

Nrf2 Deficiency Exacerbated CLP-Induced Pulmonary Injury and Inflammation through Autophagy- and NF-κB/PPARγ-Mediated Macrophage Polarization

Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Study on Patients with Sepsis
2.2. Sepsis-Induced CLP Model
2.3. Collection of Mouse Lung Tissues and BALFs
2.4. Histologic Analysis
2.5. RNA-Seq Analysis
2.6. Immunofluorescence
2.7. Macrophage Isolation and Culture
2.8. Transmission Electron Microscopy
2.9. Flow Cytometry
2.10. Induction of M1/M2 Polarization
2.11. Cell Culture and Plasmid Transfection
2.12. Drug Administration
2.13. Quantitative Real-Time PCR
2.14. Western Blot
2.15. Statistical Analysis
3. Results
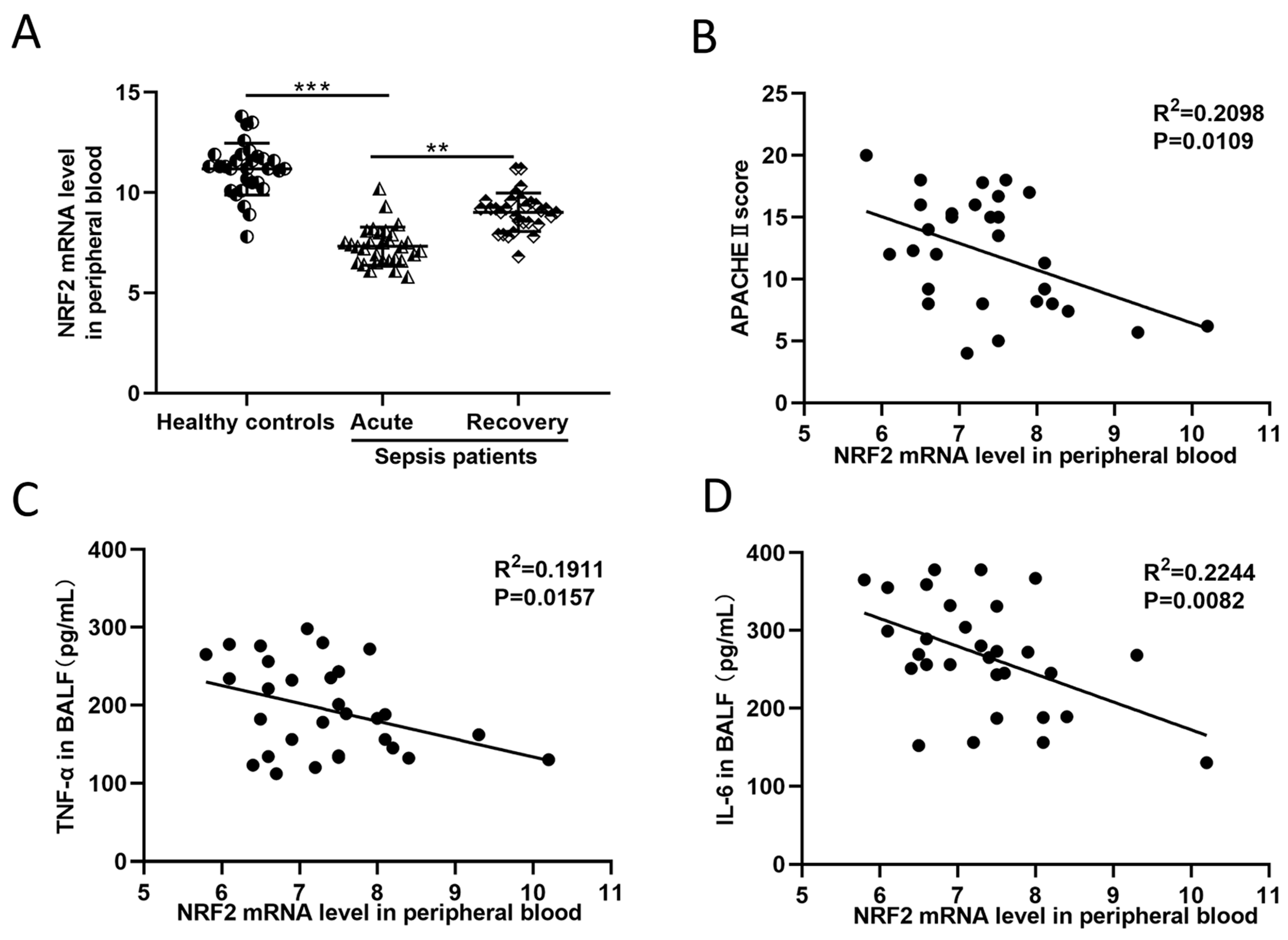
3.1. In Sepsis Patients, the NRF2 mRNA Level in Peripheral Blood Is Inversely Correlated with Inflammation and Disease Severity
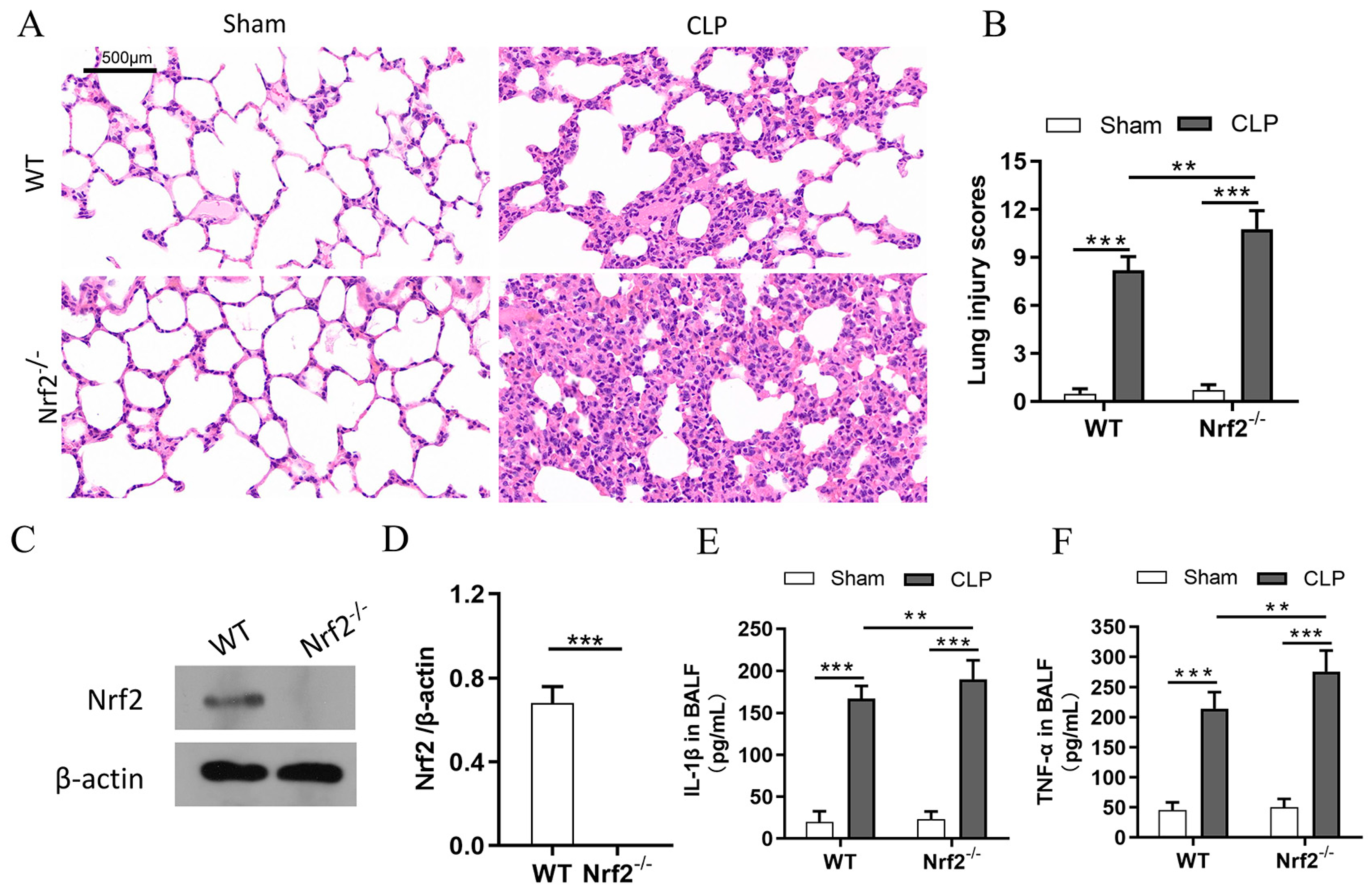
3.2. Nrf2 Deficiency Exacerbates Sepsis-Induced ALI and Promotes Inflammation in a CLP Mouse Model
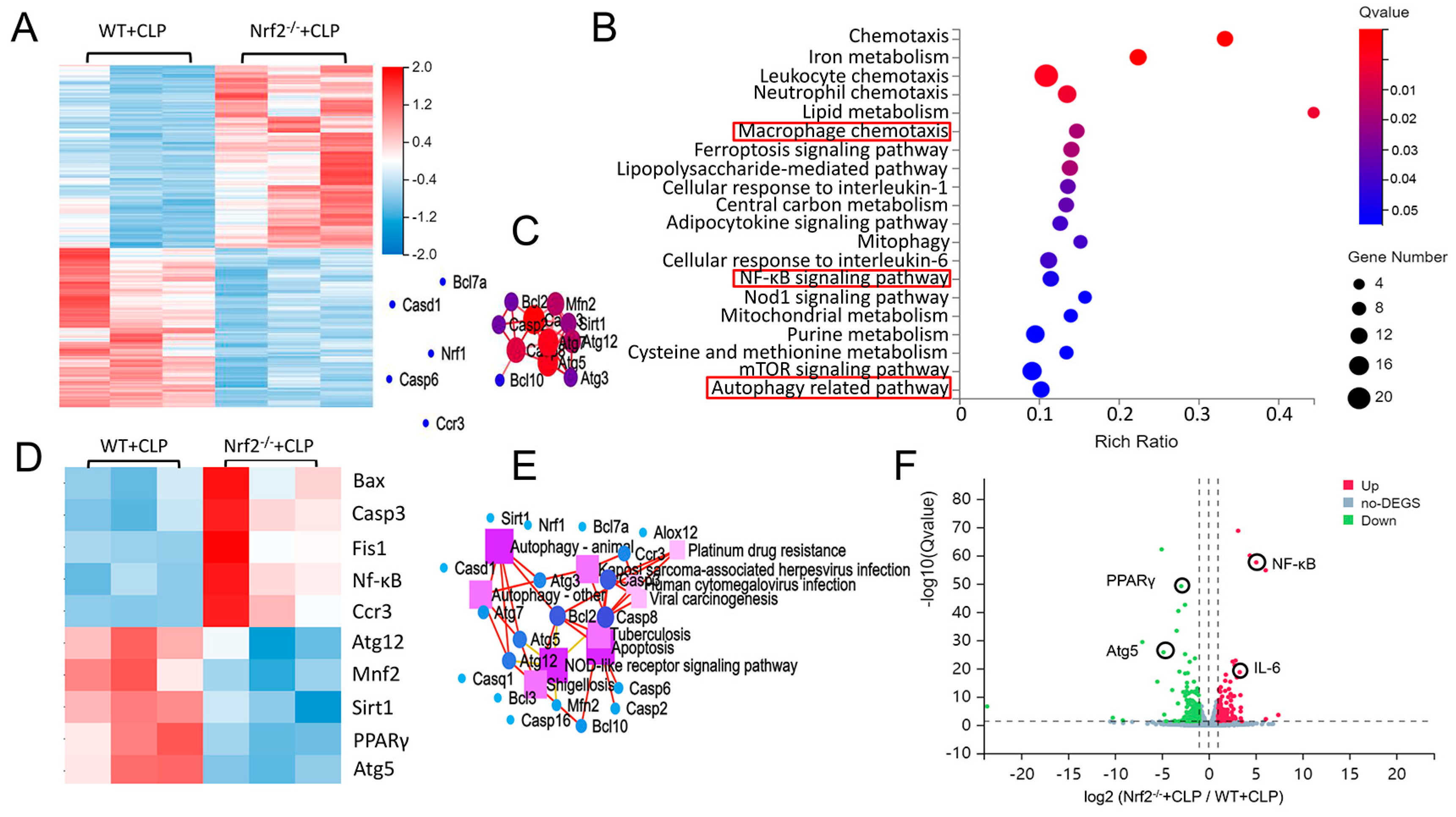
3.3. Transcriptome Sequencing Analyses of Lung Tissues in CLP-Treated WT and Nrf2−/− Mice
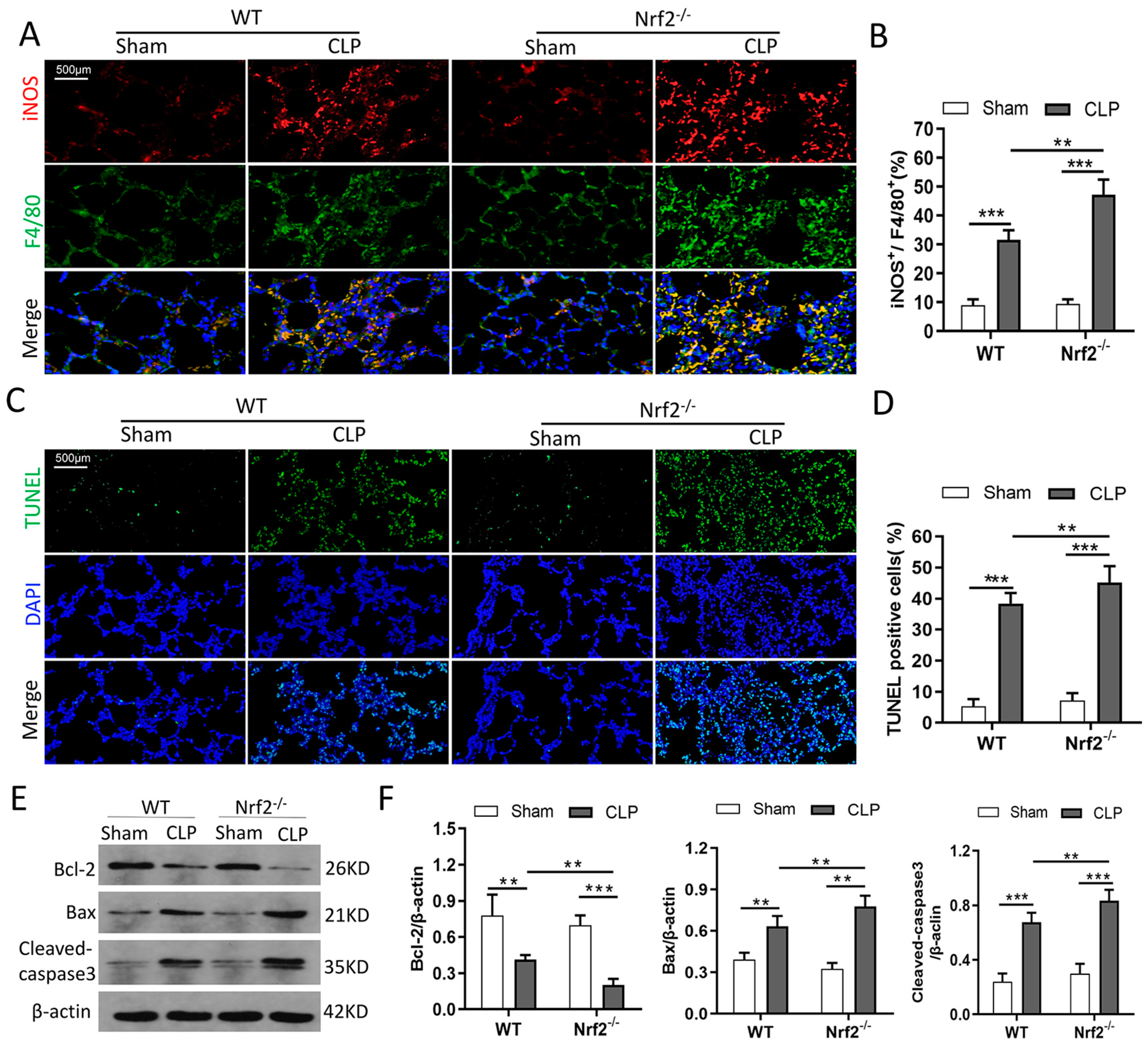
3.4. Nrf2 Deficiency Promotes CLP-Induced Increase in M1 Macrophage Polarization and Apoptosis in Lung Tissues
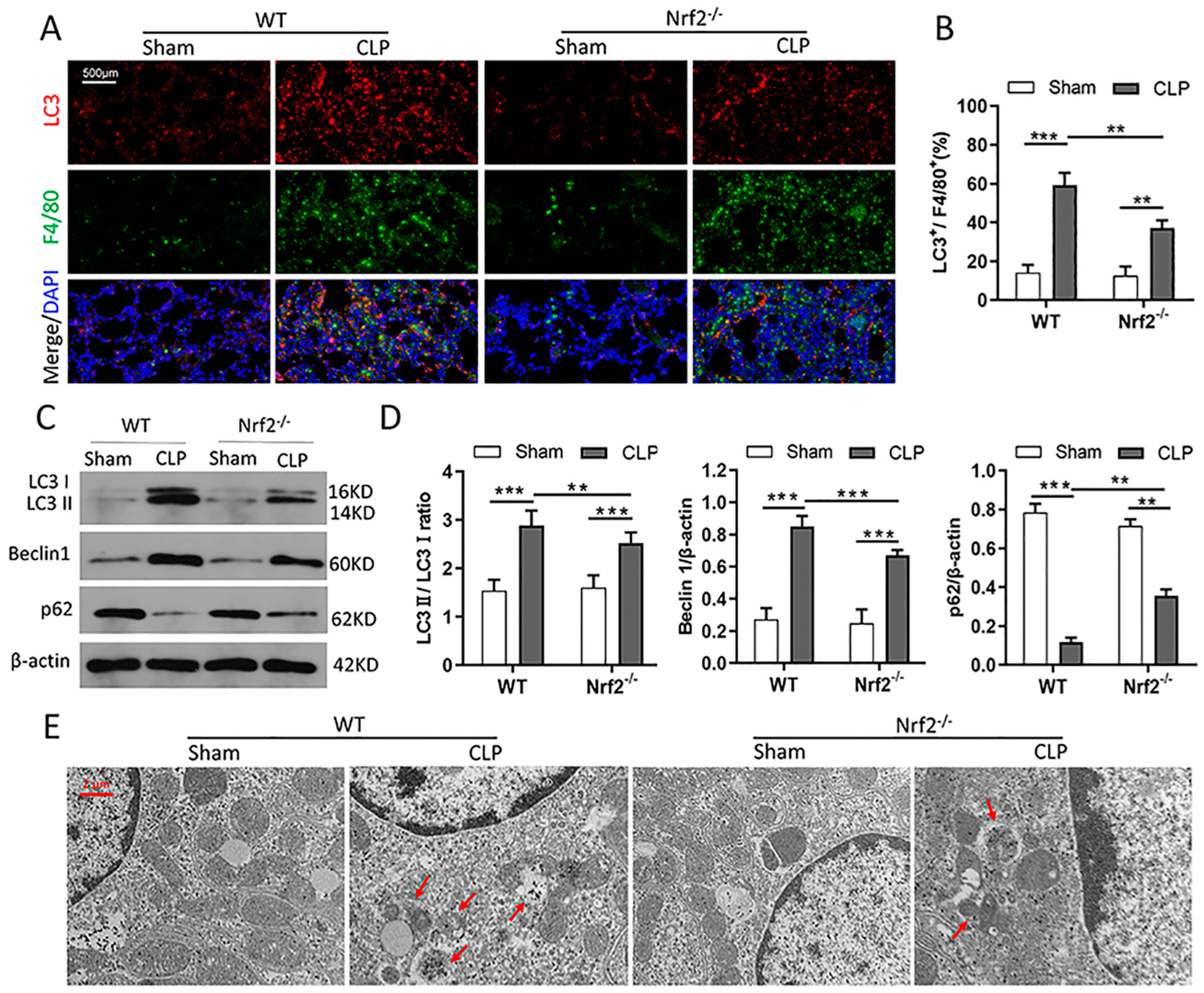
3.5. Nrf2 Deficiency Inhibits CLP-Induced Upregulation of Autophagy Level in Lung Tissues
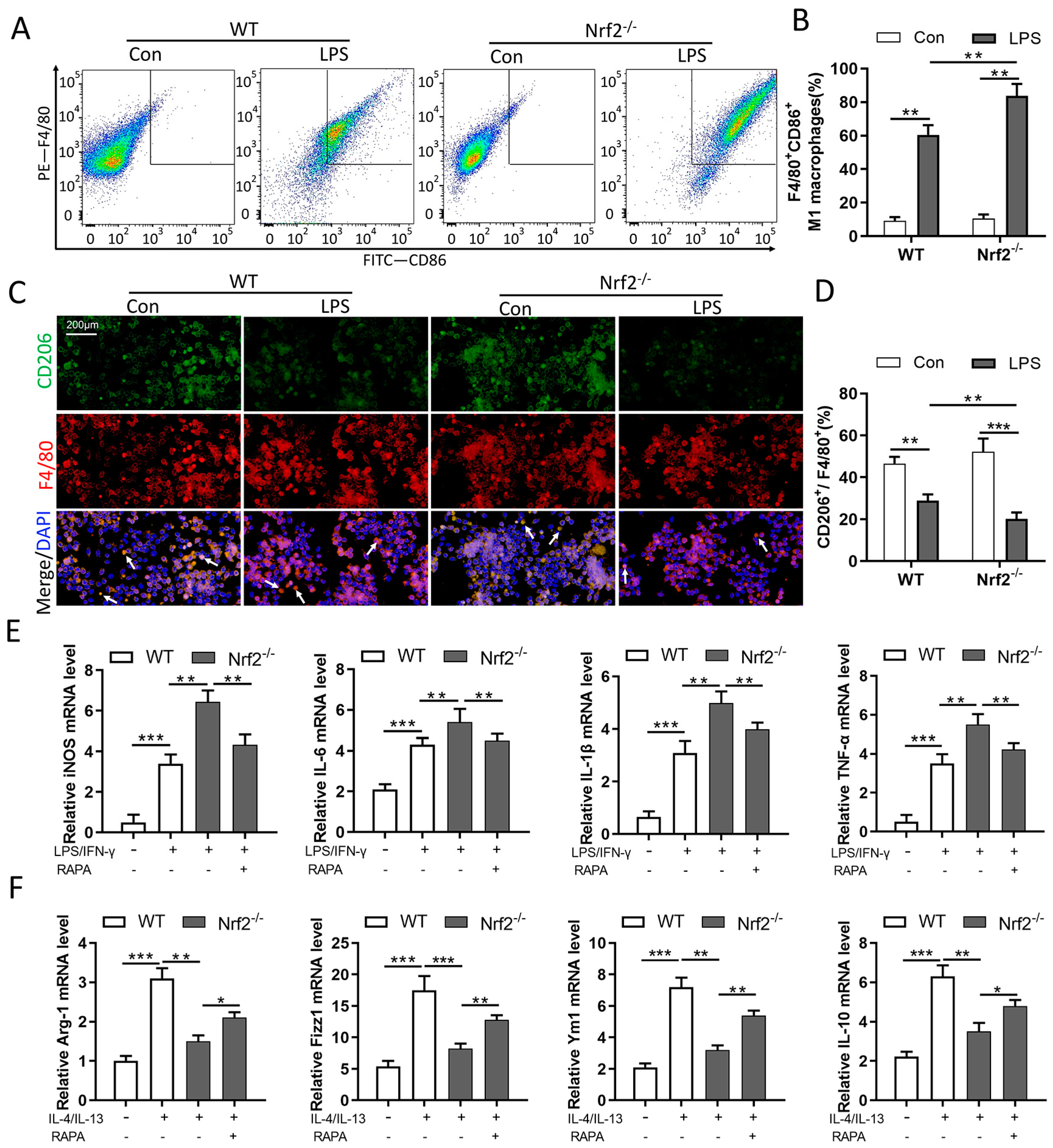
3.6. Nrf2 Deficiency Promotes M1 Macrophage Polarization and Inhibits M2 Macrophage Polarization through Autophagy Modulation
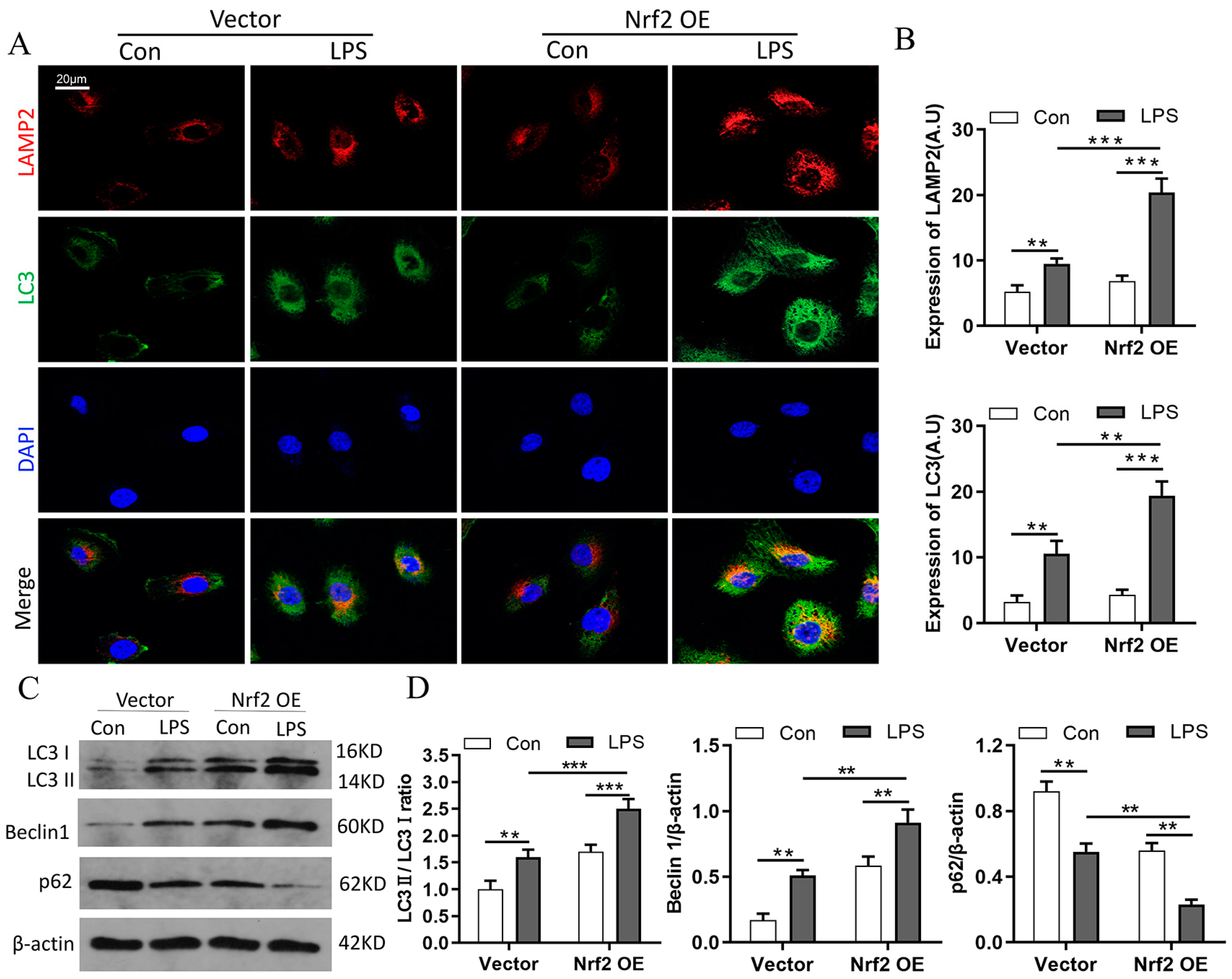
3.7. Nrf2 Overexpression Promotes LPS-Induced Upregulation of Autophagy In Vitro
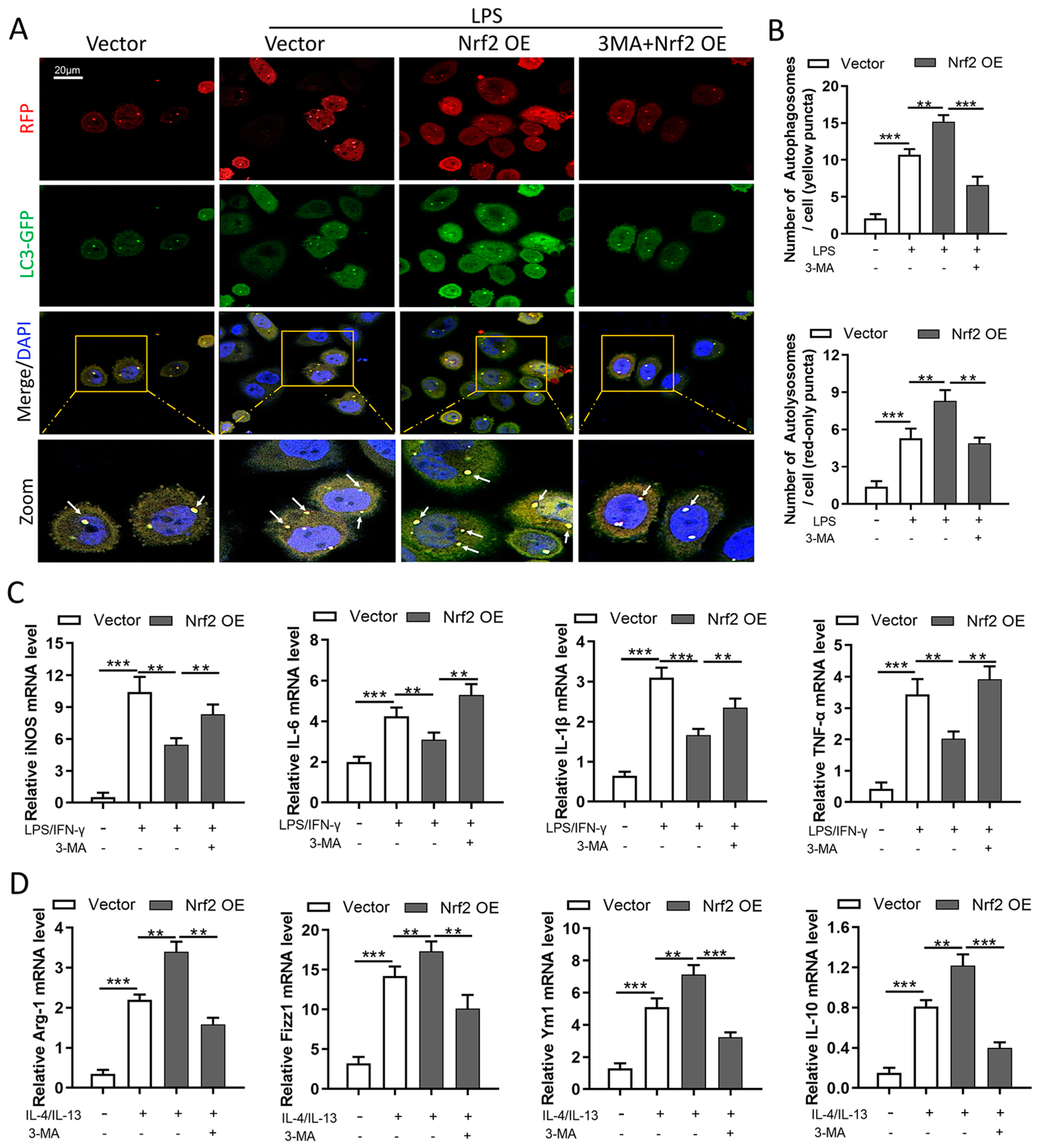
3.8. Nrf2 Overexpression Inhibits M1 Macrophage Polarization and Improves M2 Macrophage Polarization by Promoting Autophagy In Vitro
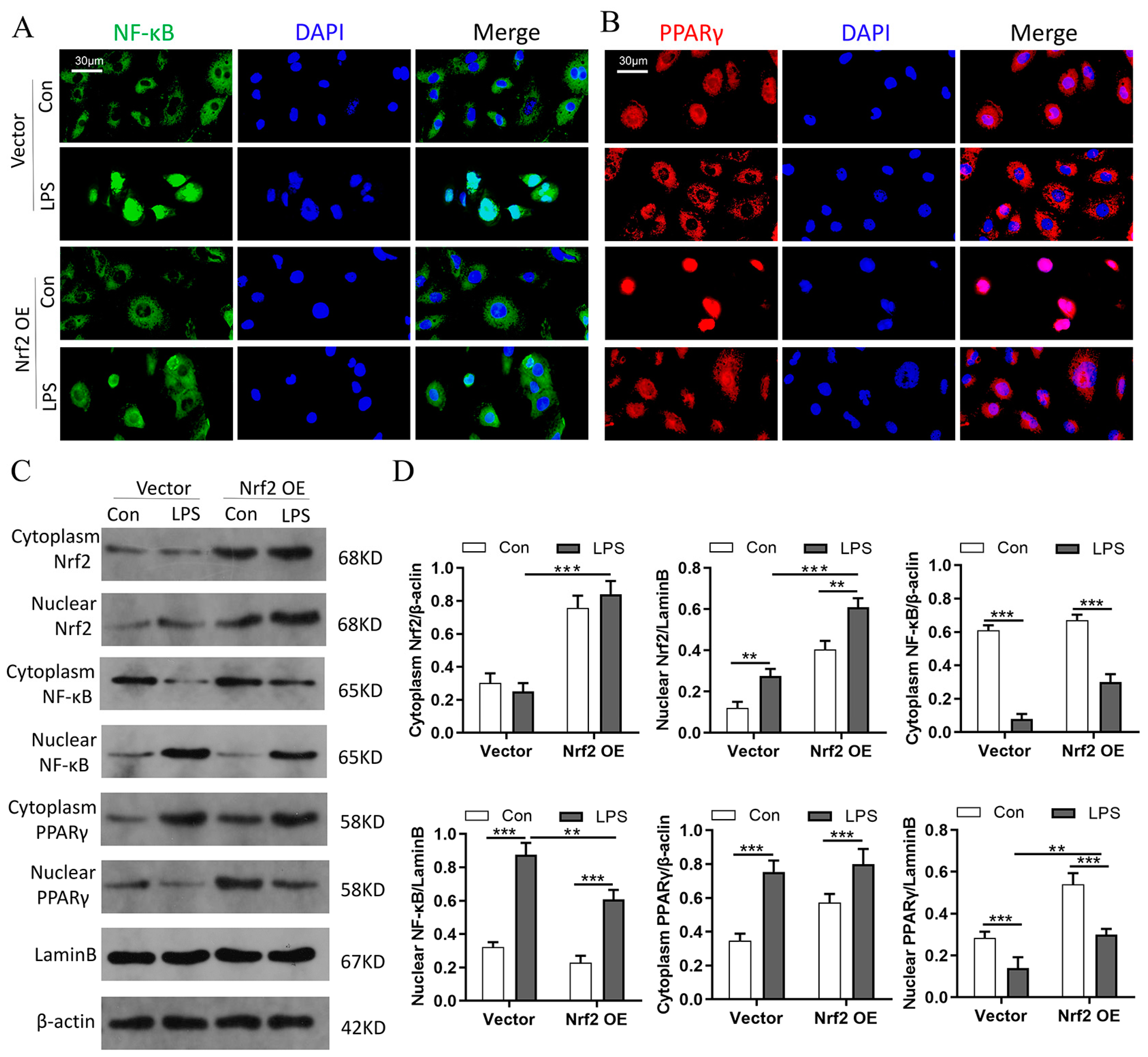
3.9. Nrf2 Overexpression In Vitro Promotes PPARγ but Inhibits NF-κB Nuclear Translocation
4. Discussion
