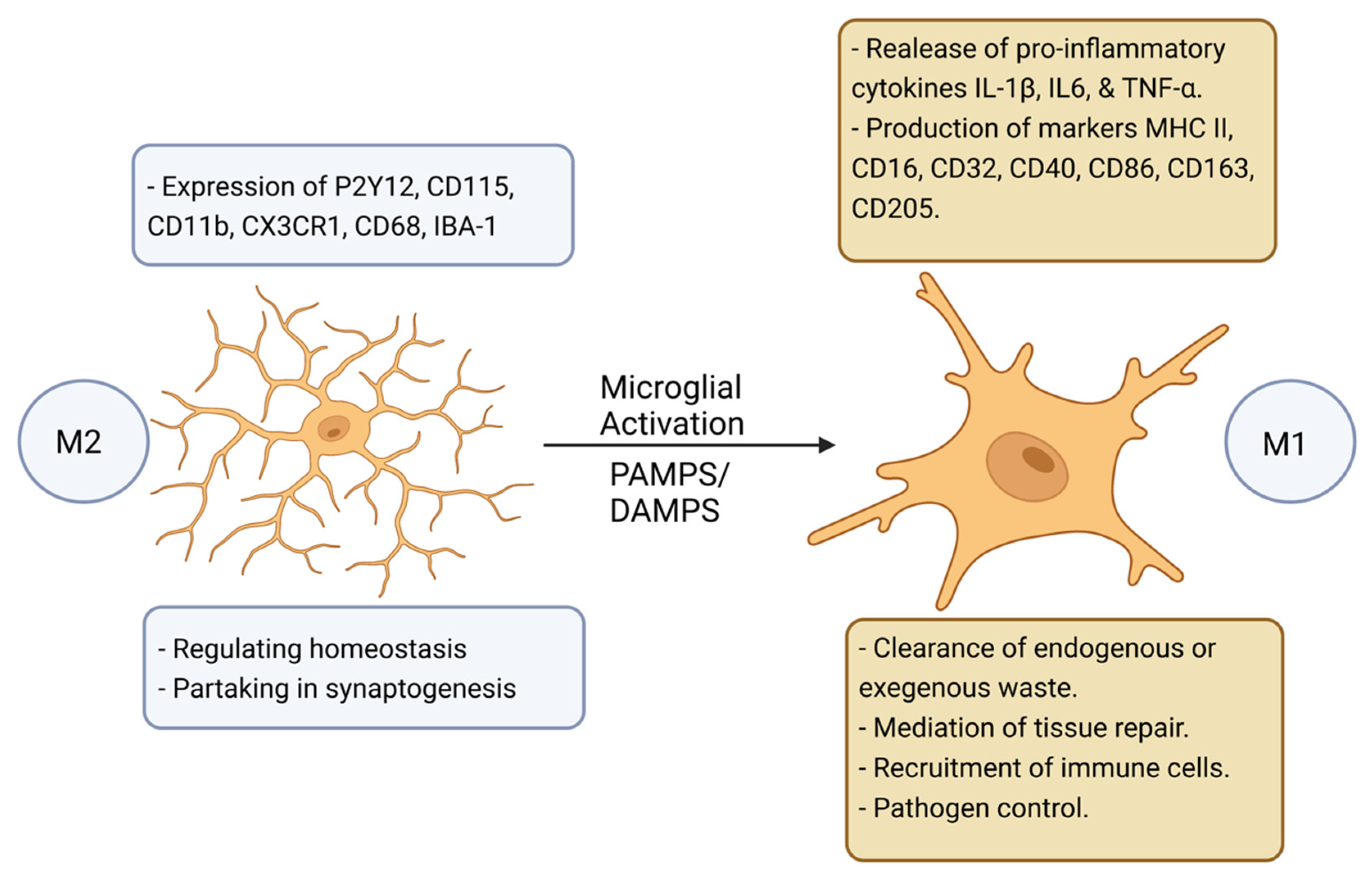
1.1. Alzheimer’s Disease: Pathological Protein Accumulation Leads to Microglial Activation and Systemic Neuroinflammation
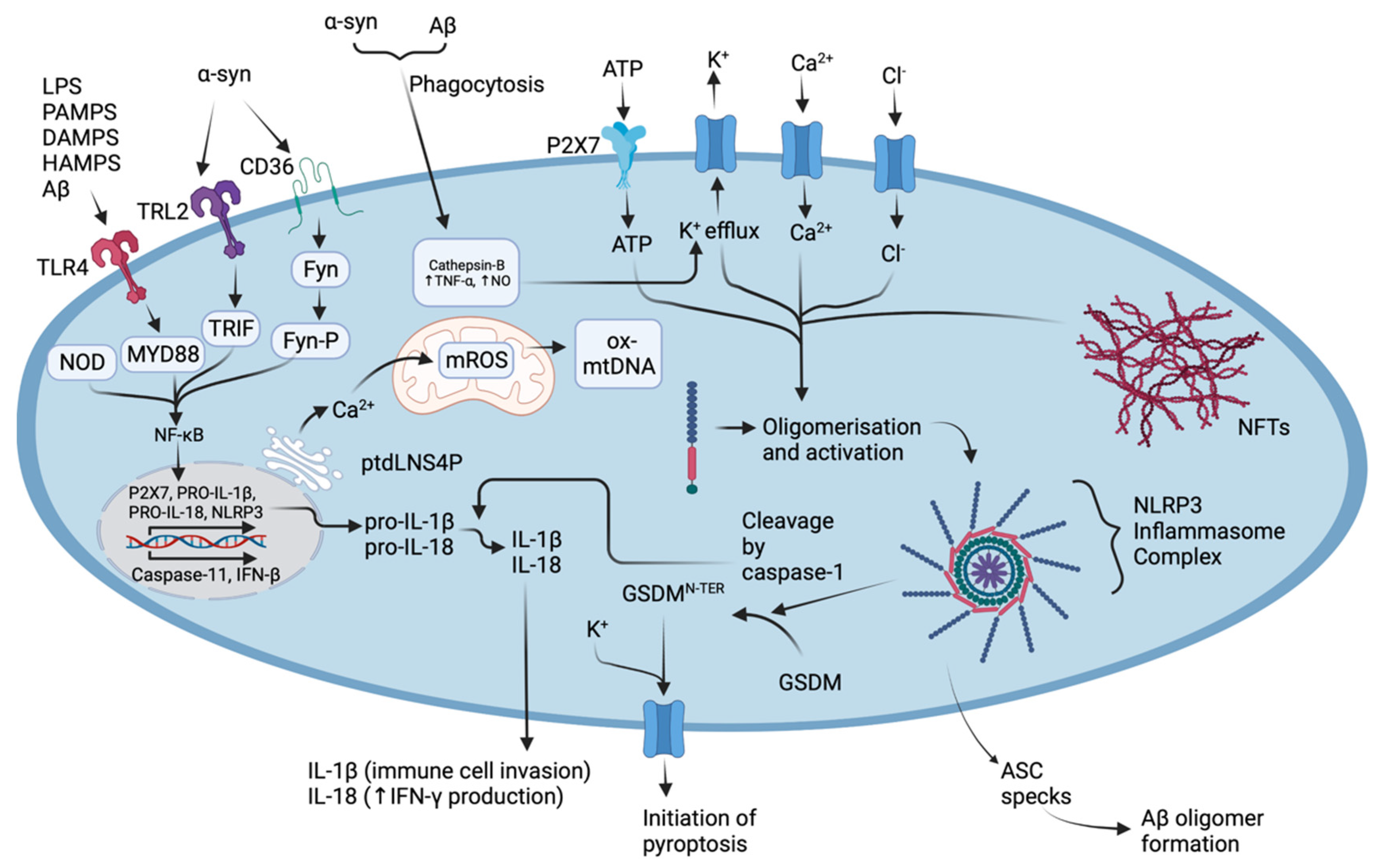
1.2. Parkinson’s Disease and the Activation of the NLRP3 Inflammasome in Activated Microglia Are Drivers of Neuroinflammation
1.3. Activated Microglia Drives an Auto-Reactive Immune Response and Neuroinflammation in Multiple Sclerosis, but Also Cleans Debris Promoting New Tissue Growth
1.4. Huntington’s Disease
2. Microglial Activation and NLRP3 Inflammasome Oligomerisation Drives Pyroptosis, Neuroinflammation, and Neurodegeneration
3. Natural Products: Drug Discovery and Clinical Trials in Neurodegenerative Disease’s
4. Mushroom Natural Products: Their Relevance in Ameliorating Neuroinflammation and Contribution to Drug Development for Neurodegenerative Diseases
4.1. Mushroom Derived Polysaccharide Natural Products and Extracts
4.2. Mushroom Derived Terpenes
4.3. Mushroom Derived Lanostanoids
4.4. Other Mushroom Derived Bioactive Natural Products
5. High-Throughput Assays Which Can Be Used to Screen Mushroom NPs and Extracts
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Peters, R. Ageing and the brain. Postgrad. Med. J. 2006, 82, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Flemming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Cataldo, A.M.; Mathews, P.M. The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: A review. Neurochem. Res. 2000, 25, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef]
- Soreq, L.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; Patani, R.; Ule, J. Major shifts in glial regional identity are a transcriptional hallmark of human brain aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Fotenos, A.F.; Snyder, A.; Girton, L.; Morris, J.; Buckner, R. Normative estimates of cross-sectional and longitudinal brain volume decline in aging and AD. Neurology 2005, 64, 1032–1039. [Google Scholar] [CrossRef]
- Cianciulli, A.; Calvello, R.; Ruggiero, M.; Panaro, M.A. Inflammaging and Brain: Curcumin and Its Beneficial Potential as Regulator of Microglia Activation. Molecules 2022, 27, 341. [Google Scholar] [CrossRef]
- Ashford, B.A.; Boche, D.; Cooper-Knock, J.; Heath, P.R.; Simpson, J.E.; Highley, J.R. Microglia in motor neuron disease. Neuropathol. Appl. Neurobiol. 2021, 47, 179–197. [Google Scholar] [CrossRef]
- Clarke, B.E.; Patani, R. The microglial component of amyotrophic lateral sclerosis. Brain 2020, 143, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, B.L.; Sicotte, N.L. Microglia in multiple sclerosis: Friend or foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jin, M.-Z.; Yang, Z.-Y.; Jin, W.-L. Microglia in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 270–280. [Google Scholar]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of general and discriminating markers of differential microglia phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Könnecke, H.; Bechmann, I. The Role of Microglia and Matrix Metalloproteinases Involvement in Neuroinflammation and Gliomas. Clin. Dev. Immunol. 2013, 2013, 914104. [Google Scholar] [CrossRef]
- Kalkman, H.O.; Feuerbach, D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 2016, 163, 82–93. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.; Nicoll, J. Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer related changes correlates with psychometrically assessed intellectual status. Neurobiol. Aging 1992, 13, S43–S44. [Google Scholar]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of Protein Aggregation in Mitochondrial Dysfunction and Neurodegeneration in Alzheimer’s and Parkinson’s Diseases. Neuromol. Med. 2003, 4, 21–35. [Google Scholar] [CrossRef]
- Novellino, F.; Saccà, V.; Donato, A.; Zaffino, P.; Spadea, M.F.; Vismara, M.; Arcidiacono, B.; Malara, N.; Presta, I.; Donato, G. Innate immunity: A common denominator between neurodegenerative and neuropsychiatric diseases. Int. J. Mol. Sci. 2020, 21, 1115. [Google Scholar] [CrossRef] [PubMed]
