


Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
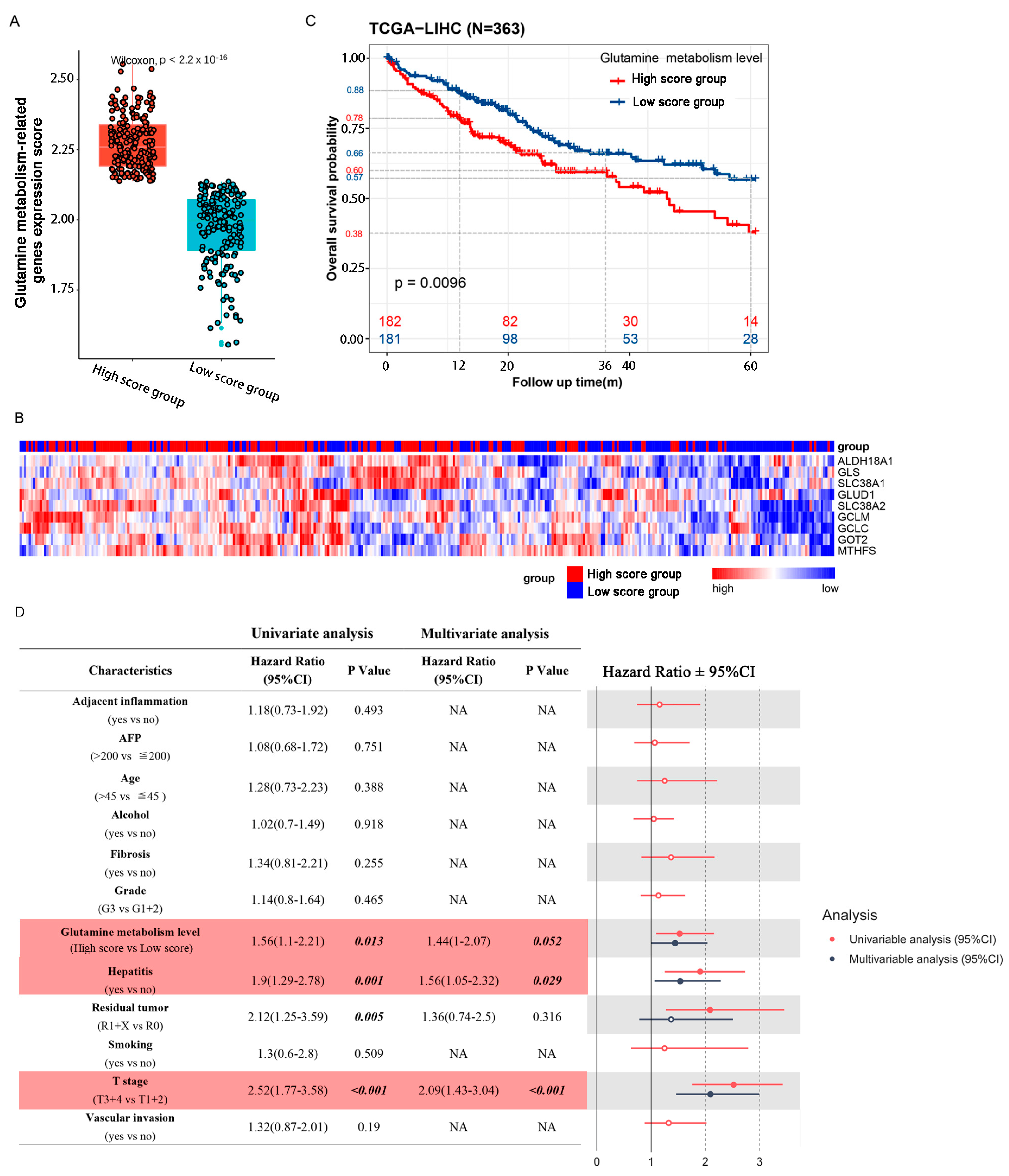
2.1. Patients with High Glutamine Metabolism-Related Gene Expression Score Suffer Worse Prognoses in TCGA Cohort
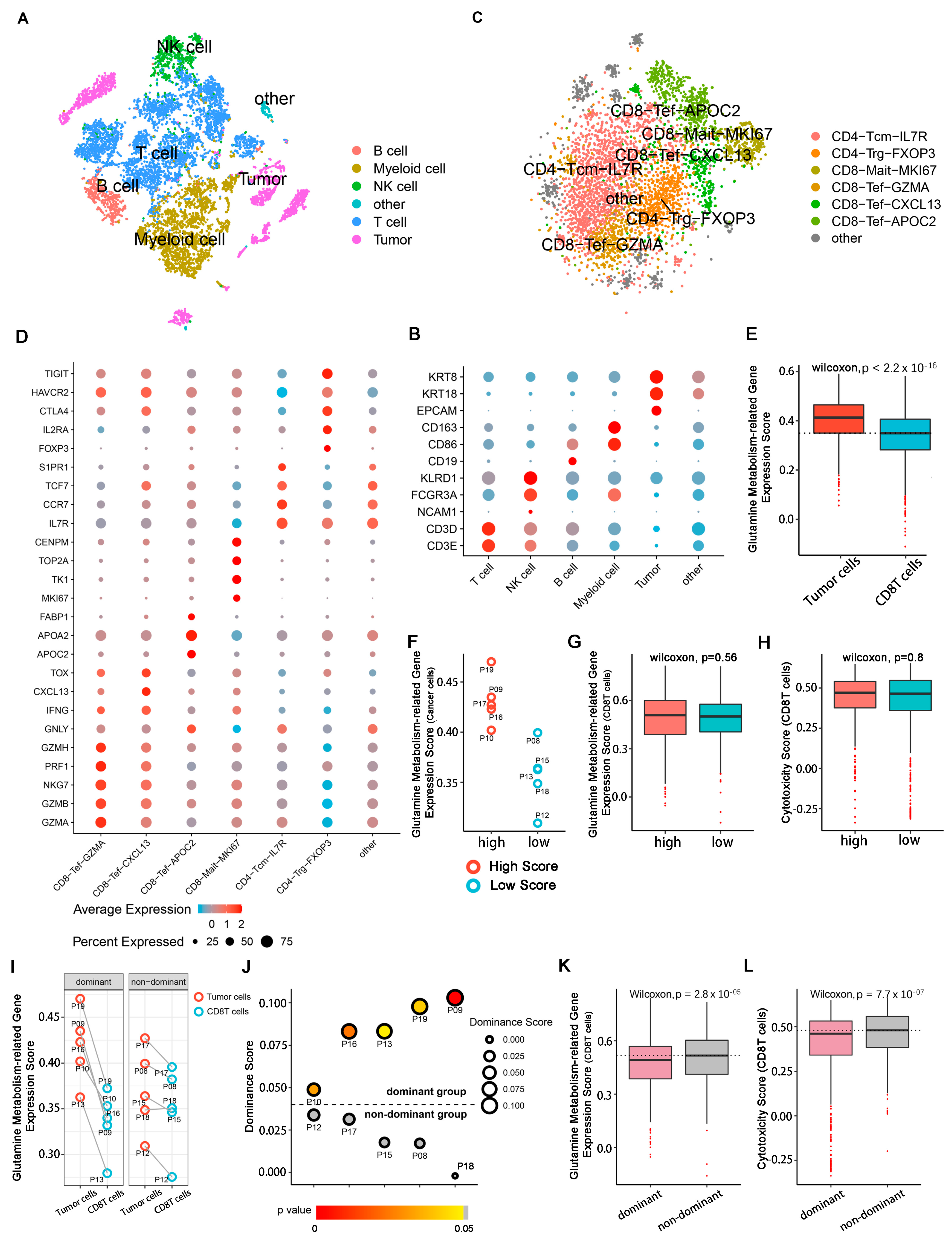
2.2. Patient Grouping and Subpopulation Annotation of Single-Cell Sequencing Data
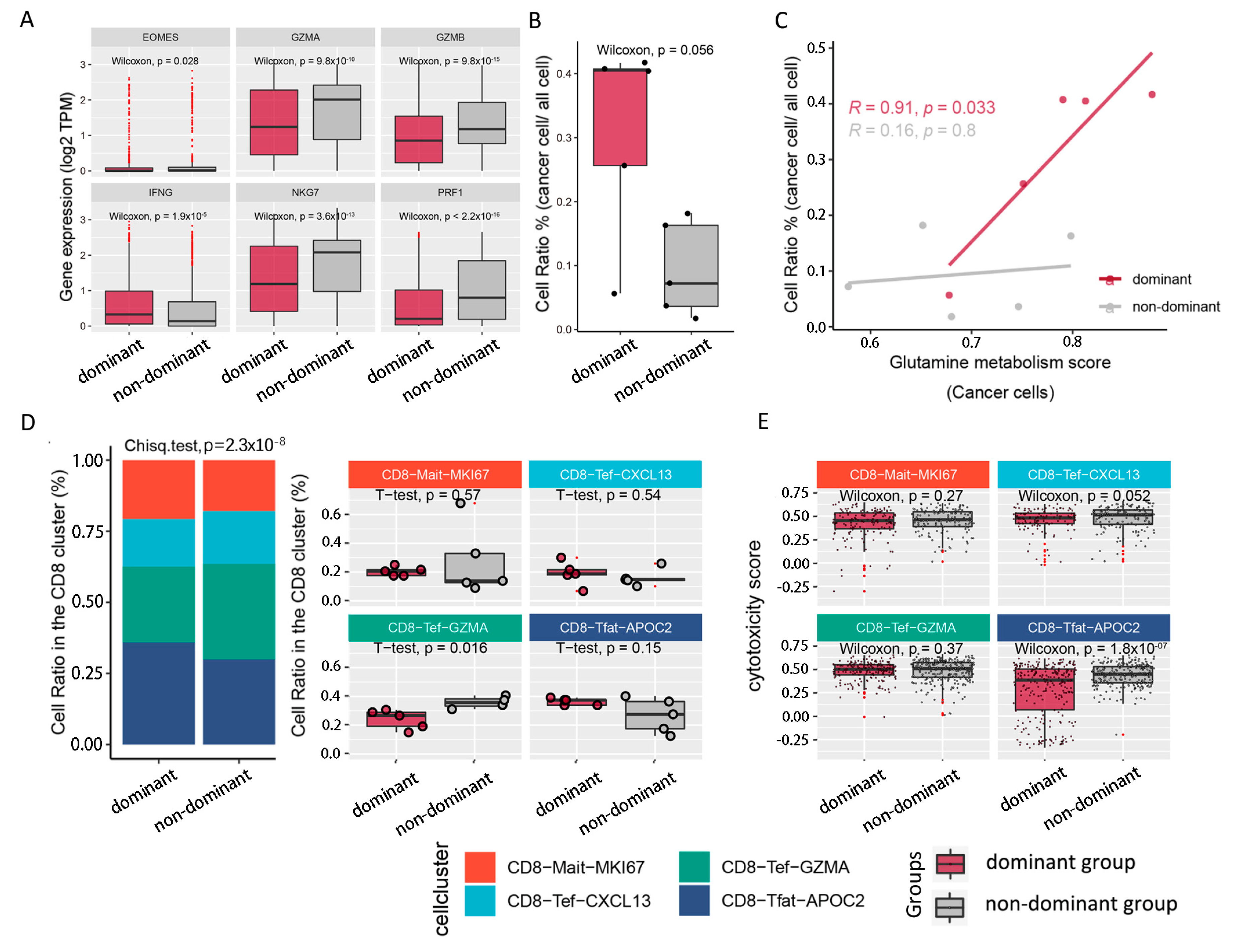
2.3. Tumoricidal Activity of CD8T Cells Is Suppressed in the Dominant Group
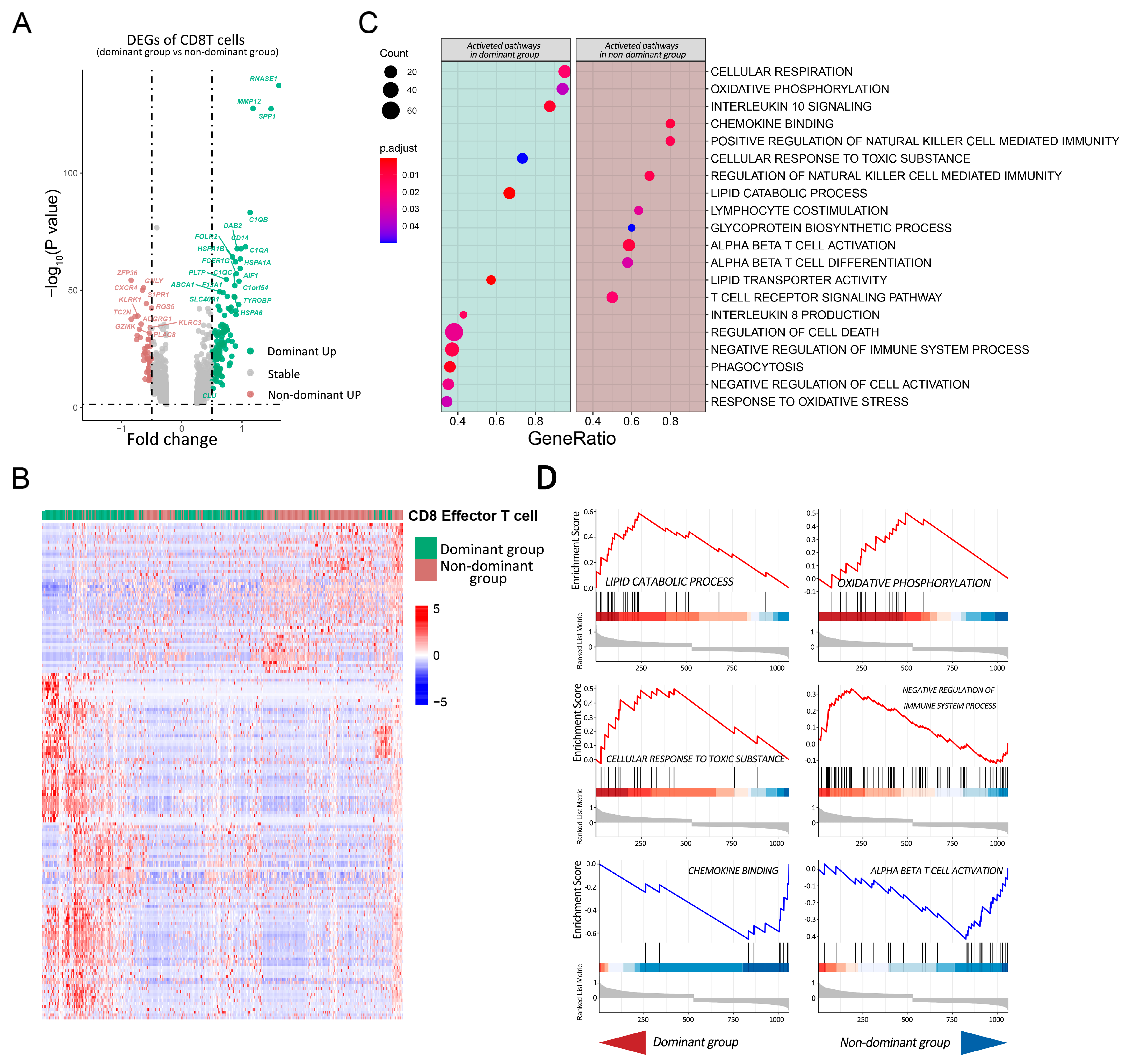
2.4. Pathway Analysis of CD8T Cells in Dominant Group vs. Nondominant Group
2.5. CD8 Effector T Cells Have Different Metabolic Programs and Various Immune Statuses under the Influence of Glutamine Metabolism in Cancer Cells
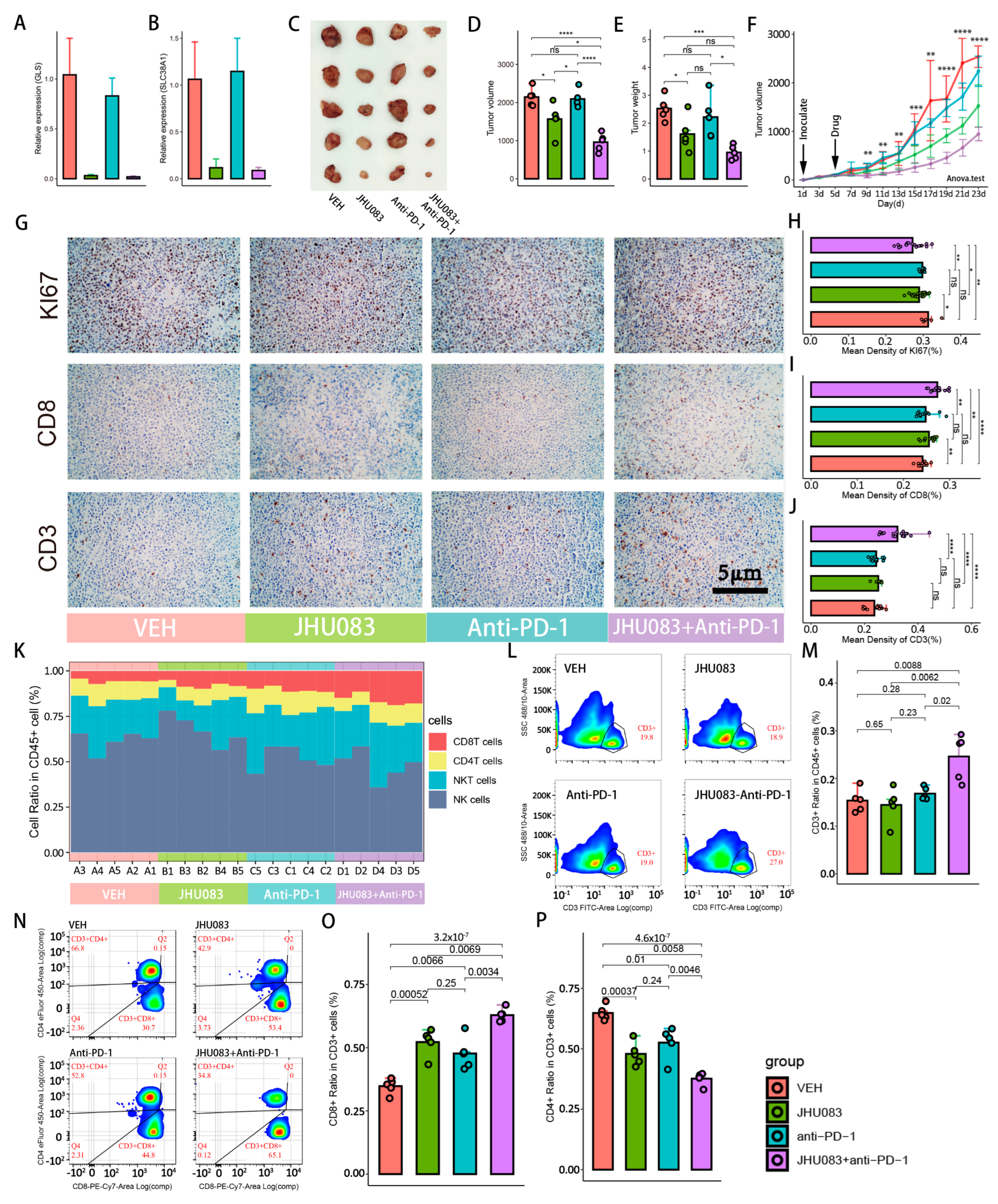
2.6. Glutamine Metabolism Inhibitor (JHU083) Enhances the Efficacy of Immune Checkpoint Inhibitors (PD-1 Inhibitor)
3. Discussion
4. Materials and Methods