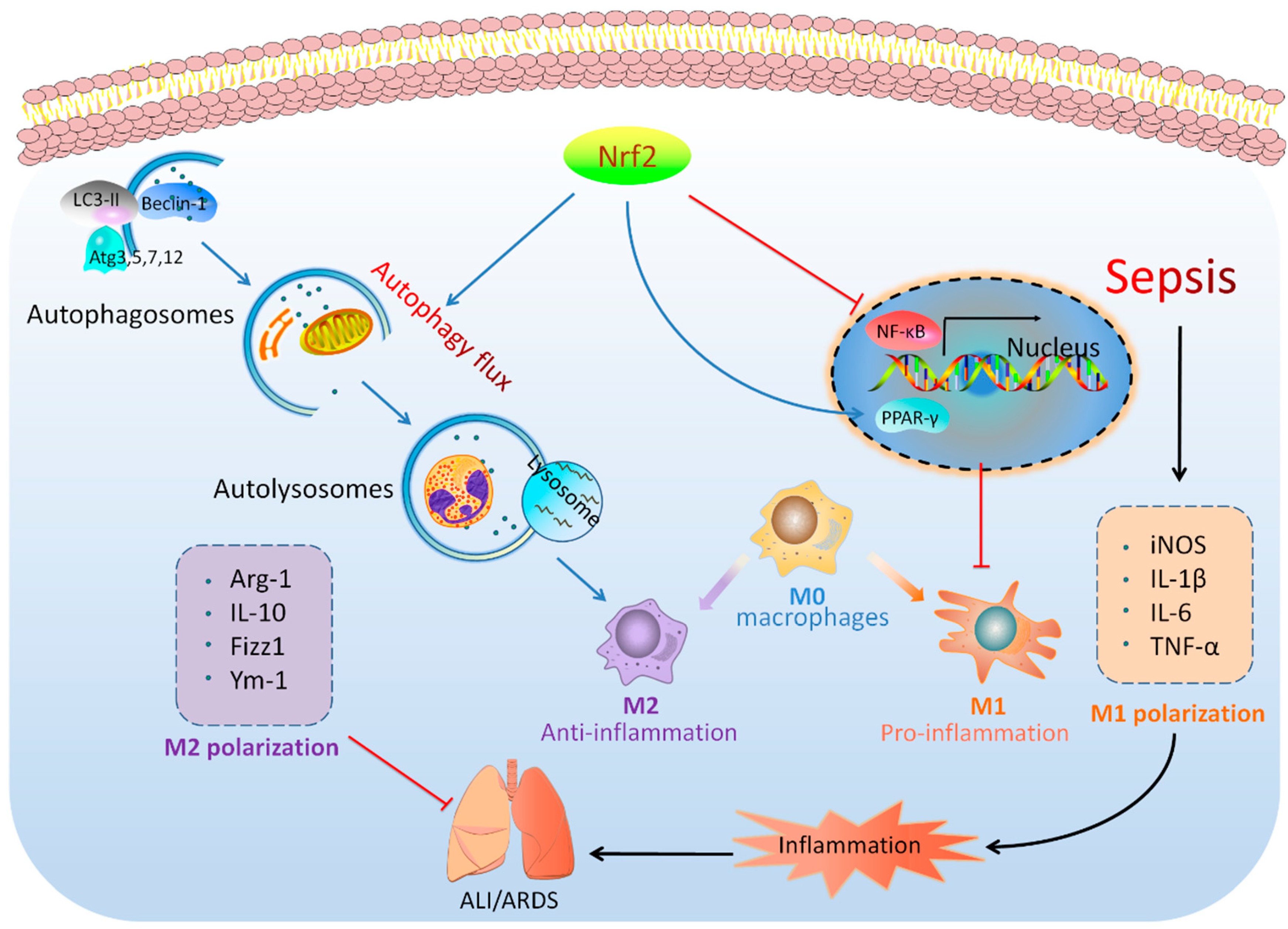
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Shashaty, M.G.S.; Reilly, J.P.; Faust, H.E.; Forker, C.M.; Ittner, C.A.G.; Zhang, P.X.; Hotz, M.J.; Fitzgerald, D.; Yang, W.; Anderson, B.J.; et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: A cohort study. Crit. Care 2019, 23, 235. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-S.; Kim, K.-H.; Park, J.; Kim, S.-M.; Cho, H.; Lee, Y.; Han, I.-O. Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. J. Biol. Chem. 2019, 294, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Hong, Z.; Huang, L.S.; Tsukasaki, Y.; Nepal, S.; Di, A.; Wu, W.; Ye, Z.; Gao, X.; Rao, G.N.; et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J. Clin. Investig. 2020, 130, 3684–3698. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, J.; Luo, Y.; Li, J.; Shang, L.; Zhou, F.; Yang, S. Honokiol alleviates LPS-induced acute lung injury by inhibiting NLRP3 inflammasome-mediated pyroptosis via Nrf2 activation in vitro and in vivo. Chin. Med. 2021, 16, 127. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.C.-F.; Lee, S.-S.; Li, Y.-C.; Ho, Y.-C.; Chen, W.-Y.; Chen, C.-J.; Lee, M.W.; Yeh, K.L.; Tsai, S.C.; Kuan, Y.H. Protective Effects of Kirenol against Lipopolysaccharide-Induced Acute Lung Injury through the Modulation of the Proinflammatory NFκB Pathway and the AMPK2-/Nrf2-Mediated HO-1/AOE Pathway. Antioxidants 2021, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Xu, Y.; Ruan, W.; Wang, H.; Zhang, Y.; Saavedra, J.M.; Zhang, L.; Huang, Z.; Pang, T. A Dual AMPK/Nrf2 Activator Reduces Brain Inflammation After Stroke by Enhancing Microglia M2 Polarization. Antioxid. Redox Signal. 2017, 28, 141–163. [Google Scholar] [CrossRef]
- Han, R.; Xiao, J.; Zhai, H.; Hao, J. Dimethyl fumarate attenuates experimental autoimmune neuritis through the nuclear factor erythroid-derived 2-related factor 2/hemoxygenase-1 pathway by altering the balance of M1/M2 macrophages. J. Neuroinflamm. 2016, 13, 97. [Google Scholar] [CrossRef]