- Candlish, M.; Hefendehl, J.K. Microglia Phenotypes Converge in Aging and Neurodegenerative Disease. Front. Neurol. 2021, 12, 660720. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.-G.; Zhou, X.-G.; Qiao, G.; Yu, L.; Tang, Y.; Yan, L.; Qiu, W.-Q.; Pan, R.; Yu, C.-L.; Law, B.Y.-K.; et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 2021, 65, 101202. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.P.; Ziegler, M.G.; Alford, M.; Hansen, L.A.; Thal, L.J.; Masliah, E. Early and persistent alterations in prefrontal cortex MAO A and B in Alzheimer’s disease. J. Neural Transm. 2003, 110, 789–801. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.; Toma, M.; Bungau, S.; Bumbu, A. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative disease and the NLRP3 inflammasome. Front. Pharmacol. 2021, 12, 193. [Google Scholar] [CrossRef]
- Si, X.-L.; Fang, Y.-J.; Li, L.-F.; Gu, L.-Y.; Yin, X.-Z.; Yan, Y.-P.; Pu, J.-L.; Zhang, B.-R. From inflammasome to Parkinson’s disease: Does the NLRP3 inflammasome facilitate exosome secretion and exosomal alpha-synuclein transmission in Parkinson’s disease? Exp. Neurol. 2021, 336, 113525. [Google Scholar] [CrossRef]
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and Environmental Factors in Parkinson’s Disease Converge on Immune Function and Inflammation. Mov. Disord. 2021, 36, 25–36. [Google Scholar] [CrossRef]
- Arawaka, S.; Saito, Y.; Murayama, S.; Mori, H. Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for α-synuclein. Neurology 1998, 51, 887–889. [Google Scholar] [CrossRef]
- Woerman, A.L.; Watts, J.C.; Aoyagi, A.; Giles, K.; Middleton, L.T.; Prusiner, S.B. alpha-Synuclein: Multiple System Atrophy Prions. Cold Spring Harb. Perspect. Med. 2018, 8, a024588. [Google Scholar] [CrossRef]
- Gai, W.P.; Power, J.H.T.; Blumbergs, P.C.; Blessing, W.W. Multiple-system atrophy: A new α-synuclein disease? Lancet 1998, 352, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Matsumoto, K.; Takayama, K.; Yoshimoto, M.; Takahashi, H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci. Lett. 1997, 239, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, S.; Patel, D.; Agrawal-Rajput, R. CD40 negatively regulates ATP-TLR4-activated inflammasome in microglia. Cell. Mol. Neurobiol. 2017, 37, 351–359. [Google Scholar] [CrossRef]
- Mohamed, I.N.; Ishrat, T.; Fagan, S.C.; El-Remessy, A.B. Role of inflammasome activation in the pathophysiology of vascular diseases of the neurovascular unit. Antioxid. Redox Signal. 2015, 22, 1188–1206. [Google Scholar] [CrossRef] [PubMed]
- Miner, A.E.; Dastgheyb, N.; Palomino, M.; Graves, J.S. The Genetics of Multiple Sclerosis. In Neuroimmunology; Springer International Publishing: Cham, Switzerland, 2021; pp. 155–172. [Google Scholar]
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.M.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxid. Med. Cell. Longev. 2020, 2020, 7191080. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Gui, L.-N.; Liu, Y.-Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Araki, S.; Tsuchiya, Y.; Watanabe, Y. Oxidative stress orchestrates mapk and nitric-oxide synthase signal. Int. J. Mol. Sci. 2020, 21, 8750. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Ann. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Moynagh, P.N. Peli1. Nat. Immunol. 2011, 12, 927. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.-H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, Ø.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Stinissen, P.; Hendriks, J.J.A. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Nogales, M.; Lucas, J.J. Altered Levels and Isoforms of Tau and Nuclear Membrane Invaginations in Huntington’s Disease. Front. Cell. Neurosci. 2020, 13, 574. [Google Scholar] [CrossRef]
- Zheng, Z.; Diamond, M.I. Huntington disease and the huntingtin protein. Prog. Mol. Biol. Transl. Sci. 2012, 107, 189–214. [Google Scholar]
- Hedreen, J.C.; Folstein, S.E. Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1995, 54, 105–120. [Google Scholar] [CrossRef]
- Mitchell, I.; Cooper, A.; Griffiths, M. The selective vulnerability of striatopallidal neurons. Prog. Neurobiol. 1999, 59, 691–719. [Google Scholar]
- Gusella, J.F.; Wexler, N.S.; Michael Conneally, P.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef]
- Vuono, R.; Kouli, A.; Legault, E.M.; Chagnon, L.; Allinson, K.S.; La Spada, A.; Biunno, I.; Barker, R.A.; Drouin-Ouellet, J.; REGISTRY Investigators of the European Huntington’s Disease Network. Association between Toll-Like Receptor 4 (TLR4) and Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) Genetic Variants and Clinical Progression of Huntington’s Disease. Mov. Disord. 2020, 35, 401–408. [Google Scholar] [CrossRef]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) Diseases: Genetics to Treatments; SAGE Publications: Los Angeles, CA, USA, 2014; pp. 441–458. [Google Scholar]