- Wei, J.; Chen, G.; Shi, X.; Zhou, H.; Liu, M.; Chen, Y.; Feng, D.; Zhang, P.; Wu, L.; Lv, X. Nrf2 activation protects against intratracheal LPS induced mouse/murine acute respiratory distress syndrome by regulating macrophage polarization. Biochem. Biophys. Res. Commun. 2018, 500, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-X.; Jiang, F.-J.; Liu, G.; Wang, Y.-Y.; Gao, Z.-Q.; Jin, S.-H.; Nie, Y.-J.; Chen, D.; Chen, J.-L.; Pang, Q.-F. Dehydrocostus Lactone Attenuates Methicillin-Resistant Staphylococcus aureus-Induced Inflammation and Acute Lung Injury via Modulating Macrophage Polarization. Int. J. Mol. Sci. 2021, 22, 9754. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, U213–U217. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Fan, W.; Tang, Z.; Chen, D.; Moughon, D.; Ding, X.; Chen, S.; Zhu, M.; Zhong, Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010, 6, 614–621. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; de la Vega, M.R.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Tang, Z.; Hu, B.; Zang, F.; Wang, J.; Zhang, X.; Chen, H. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell. Death Dis. 2019, 10, 510. [Google Scholar] [CrossRef]
- Lazaro, I.; Lopez-Sanz, L.; Bernal, S.; Oguiza, A.; Recio, C.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Madrigal-Matute, J.; Gomez-Guerrero, C. Nrf2 Activation Provides Atheroprotection in Diabetic Mice through Concerted Upregulation of Antioxidant, Anti-inflammatory, and Autophagy Mechanisms. Front. Pharmacol. 2018, 9, 819. [Google Scholar] [CrossRef]
- Yan, J.; Li, J.; Zhang, L.; Sun, Y.; Jiang, J.; Huang, Y.; Xu, H.; Jiang, H.; Hu, R. Nrf2 protects against acute lung injury and inflammation by modulating TLR4 and Akt signaling. Free Radic. Biol. Med. 2018, 121, 78–85. [Google Scholar] [CrossRef]