- Rocha, N.P.; Ribeiro, F.M.; Furr-Stimming, E.; Teixeira, A.L. Neuroimmunology of Huntington’s Disease: Revisiting Evidence from Human Studies. Mediat. Inflamm. 2016, 2016, 8653132. [Google Scholar] [CrossRef] [PubMed]
- Vuono, R.; Winder-Rhodes, S.; De Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A.; REGISTRY Investigators of the European Huntington’s Disease Network. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain 2015, 138, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.C.; Farag, S.H.; Casey, C.S.; Wood-Kaczmar, A.; Pocock, J.M.; Tabrizi, S.J.; Andre, R. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J. Neuroinflamm. 2021, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Quek, H.; Cuní-López, C.; Stewart, R.; Colletti, T.; Notaro, A.; Nguyen, T.H.; Sun, Y.; Guo, C.C.; Lupton, M.K.; Roberts, T.L.; et al. ALS monocyte-derived microglia-like cells reveal cytoplasmic TDP-43 accumulation, DNA damage, and cell-specific impairment of phagocytosis associated with disease progression. J. Neuroinflamm. 2022, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Sahashi, K.; Iguchi, Y.; Hashizume, A. Preclinical progression of neurodegenerative diseases. Nagoya J. Med. Sci. 2018, 80, 289. [Google Scholar]
- Guan, Y.; Han, F. Key mechanisms and potential targets of the NLRP3 inflammasome in neurodegenerative diseases. Front. Integr. Neurosci. 2020, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chi, J.; Huang, D.; Ding, L.; Zhao, X.; Jiang, L.; Yu, Y.; Gao, F. α-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp. Ther. Med. 2020, 19, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.; Holley, C.L.; Bierschenk, D.; Stacey, K.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Jha, S. Comprehensive review of ASC structure and function in immune homeostasis and disease. Mol. Biol. Rep. 2020, 47, 3077–3096. [Google Scholar] [CrossRef] [PubMed]
- Hochheiser, I.; Pilsl, M.; Hagelueken, G.; Moecking, J.; Marleaux, M.; Brinkschulte, R.; Latz, E.; Engel, C.; Geyer, M. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 2022, 604, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Jeltema, D.; Wang, J.; Cai, J.; Kelley, N.; Yang, Z.; He, Y. A Single Amino Acid Residue Defines the Difference in NLRP3 Inflammasome Activation between NEK7 and NEK6. J. Immunol. 2022, 208, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef]
- Dick, M.S.; Sborgi, L.; Rühl, S.; Hiller, S.; Broz, P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 2016, 7, 11929. [Google Scholar] [CrossRef]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Akther, M.; Haque, E.; Park, J.; Kang, T.-B.; Lee, K.-H. Nlrp3 ubiquitination—A new approach to target nlrp3 inflammasome activation. Int. J. Mol. Sci. 2021, 22, 8780. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; Di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 2020, 369, 1448. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kuang, W.; Chen, W.; Xu, W.; Zhang, L.; Li, Y.; Li, H.; Peng, Y.; Chen, Y.; Wang, B.; et al. A phase II randomized trial of sodium oligomannate in Alzheimer’s dementia. Alzheimer’s Res. Ther. 2020, 12, 110. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Takeda, S.; Cho, H.; Wegmann, S.; Shoup, T.M.; Takahashi, K.; Irimia, D.; Elmaleh, D.R.; Hyman, B.T.; Hudry, E. A Food and Drug Administration-approved Asthma Therapeutic Agent Impacts Amyloid β in the Brain in a Transgenic Model of Alzheimer Disease. J. Biol. Chem. 2015, 290, 1966–1978. [Google Scholar] [CrossRef]
- Brazier, D.; Perry, R.; Keane, J.; Barrett, K.; Elmaleh, D.R. Pharmacokinetics of Cromolyn and Ibuprofen in Healthy Elderly Volunteers. Clin. Drug Investig. 2017, 37, 1025–1034. [Google Scholar] [CrossRef]
- Goldberg, J.; Currais, A.; Prior, M.; Fischer, W.; Chiruta, C.; Ratliff, E.; Daugherty, D.; Dargusch, R.; Finley, K.; Esparza-Moltó, P.B.; et al. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 2018, 17, e12715. [Google Scholar] [CrossRef]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 2019, 8, e47866. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trial: Study to Assess the Safety, Tolerability and Pharmacokinetics of Single Ascending Oral Doses of J147. In US Fed News Service, Including US State News; HT Digital Streams Limited: Washington, DC, USA, 2019.
- Menegazzi, M.; Campagnari, R.; Bertoldi, M.; Crupi, R.; Di Paola, R.; Cuzzocrea, S. Protective effect of epigallocatechin-3-gallate (EGCG) in diseases with uncontrolled immune activation: Could such a scenario be helpful to counteract COVID-19? Int. J. Mol. Sci. 2020, 21, 5171. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wang, Y.; Yuan, Q.; Luan, Q. The effect of (−)-epigallocatechin gallate as an adjunct to non-surgical periodontal treatment: A randomized clinical trial. Trials 2022, 23, 368. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Cano, A.; Manzine, P.R.; Busquets, O.; Verdaguer, E.; Castro-Torres, R.D.; García, M.L.; Beas-Zarate, C.; Olloquequi, J.; Auladell, C.; et al. Epigallocatechin-3-Gallate (EGCG) Improves Cognitive Deficits Aggravated by an Obesogenic Diet Through Modulation of Unfolded Protein Response in APPswe/PS1dE9 Mice. Mol. Neurobiol. 2019, 57, 1814–1827. [Google Scholar] [CrossRef]
- Bao, J.; Liu, W.; Zhou, H.-Y.; Gui, Y.-R.; Yang, Y.-H.; Wu, M.-J.; Xiao, Y.-F.; Shang, J.-T.; Long, G.-F.; Shu, X.-J. Epigallocatechin-3-gallate Alleviates Cognitive Deficits in APP/PS1 Mice. Curr. Med. Sci. 2020, 40, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Obregon, D.; Rezai-Zadeh, K.; Bai, Y.; Sun, N.; Hou, H.; Zeng, J.; Mori, T.; Arndash, G.; Shytle, D.; et al. P4-267: ADAM10 activation is required for green tea EGCG-induced alpha-secretase cleavage of amyloid precursor protein. Alzheimer’s Dement. 2006, 2, S595. [Google Scholar] [CrossRef]
- Press Release: Alzheon Appoints Susan Flint as Vice President of Clinical Operations, Patrick Kesslak, PhD, as Vice President of Clinical Development & Medical Affairs, and Erwan de Naurois as Vice President of Finance; Dow Jones & Company Inc.: New York, NY, USA, 2022.
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Alzheon to Present ALZ-801 (Valiltramiprosate) Phase 3 Program Update and Industry-Leading Disease Modifying Effects from Phase 2 Trial in Patients with Early Alzheimer’s Disease at AD/PD and NDDS Scientific Conferences; Business Wire, Inc.: New York, NY, USA, 2022.
- Li, I.C.; Chang, H.-H.; Lin, C.-H.; Chen, W.-P.; Lu, T.-H.; Lee, L.-Y.; Chen, Y.-W.; Chen, Y.-P.; Chen, C.-C.; Lin, D.P.-C. Prevention of Early Alzheimer’s Disease by Erinacine A-Enriched Hericium erinaceus Mycelia Pilot Double-Blind Placebo-Controlled Study. Front. Aging Neurosci. 2020, 12, 155. [Google Scholar] [CrossRef]
- Lozupone, M.; Solfrizzi, V.; D’Urso, F.; Di Gioia, I.; Sardone, R.; Dibello, V.; Stallone, R.; Liguori, A.; Ciritella, C.; Daniele, A.; et al. Anti-amyloid-β protein agents for the treatment of Alzheimer’s disease: An update on emerging drugs. Expert Opin. Emerg. Drugs 2020, 25, 319–335. [Google Scholar] [CrossRef]
- Prior, M.; Goldberg, J.; Chiruta, C.; Farrokhi, C.; Kopynets, M.; Roberts, A.J.; Schubert, D. Selecting for neurogenic potential as an alternative for Alzheimer’s disease drug discovery. Alzheimer’s Dement. 2016, 12, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Goldberg, J.; Farrokhi, C.; Chang, M.; Prior, M.; Dargusch, R.; Daugherty, D.; Armando, A.; Quehenberger, O.; Maher, P.; et al. A comprehensive multiomics approach toward understanding the relationship between aging and dementia. Aging 2015, 7, 937–955. [Google Scholar] [CrossRef] [PubMed]
- Prior, M.; Dargusch, R.; Ehren, J.L.; Chiruta, C.; Schubert, D. The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimer’s Res. Ther. 2013, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Prior, M.; Dargusch, R.; Roberts, A.; Riek, R.; Eichmann, C.; Chiruta, C.; Akaishi, T.; Abe, K.; Maher, P.; et al. A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease. PLoS ONE 2011, 6, e27865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Green tea (−)-epigallocatechin-3-gallate inhibits β-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-κB pathways in mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-L.; Chen, T.-F.; Chiu, M.-J.; Way, T.-D.; Lin, J.-K. Epigallocatechin gallate (EGCG) suppresses β-amyloid-induced neurotoxicity through inhibiting c-Ab1/FE65 nuclear translocation and GSK3β activation. Neurobiol. Aging 2009, 30, 81–92. [Google Scholar] [CrossRef]
- Liu, M.; Chen, F.; Sha, L.; Wang, S.; Tao, L.; Yao, L.; He, M.; Yao, Z.; Liu, H.; Zhu, Z.; et al. (−)-Epigallocatechin-3-Gallate Ameliorates Learning and Memory Deficits by Adjusting the Balance of TrkA/p75NTR Signaling in APP/PS1 Transgenic Mice. Mol. Neurobiol. 2013, 49, 1350–1363. [Google Scholar] [CrossRef]
- Singh, N.A.; Bhardwaj, V.; Ravi, C.; Ramesh, N.; Mandal, A.K.A.; Khan, Z.A. EGCG nanoparticles attenuate aluminum chloride induced neurobehavioral deficits, beta amyloid and tau pathology in a rat model of Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 244. [Google Scholar] [CrossRef]
- Yufang, G.Z., Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. Researchers from Xi’an Medical University Report on Findings in Alzheimer Disease, (-)-Epigallocatechin-3-Gallate Ameliorates Memory Impairment and Rescues the Abnormal Synaptic Protein Levels in the Frontal Cortex and Hippocampus in a Mouse. Available online: https://pubmed.ncbi.nlm.nih.gov/28520620/ (accessed on 1 November 2022).