- Angus, D.C.; van der Poll, T. Severe Sepsis and Septic Shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Knaus, W.A.; Draper, E.A.; Wagner, D.P.; Zimmerman, J.E. Apache-Ii—A Severity of Disease Classification-System. Crit. Care Med. 1985, 13, 818–829. [Google Scholar] [CrossRef]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef]
- Zhu, R.; Lei, Y.-Q.; Zhao, D.-C. Overexpression of CXCL14 Alleviates Ventilator-Induced Lung Injury through the Downregulation of PKM2-Mediated Cytokine Production. Mediat. Inflamm. 2020, 2020, 7650978. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Xiao, J.; Zhang, H.; Yang, C.; Wei, Z.; Chen, W.; Du, X.; Liu, J. Modulating Neuro-Immune-Induced Macrophage Polarization with Topiramate Attenuates Experimental Abdominal Aortic Aneurysm. Front. Pharmacol. 2020, 11, 565461. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef]
- He, Z.-H.; Zou, S.-Y.; Li, M.; Liao, F.-L.; Wu, X.; Sun, H.-Y.; Zhao, X.-Y.; Hu, Y.-J.; Li, D.; Xu, X.-X.; et al. The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox. Biol. 2020, 28, 101364. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef]
- Chan, K.; Kan, Y.W. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12731–12736. [Google Scholar] [CrossRef] [PubMed]
- Papaiahgari, S.; Kleeberger, S.R.; Cho, H.-Y.; Kalvakolanu, D.V.; Reddy, S.P. NADPH Oxidase and ERK Signaling Regulates Hyperoxia-induced Nrf2-ARE Transcriptional Response in Pulmonary Epithelial Cells. J. Biol. Chem. 2004, 279, 42302–42312. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Dodson, M.; Gross, C.; Mansour, H.M.; Lantz, R.C.; Chapman, E.; Wang, T.; Black, S.M.; Garcia, J.G.N.; Zhang, D.D. Role of Nrf2 and Autophagy in Acute Lung Injury. Curr. Pharmacol. Rep. 2016, 2, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Audousset, C.; McGovern, T.; Martin, J.G. Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches—Pulmonary Disease/Asthma. Front. Physiol. 2021, 12, 727806. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Gong, Q.; Xue, Y.; Li, X.; Song, L.; Zhu, L. DL-3-n-butylphthalide attenuates lipopolysaccharide-induced acute lung injury via SIRT1-dependent and -independent regulation of Nrf2. Int. Immunopharmacol. 2019, 74, 105658. [Google Scholar] [CrossRef]