- Du, K.; Liu, M.; Zhong, X.; Yao, W.; Xiao, Q.; Wen, Q.; Yang, B.; Wei, M. Epigallocatechin Gallate Reduces Amyloid β-Induced Neurotoxicity via Inhibiting Endoplasmic Reticulum Stress-Mediated Apoptosis. Mol. Nutr. Food Res. 2022, 66, e2270007. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Choi, D.-Y.; Yun, Y.-P.; Han, S.B.; Oh, K.-W.; Hong, J.T. Epigallocatechin-3-gallate prevents systemic inflammation-induced memory deficiency and amyloidogenesis via its anti-neuroinflammatory properties. J. Nutr. Biochem. 2013, 24, 298–310. [Google Scholar] [CrossRef]
- Lee, Y.K.; Yuk, D.Y.; Lee, J.W.; Lee, S.Y.; Ha, T.Y.; Oh, K.W.; Yun, Y.P.; Hong, J.T. (−)-Epigallocatechin-3-gallate prevents lipopolysaccharide-induced elevation of beta-amyloid generation and memory deficiency. Brain Res. 2008, 1250, 164–174. [Google Scholar] [CrossRef]
- de la Torre, R.P.; de Sola, S.P.; Hernandez, G.M.D.; Farré, M.M.D.; Pujol, J.M.D.; Rodriguez, J.M.; Espadaler, J.M.M.D.; Langohr, K.P.; Cuenca-Royo, A.P.; Principe, A.M.D.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin. Pharmacokinet. 2017, 57, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Silva, R.; Pinto, M.M.M.; Sousa, M.E. Marine natural products, multitarget therapy and repurposed agents in Alzheimer’s disease. Pharmaceuticals 2020, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A.; Sahlgrenska, A. Elucidating the Aβ42 Anti-Aggregation Mechanism of Action of Tramiprosate in Alzheimer’s Disease: Integrating Molecular Analytical Methods, Pharmacokinetic and Clinical Data. CNS Drugs 2017, 31, 495–509. [Google Scholar] [CrossRef]
- Alzheon Inc.—Pharmaceuticals and Healthcare Product Pipeline Summary. Available online: https://www.proquest.com/docview/2454571667?parentSessionId=NFLFBd%2BggXpJUQCJsUqh4cnhq4DGQyyeyLw1VppAPy4%3D&pq-origsite=summon&accountid=14543 (accessed on 20 November 2022).
- Yang, W.; Fang, Y.; Liang, J.; Hu, Q. Optimization of ultrasonic extraction of Flammulina velutipes polysaccharides and evaluation of its acetylcholinesterase inhibitory activity. Food Res. Int. 2011, 44, 1269–1275. [Google Scholar] [CrossRef]
- Yang, W.; Yu, J.; Zhao, L.; Ma, N.; Fang, Y.; Pei, F.; Mariga, A.M.; Hu, Q. Polysaccharides from Flammulina velutipes improve scopolamine-induced impairment of learning and memory of rats. J. Funct. Foods 2015, 18, 411–422. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, L.; Chen, Y.; Chen, X.; Dong, Y.; Zheng, S.; Zhang, L.; Li, W.; Du, J.; Li, H. A Maitake (Grifola frondosa) polysaccharide ameliorates Alzheimer’s disease-like pathology and cognitive impairments by enhancing microglial amyloid-β clearance. RSC Adv. 2019, 9, 37127–37135. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Dong, Y.; Qin, F.-Y.; Cheng, Y.-X. Australeols A−F, neuroprotective meroterpenoids from Ganoderma australe. Fitoterapia 2019, 134, 250–255. [Google Scholar] [CrossRef]
- Wei, J.; Guo, W.-H.; Cao, C.-Y.; Kou, R.-W.; Xu, Y.-Z.; Gorecki, M.; Di Bari, L.; Pescitelli, G.; Gao, J.-M. Polyoxygenated cyathane diterpenoids from the mushroom Cyathus africanus, and their neurotrophic and anti-neuroinflammatory activities. Sci. Rep. 2018, 8, 2175–2715. [Google Scholar] [CrossRef]
- Wei, J.; Cheng, Y.; Guo, W.-H.; Wang, D.-C.; Zhang, Q.; Li, D.; Rong, J.; Gao, J.-M. Molecular Diversity and Potential Anti-neuroinflammatory Activities of Cyathane Diterpenoids from the Basidiomycete Cyathus africanus. Sci. Rep. 2017, 7, 8883. [Google Scholar] [CrossRef]
- Yin, X.; Wei, J.; Wang, W.-W.; Gao, Y.-Q.; Stadler, M.; Kou, R.-W.; Gao, J.-M. New cyathane diterpenoids with neurotrophic and anti-neuroinflammatory activity from the bird’s nest fungus Cyathus africanus. Fitoterapia 2019, 134, 201–209. [Google Scholar] [CrossRef]
- Yoon, C.-S.; Kim, D.-C.; Park, J.-S.; Kim, K.-W.; Kim, Y.-C.; Oh, H. Isolation of novel sesquiterpeniods and anti-neuroinflammatory metabolites from nardostachys Jatamansi. Molecules 2018, 23, 2367. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Y.; Huang, C.-S.; Chen, Y.-H.; Chen, C.-C.; Chen, C.-C.; Chuang, C.-H. Anti-inflammatory effect of erinacine C on NO production through down-regulation of NF-κB and activation of Nrf2-mediated HO-1 in BV2 microglial cells treated with LPS. Molecules 2019, 24, 3317. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-L.; Hsu, J.-Y.; Chen, T.-C.; Huang, C.-C.; Wu, T.-Y.; Chin, T.-Y. Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 810. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.-W.; Han, R.; Gao, Y.-Q.; Li, D.; Yin, X.; Gao, J.-M. Anti-neuroinflammatory polyoxygenated lanostanoids from Chaga mushroom Inonotus obliquus. Phytochemistry 2021, 184, 112647. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Yu, C.; Tuong, T.M.; Kou, R.-W.; Yang, A.-A.; Chen, X.; Wang, W.J.; Gao, Y.-Q.; Gao, J.-M. Structures of ganorbifates C–I, seven previously undescribed lanostanoids from the mushroom Ganoderma orbiforme, and insights of computed biosynthesis with DFT. Phytochemistry 2022, 194, 113004. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.-W.; Xia, B.; Wang, Z.-J.; Li, J.-N.; Yang, J.-R.; Gao, Y.-Q.; Yin, X.; Gao, J.-M. Triterpenoids and meroterpenoids from the edible Ganoderma resinaceum and their potential anti-inflammatory, antioxidant and anti-apoptosis activities. Bioorganic Chem. 2022, 121, 105689. [Google Scholar] [CrossRef]
- Chang, Y.; Bai, M.; Xue, X.-B.; Zou, C.-X.; Huang, X.-X.; Song, S.-J. Isolation of chemical compositions as dietary antioxidant supplements and neuroprotectants from Chaga mushroom (Inonotus obliquus). Food Biosci. 2022, 47, 101623. [Google Scholar] [CrossRef]
- Song, H.; Huang, L.-P.; Li, Y.; Liu, C.; Wang, S.; Meng, W.; Wei, S.; Liu, X.-P.; Gong, Y.; Yao, L.-H. Neuroprotective effects of cordycepin inhibit Aβ-induced apoptosis in hippocampal neurons. Neurotoxicology 2018, 68, 73–80. [Google Scholar] [CrossRef]
- Zeng, X.; Suwandi, J.; Fuller, J.; Doronila, A.; Ng, K. Antioxidant capacity and mineral contents of edible wild Australian mushrooms. Food Sci. Technol. Int. 2012, 18, 367–379. [Google Scholar] [CrossRef]
- Hayton, J.B.; Ward, B.T.; Elnaas, A.R.; Zunk, M.; Holland, D.C.; May, T.W.; Voser, T.M.; Abitbol, A.; Cooper, O.; Tiralongo, J.; et al. Isolation of the 3′R and 3′S diastereomers of fasciculic acid C from the Australian mushroom Hypholoma australianum. Tetrahedron Lett. 2021, 78, 153294. [Google Scholar] [CrossRef]
- Aguilera, G.; Colín-González, A.L.; Rangel-López, E.; Chavarría, A.; Santamaría, A. Redox Signaling, Neuroinflammation, and Neurodegeneration. Antioxid. Redox Signal. 2018, 28, 1626–1651. [Google Scholar] [CrossRef]
- Rai, S.N.; Mishra, D.; Singh, P.; Vamanu, E.; Singh, M. Therapeutic applications of mushrooms and their biomolecules along with a glimpse of in silico approach in neurodegenerative diseases. Biomed. Pharmacother. 2021, 137, 111377. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G. Biomarker-Driven Analysis Using High-Throughput Approaches in Neuroinflammation and Neurodegenerative Diseases. In GeNeDis 2020; Springer International Publishing: Cham, Switzerland, 2022; pp. 51–58. [Google Scholar]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. In Biotechnology Annual Review; Elsevier Science & Technology: Amsterdam, The Netherlands, 2005; pp. 127–152. [Google Scholar]
- Pei, Y.; Peng, J.; Behl, M.; Sipes, N.S.; Shockley, K.R.; Rao, M.S.; Tice, R.R.; Zeng, X. Comparative neurotoxicity screening in human iPSC-derived neural stem cells, neurons and astrocytes. Brain Res. 2015, 1638 Pt A, 57–73. [Google Scholar] [CrossRef]
- Chiswick, E.L.; Duffy, E.; Japp, B.; Remick, D. Detection and quantification of cytokines and other biomarkers. In Leucocytes; Springer: Berlin, Germany, 2012; pp. 15–30. [Google Scholar]
- Wu, J.; Han, C.; Cao, X.; Lv, Z.; Wang, C.; Huo, X.; Feng, L.; Zhang, B.; Tian, X.; Ma, X. Mitochondria targeting fluorescent probe for MAO-A and the application in the development of drug candidate for neuroinflammation. Anal. Chim. Acta 2022, 1199, 339573. [Google Scholar] [CrossRef] [PubMed]
- Nizami, S.; Millar, V.; Arunasalam, K.; Zarganes-Tzitzikas, T.; Brough, D.; Tresadern, G.; Brennan, P.E.; Davis, J.B.; Ebner, D.; Di Daniel, E. A phenotypic high-content, high-throughput screen identifies inhibitors of NLRP3 inflammasome activation. Sci. Rep. 2021, 11, 15319. [Google Scholar] [CrossRef] [PubMed]
- McClure, R.; Redha, R.; Vinson, P.; Pham, W. A Robust and Scalable High-Throughput Compatible Assay for Screening Amyloid-β-Binding Compounds. J. Alzheimer’s Dis. 2019, 70, 187–197. [Google Scholar] [CrossRef]
- Tewari, D.; Sah, A.N.; Bawari, S.; Nabavi, S.F.; Dehpour, A.R.; Shirooie, S.; Braidy, N.; Fiebich, B.L.; Vacca, R.A.; Nabavi, S.M. Role of Nitric Oxide in Neurodegeneration: Function, Regulation, and Inhibition. Curr. Neuropharmacol. 2021, 19, 114–126. [Google Scholar] [CrossRef]
- Martín, A.S.; Arce-Molina, R.; Galaz, A.; Pérez-Guerra, G.; Barros, L.F. Nanomolar nitric oxide concentrations quickly and reversibly modulate astrocytic energy metabolism. J. Biol. Chem. 2017, 292, 9432–9438. [Google Scholar] [CrossRef]