- Ye, J.; Guan, M.; Lu, Y.; Zhang, D.; Li, C.; Zhou, C. Arbutin attenuates LPS-induced lung injury via Sirt1/Nrf2/NF-κBp65 pathway. Pulm. Pharmacol. Ther. 2019, 54, 53–59. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R.M.; El-Agamy, D.S.; Elsaed, W.M.; Sirwi, A.; Asfour, H.Z.; Koshak, A.E.; Elhady, S.S. Terretonin as a New Protective Agent against Sepsis-Induced Acute Lung Injury: Impact on SIRT1/Nrf2/NF-κBp65/NLRP3 Signaling. Biology 2021, 10, 1219. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef]
- Kanayama, M.; He, Y.-W.; Shinohara, M.L. The Lung Is Protected from Spontaneous Inflammation by Autophagy in Myeloid Cells. J. Immunol. 2015, 194, 5465. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Maazi, H.; Sankaranarayanan, I.; Lam, J.; Khoo, B.; Soroosh, P.; Barbers, R.G.; Ou, J.-H.J.; Jung, J.U.; Akbari, O. Lack of autophagy induces steroid-resistant airway inflammation. J. Allergy Clin. Immunol. 2016, 137, 1382–1389.e1389. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. 2017, 198, 3205–3213. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; Yuan, S.-S.F.; Hsu, C.; Cheng, Y.-J.; Chang, Y.-F.; Hsueh, H.-W.; Lee, P.-H.; Hsieh, Y.-C. Lc3 Over-Expression Improves Survival and Attenuates Lung Injury Through Increasing Autophagosomal Clearance in Septic Mice. Ann. Surg. 2013, 257, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of Cigarette Smoke–Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease–Emphysema Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2014, 53, 159–173. [Google Scholar] [CrossRef]
- Chen, C.-Z.; Ou, C.-Y.; Wang, R.-H.; Lee, C.-H.; Lin, C.-C.; Chang, H.-Y.; Hsiue, T.-R. Association of Egr-1 and autophagy-related gene polymorphism in men with chronic obstructive pulmonary disease. J. Formos. Med. Assoc. 2015, 114, 750–755. [Google Scholar] [CrossRef]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, H.; Strulovici-Barel, Y.; Al-Hijji, M.; Ou, X.; Salit, J.; Walters, M.S.; Staudt, M.; Kaner, R.J.; Crystal, R.G. Role of OSGIN1 in mediating smoking-induced autophagy in the human airway epithelium. Autophagy 2017, 13, 1205–1220. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an Autophagy Defect in Smokers’ Alveolar Macrophages. J. Immunol. 2010, 185, 5425. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Lu, J.-H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lu, S.; Gao, Y.; Yang, K.; Wu, D.; Xu, X.; Sun, G.; Sun, X. Araloside C attenuates atherosclerosis by modulating macrophage polarization via Sirt1-mediated autophagy. Aging 2020, 12, 1704–1724. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.-N.; Chen, Z.-Y.; Yang, X.-B.; Chen, L.; Yang, Y.-Y.; Pan, S.-W.; Wang, Y.-X.; Xu, J.-Q.; Zhou, T.; Xiao, H.-R.; et al. Trichostatin A modulates the macrophage phenotype by enhancing autophagy to reduce inflammation during polymicrobial sepsis. Int. Immunopharmacol. 2019, 77, 105973. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; E Tanaka, K.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Weng, D.; Zhou, F.; Owen, Y.D.; Qin, H.; Zhao, J.; Yu, W.; Huang, Y.; Chen, J.; Fu, H.; et al. Activation of PPARγ by a Natural Flavonoid Modulator, Apigenin Ameliorates Obesity-Related Inflammation via Regulation of Macrophage Polarization. EBioMedicine 2016, 9, 61–76. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Escoté, X.; Ortega, F.; Serino, M.; Campbell, M.; Michalski, M.-C.; Laville, M.; Xifra, G.; Luche, E.; Domingo, P.; et al. A role for adipocyte-derived lipopolysaccharide-binding protein in inflammation- and obesity-associated adipose tissue dysfunction. Diabetologia 2013, 56, 2524–2537. [Google Scholar] [CrossRef]