- Bal-Price, A.; Brown, G.C. Inflammatory Neurodegeneration Mediated by Nitric Oxide from Activated Glia-Inhibiting Neuronal Respiration, Causing Glutamate Release and Excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef]
- Borutaite, V.; Brown, G. What else has to happen for nitric oxide to induce cell death? Biochem. Soc. Tran. 2005, 33, 1394–1396. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Hwang, S.-Y.; Oh, E.-S.; Oh, S.; Han, I.-O. IL-1beta, an immediate early protein secreted by activated microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK and NF-kappaB pathways. J. Neurosci. Res. 2006, 84, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Shin, M.; McInnis, M.G.; Bostwick, J.R. Combination Therapy with Monoamine Oxidase Inhibitors and Other Antidepressants or Stimulants: Strategies for the Management of Treatment-Resistant Depression. Pharmacotherapy 2015, 35, 433–449. [Google Scholar] [CrossRef]
- Chajkowski-Scarry, S.; Rimoldi, J.M. Monoamine oxidase A and B substrates: Probing the pathway for drug development. Future Med. Chem. 2014, 6, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.; Eatmon, C.V.; Slevin, J.T. Drug treatment strategies for depression in Parkinson disease. Expert Opin. Pharmacother. 2019, 20, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Sarkar, C.; Rawat, V.S.; Kalita, D.; Deka, S.; Agnihotri, A. Promise of the nlrp3 inflammasome inhibitors in in vivo disease models. Molecules 2021, 26, 4996. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Class, Composition and Origin | Mechanism of Action | Clinical Trial Status | Ref. |
|---|---|---|---|---|
| GV-917 (sodium oligomannate) | Acidic linear oligosaccharides that are found in marine brown algae | Suggested to recondition gut microbiota and alter peripheral immune system response underlying AD pathogenesis and penetrates BBB through GLUT1 destabilising Aβ fibril formation via forming non-toxic monomers | Active (phase 3) | [78,79] |
| ALZT-OP1 | Family of chromones. The drug is a mixture of Cromolyn (a synthetic derivative of the natural product khellin) and ibuprofen | Mast cell stabilizer suggested by decreasing Ca2+ efflux driven granulation and microglial activation modulator | Completed (phase 1) | [80,81] |
| J-147 | Phenyl hydrazide derivative of natural product curcumin | Activation of AMPK and stabilisation of AMPK/ACC1 signaling in mitochondria | Completed (phase 1) | [82,83,84] |
| Epigallocatechin-gallate (EGCG) | Polyphenol/catechin is commonly found in green tea | Induces α-secretase expression and decreased neuroinflammation by decreasing the expression of TLR4 in animal models | Completed (phase 4) | [85,86,87,88,89] |
| ALZ-801 (Valiltramiprosate) | Homotaurine is a modified amino acid commonly found in seaweed | The prodrug acts as an Aβ aggregation inhibitor with efficacy in APOE4 variants (heterozygotes < homozygotes) | Active (phase 2/3) | [90,91,92] |
| Erinacine A | Cyathin diterpenoid isolated from the mycelia of the mushroom H. erinaceus | Upregulate NGF gene expression, neurotrophic and neuroprotective activities | Completed (N/A) | [93] |
| Compound/Extract | Bioactivity | Cells/Model/Assay | Ref. |
|---|---|---|---|
| Polysaccharides | |||
| Polysaccharide extracts | ↑Spatial memory and cognition | MWM test in rats | [112,113] |
| Restoring AChE levels | AChE activity assay kit | ||
| ↑Connexin 36 & p-CaMKII expression | Anti-antibody detection kit | ||
| Derived polysaccharide extract from maitake (PGM) | PGM (5 mg–10 mg/kg) ↑escape latency time and cognition | APP-PS1 mice | [114] |
| PGM ameliorated histological and necrotic morphology, ↓Aβ/mm2 pathology, ↑microglial and astrocyte activation, and microglial mediated Aβ clearance | APP/PS1 mice isolated hippocampal cells | ||
| Terpenes | |||
| Ganomycin C (1), ganoresinain A (2), ganotheaecoloid G (3) | 1, 2, and 3 ↓glutamate-induced neurotoxicity | SH-SY5Y cells | [115] |
| New neocyathins K–R (4–11), & 3 known congeners: cyathin V, (12 S)-11α,14α-epoxy-13α,14β,15-trihydroxycyath-3-ene, & allocyathin B2 (12–14) | 4–14 no cytotoxicity (10 μM) | BV2 microglia & PC-12 cells. | [116] |
| 4–14 ↑In neurite-bearing cells (1–25 μM) with NGF (20 ng/mL) in PC-12 cells | PC-12 cells | ||
| 14 ↓iNOS (IC50 = 19.8 μM) | BV2 microglia & molecular docking | ||
| Cyanthane I (15), (12R)-11α,14α-epoxy-13α,14β,15-trihydroxycyath-3-ene (16), cyathin O (17), allocyafrin B4 (18) | 15–18 ↓NO suppression via iNOS & no cytotoxicity | Aβ1-42-induced & LPS-induced BV2 microglia, molecular docking, and Western blotting | [117] |