- Wang, X.; Ribeiro, M.D.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-Specific Hypoxia-Inducible Factor-1α Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 545–563. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Jia, L.; Pan, L.; Li, H.; Du, J. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II. Free Radic. Biol. Med. 2014, 69, 108–115. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Fan, L.; Li, Y.; Zou, Z.; Scott, M.J.; Xiao, G.; Li, S.; Billiar, T.R.; Wilson, M.A.; Shi, X.; et al. Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: Role in posthemorrhagic shock acute lung inflammation. J. Immunol. 2014, 193, 4623–4633. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Healthy Controls (n = 30) | Sepsis Patients (Acute Stage, n = 30) | p Value |
|---|---|---|---|
| Age (years; mean ± SD) | 56.23 ± 11.23 | 54.58 ± 13.87 | 0.615 a |
| Male gender (n, %) | 21 (70.00) | 23 (76.67) | 0.559 b |
| Comorbidities (n, %) | |||
| Hypertension | 7 (23.33) | 10 (33.33) | 0.390 b |
| Coronary heart diseases | 6 (20.00) | 3 (10.00) | 0.608 b |
| Diabetes | 2 (0.07) | 5 (0.17) | 0.421 b |
| Complete blood count (mean ± SD or median (interquartile range)) | |||
| WBC (×109/L) | 8.31 ± 4.61 | 11.78 ± 7.49 | 0.034 a |
| LYM (×109/L) | 0.51 (0.32–0.91) | 0.42 (0.44–0.79) | 0.331 c |
| NEU (×109/L) | 5.39 (3.81–6.73) | 13.51 (7.11–18.78) | 0.017 c |
| MONO (×109/L) | 0.39 ± 0.33 | 0.50 ± 0.38 | 0.236 a |
| HGB (g/L) | 112.32 ± 21.87 | 105.57 ± 26.58 | 0.287 a |
| PLT (×109/L) | 150.73 ± 83.45 | 145.28 ± 90.92 | 0.809 a |
| Blood gas analysis (mean ± SD) | |||
| SaO2 | 0.98 ± 0.02 | 0.87 ± 0.23 | 0.012 a |
| PCO2 (mmHg) | 35.65 ± 8.45 | 58.53 ± 15.37 | <0.001 a |
| PO2 (mmHg) | 146.91 ± 56.34 | 72.35 ± 35.34 | <0.001 a |
| PH | 7.38 ± 0.12 | 7.24 ± 0.23 | 0.004 a |
| BE (mmol/L) | −1.29 ± 1.88 | −5.47 ± 6.70 | 0.003 a |
| HCO3− (mmol/L) | 18.26 ± 4.33 | 17.57 ± 8.13 | 0.683 a |
| Lac (mmol/L) | 1.81 ± 0.85 | 2.77 ± 1.92 | 0.017 a |
| Inflammatory profile (median (interquartile range)) | |||
| PCT (ng/mL) | 0.72 (0.21–2.83) | 3.58 (2.45–8.39) | 0.007 c |
| CRP (mg/L) | 82.65 (23.45–130.39) | 145.77 (56.21–312.40) | 0.008 c |
| Biochemical test (mean ± SD or median (interquartile range)) | |||
| TP (g/L) | 65.38 ± 8.71 | 62.67 ± 6.34 | 0.174 a |
| GLB (g/L) | 31.45 ± 7.32 | 30.22 ± 8.19 | 0.542 a |
| ALB (g/L) | 35.73 ± 5.75 | 33.30 ± 6.36 | 0.126 a |
| ALT (U/L) | 38.41 ± 12.15 | 40.33 ± 9.16 | 0.492 a |
| AST (U/L) | 55.97 ± 6.31 | 53.53 ± 7.52 | 0.807 a |
| BUN (mmol/L) | 9.32 ± 3.75 | 10.54 ± 2.33 | 0.136 a |
| Cr (μmol/L) | 94.22 ± 15.76 | 101.75 ± 22.97 | 0.144 a |
| UA (μmol/L) | 306.19 ± 98.23 | 345.74 ± 112.13 | 0.152 a |
| LDH (U/L) | 152.42 ± 34.16 | 157.83 ± 22.74 | 0.473 a |
| BNP (pg/mL) | 126.21 ± 32.67 | 143.37 ± 26.75 | 0.030 a |
| CK-MB (U/L) | 30.51 (11.01–23.26) | 31.87 (15.71–44.61) | 0.271 c |
| cTnI (pg/mL) | 0.01 (0.00–0.13) | 0.01 (0.00–0.21) | 0.890 c |
| K+ (mmol/L) | 4.31 ± 0.75 | 4.27 ± 0.81 | 0.843 a |
| Na+ (mmol/L) | 137.23 ± 15.21 | 134.56 ± 14.23 | 0.485 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Wang, J.; Zhang, J.; Sang, A.; Ye, X.; Cheng, Z.; Li, X. Nrf2 Deficiency Exacerbated CLP-Induced Pulmonary Injury and Inflammation through Autophagy- and NF-κB/PPARγ-Mediated Macrophage Polarization. Cells 2022, 11, 3927. https://doi.org/10.3390/cells11233927
Luo J, Wang J, Zhang J, Sang A, Ye X, Cheng Z, Li X. Nrf2 Deficiency Exacerbated CLP-Induced Pulmonary Injury and Inflammation through Autophagy- and NF-κB/PPARγ-Mediated Macrophage Polarization. Cells. 2022; 11(23):3927. https://doi.org/10.3390/cells11233927
Chicago/Turabian StyleLuo, Jing, Jin Wang, Jing Zhang, Aming Sang, Xujun Ye, Zhenshun Cheng, and Xinyi Li. 2022. "Nrf2 Deficiency Exacerbated CLP-Induced Pulmonary Injury and Inflammation through Autophagy- and NF-κB/PPARγ-Mediated Macrophage Polarization" Cells 11, no. 23: 3927. https://doi.org/10.3390/cells11233927
APA StyleLuo, J., Wang, J., Zhang, J., Sang, A., Ye, X., Cheng, Z., & Li, X. (2022). Nrf2 Deficiency Exacerbated CLP-Induced Pulmonary Injury and Inflammation through Autophagy- and NF-κB/PPARγ-Mediated Macrophage Polarization. Cells, 11(23), 3927. https://doi.org/10.3390/cells11233927