| 15, 16, & 18 abolished iNOS expression | Aβ1–42-induced BV2 microglia and molecular docking | ||
| 15 & 18 ↓COX-2 expression in BV2 cells supported by molecular docking | |||
| Cyafricanins A–K (19–29) | 19–29 (5–100 μM) + NGF (20 μg/mL) increased neurite-bearing cells & had no cytotoxicity | PC-12 cells | [118] |
| 29 ↓COX-2 expression, 20 ↓iNOS expression, & 19 & 20 ↓NO production | LPS-induced BV2 cells | ||
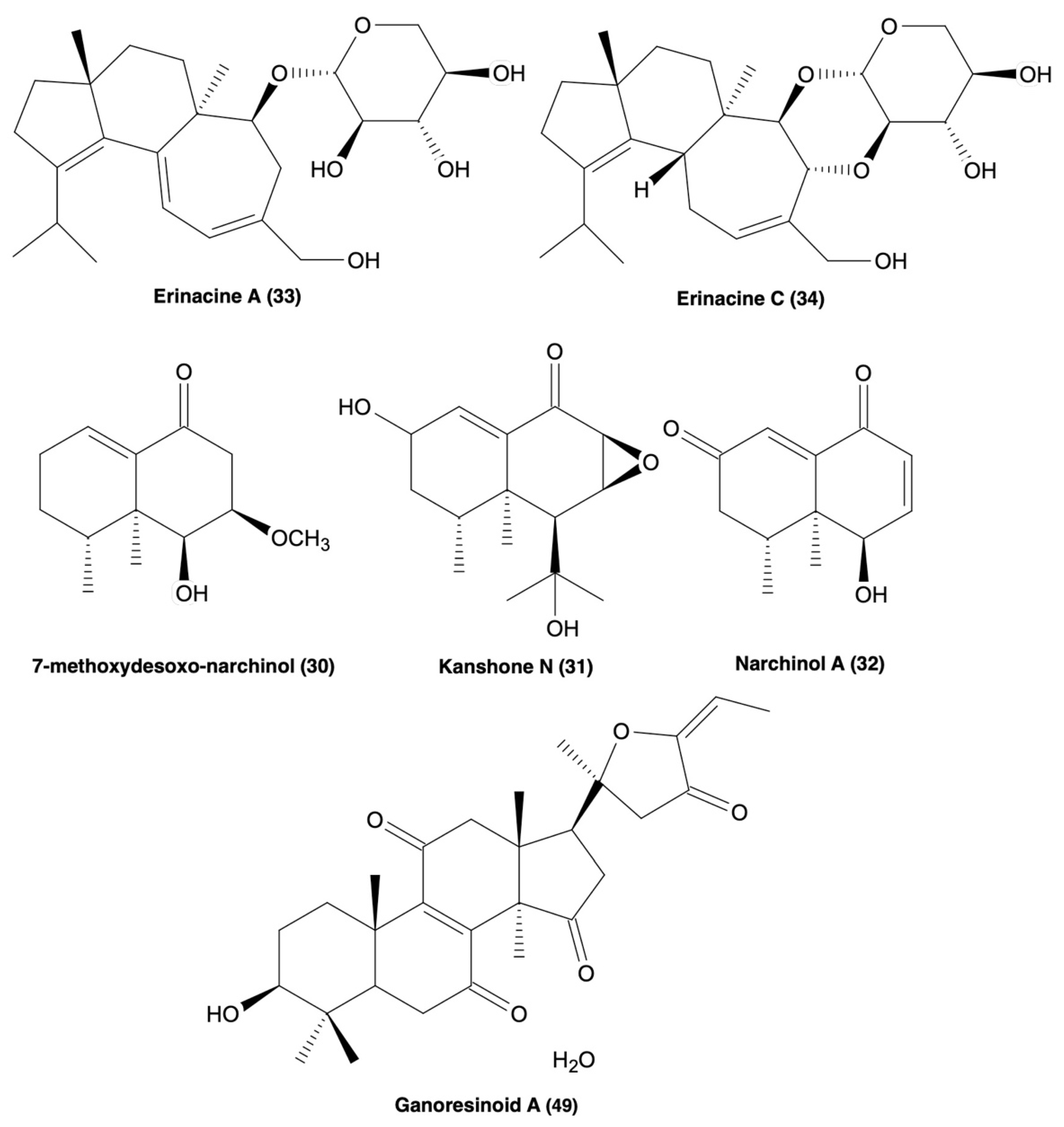
| 7-methoxydesoxo-narchinol (30), Kanshone N (31), narchinol A (32) | 30–32 ↓iNOS, PGE2, COX-2, IL-12, IL-1β, TNF-α expression & ↑IL-10, blocked p65/p50 translocation and phosphorylation of Iκ-B-α & displayed no cytotoxicity | LPS-stimulated BV2 microglia | [119] |
| Erinacine A (33), erinacine C (34) | 33 ↓iNOS & NO (20 μM) | LPS-induced BV2 microglia | [93,120,121] |
| LPS-stimulated astrocytes | |||
| 33 ↓TNF-α expression | N2a cells | ||
| 33 showed no cytotoxicity (1 mg/mL), ↓tyrosine hydroxylase, JNA, and NF-κB expression | |||
| 33 ↓inflammatory cytokine expression and ↑ motor and cognitive ability | LPS-induced mouse model | ||
| 34 ↓cell viability <50% (10 μM), but not at 0.1–2.5 μM. ↓iNOS, NO, IL-6, & TNF-α, P-IκB-α, and ↑Nrf2 expression | LPS-induced BV2 microglia | ||
| Lanostanoids | |||
| Inonotusols H–N (35–41) | 39–40 no cytotoxicity (25 μM) | BV2 microglia | [122] |
| 35, 36, 39, & 40 ↓NO (IC50 = 2.32–9.17 μM) | LPS-induced BV2 microglia | ||
| 36 & 39 ↓iNOS (50 μM) | LPS-induced BV2 microglia, Western blotting, and molecular docking | ||
| Ganorbifates C–I (42–48) | 89–95 ↓NO, 89 had the strongest (IC50 = 4.37 μM) | LPS-induced BV2 microglia | [123] |
| New: Ganoresinoids A & B (49 & 50) | 96 & 97 ↓NO | LPS-induced BV2 cells | [124] |
| 96 has no cytotoxicity at 10 μM, ↓TNF-α, IL-1β, IL-6, iNOS, COX2, TLR4, and NF-κB expression, and ameliorated ROS-induced MMP dysfunction and apoptosis | LPS-induced BV2 microglia | ||
| 96 ↑P-Akt and P-GSK-3β and [125] ↑HO-1, NQO-1, and Nrf2 expression | SH-SY5Y cells | ||
| Misc/extracts | |||
| Cordycepin | Cordyecepin ↓apoptosis, ROS-induced neuronal death, Ca2+ efflux, ICa dysfunction and resultant neurotoxicity via A1-R, AChE activity, and p-tau formation | Aβ25–35-induced rat hippocampal neurons | [126] |
| Phellxinye A (51), Inonotphenol A (52) | 51 & 52 have antioxidative capacity | DPPH and FRAP assay | [125] |
| 52 ↓apoptosis and MMP dysfunction | H2O2-induced apoptosis model using SH-SY5Y cells and fluorescent markers Hoechst 33258 and JC-1, respectively | ||
| MeOH extracts (53–57) | Weak ferrous ion chelating activity | FCA assay | [127] |
| Antioxidant capacity | Trolox equivalent assay | ||
| Ferric reducing antioxidant power | Ferric ion reducing antioxidant power assay | ||
| Assay | Sample Type | Target | Limitations | Size/High-Throughput | Ref. |
|---|---|---|---|---|---|
| MTT assay | Cells | Mitochondrial dehydrogenase dysfunction | Cells only, cannot be performed on tissue. | 96-well plate. | [133] |
| ELISA-based cytokine expression assay | Cells, tissues | Detection of cytokine expression | Coefficient of variation of 15% | 96-well plate. | [135] |
| Greiss Test | Cells, tissues | NO production | Limited by NO concentration > 5 μM | 96-well plate. | [15] |
| HCCP assay | Cells, tissues | Mitochondrial MAO-A | Background fluorescence interference | 96-well plate. | [136] |
| ASC speck detection assay | Cells | NLRP3-dependant ASC speck formation | Cytotoxicity in long treatments | 384-well plate. | [137] |
| HATCO assay | Brain lysate containing Aβ peptide | Aβ-binding molecules | DMSO solvent up to 5% of the assay’s volume | 384-well plate. | [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abitbol, A.; Mallard, B.; Tiralongo, E.; Tiralongo, J. Mushroom Natural Products in Neurodegenerative Disease Drug Discovery. Cells 2022, 11, 3938. https://doi.org/10.3390/cells11233938
Abitbol A, Mallard B, Tiralongo E, Tiralongo J. Mushroom Natural Products in Neurodegenerative Disease Drug Discovery. Cells. 2022; 11(23):3938. https://doi.org/10.3390/cells11233938
Chicago/Turabian StyleAbitbol, Arjuna, Brody Mallard, Evelin Tiralongo, and Joe Tiralongo. 2022. "Mushroom Natural Products in Neurodegenerative Disease Drug Discovery" Cells 11, no. 23: 3938. https://doi.org/10.3390/cells11233938
APA StyleAbitbol, A., Mallard, B., Tiralongo, E., & Tiralongo, J. (2022). Mushroom Natural Products in Neurodegenerative Disease Drug Discovery. Cells, 11(23), 3938. https://doi.org/10.3390/cells11233938