4.1. Data Resources for scRNA-Seq and Bluk RNA-Seq
4.2. Data Processing and Analysis of TCGA Cohort
4.3. Data Processing and Subgroup Annotation of Single-Cell RNA-Seq
4.4. Calculating Pathway Enrichment Scores and Patient Grouping
4.5. Immunophenotype Remodeling and Metabolism Reprogramming of Tumor-Infiltrated CD8 T Cells in Dominant and Nondominant Groups
4.6. Gene Set Enrichment Analysis of CD8T Cells
4.7. Animal Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Metabolic control of arthritis: Switch pathways to treat. Sci. Transl. Med. 2016, 8, 331fs338. [Google Scholar] [CrossRef] [PubMed]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Rodas, P.C.; Rooyackers, O.; Hebert, C.; Norberg, A.; Wernerman, J. Glutamine and glutathione at ICU admission in relation to outcome. Clin. Sci. 2012, 122, 591–597. [Google Scholar] [CrossRef]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Investig. 2021, 131, 4. [Google Scholar] [CrossRef]
- Wei, Y.; Tang, X.; Ren, Y.; Yang, Y.; Song, F.; Fu, J.; Liu, S.; Yu, M.; Chen, J.; Wang, S.; et al. An RNA-RNA crosstalk network involving HMGB1 and RICTOR facilitates hepatocellular carcinoma tumorigenesis by promoting glutamine metabolism and impedes immunotherapy by PD-L1+ exosomes activity. Signal Transduct. Target. Ther. 2021, 6, 421. [Google Scholar] [CrossRef]
- Cheng, H.; Wu, Z.; Wu, C.; Wang, X.; Liow, S.S.; Li, Z.; Wu, Y.L. Overcoming STC2 mediated drug resistance through drug and gene co-delivery by PHB-PDMAEMA cationic polyester in liver cancer cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 83, 210–217. [Google Scholar] [CrossRef]
- Cheng, H.; Yang, X.; Liu, G. Superstable homogeneous iodinated formulation technology: Revolutionizing transcatheter arterial chemoembolization. Sci. Bull. 2020, 65, 1685–1687. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, J.; Li, H.Y.; Wang, Z.H.; Wu, J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim Biophys. Acta Rev. Cancer 2020, 1874, 188441. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; Garcia-Canaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting arginine-dependent cancers with arginine-degrading enzymes: Opportunities and challenges. Cancer Res. Treat. 2013, 45, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842 e813. [Google Scholar] [CrossRef]
- Corrado, M.; Edwards-Hicks, J.; Villa, M.; Flachsmann, L.J.; Sanin, D.E.; Jacobs, M.; Baixauli, F.; Stanczak, M.; Anderson, E.; Azuma, M.; et al. Dynamic Cardiolipin Synthesis Is Required for CD8(+) T Cell Immunity. Cell Metab. 2020, 32, 981–995 e987. [Google Scholar] [CrossRef]
- Brown, R.E.; Steele, R.W.; Marmer, D.J.; Hudson, J.L.; Brewster, M.A. Fatty acids and the inhibition of mitogen-induced lymphocyte transformation by leukemic serum. J. Immunol. 1983, 131, 1011–1016. [Google Scholar]
- Kleinfeld, A.M.; Okada, C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J. Lipid Res. 2005, 46, 1983–1990. [Google Scholar] [CrossRef]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1alpha Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e4. [Google Scholar] [CrossRef]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef]
- Yuan, Y.; Adam, A.; Zhao, C.; Chen, H. Recent Advancements in the Mechanisms Underlying Resistance to PD-1/PD-L1 Blockade Immunotherapy. Cancers 2021, 13, 663. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Chen, M.; Zhang, R.; Zhang, W.; Wang, C.; Zhang, Y.; Wei, X.; Guan, Y.; Liu, J.; Feng, K.; et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020, 30, 966–979. [Google Scholar] [CrossRef] [PubMed]
- O′Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Chisolm, D.A.; Savic, D.; Moore, A.J.; Ballesteros-Tato, A.; Leon, B.; Crossman, D.K.; Murre, C.; Myers, R.M.; Weinmann, A.S. CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 2017, 47, 251–267.e7. [Google Scholar] [CrossRef]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Velica, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, Y.; Sun, F.; Hou, L.; Zhang, C.; Ge, T.; Yu, H.; Wu, C.; Zhu, Y.; Duan, L.; et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020, 21, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Holgersen, E.M.; Gandhi, S.; Zhou, Y.; Kim, J.; Vaz, B.; Bogojeski, J.; Bugno, M.; Shalev, Z.; Cheung-Ong, K.; Gonçalves, J.; et al. Transcriptome-Wide Off-Target Effects of Steric-Blocking Oligonucleotides. Nucleic Acid Ther. 2021, 31, 392–403. [Google Scholar] [CrossRef]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Wang, R.; Liu, Z.; Fan, J.; Liu, S.; Tan, S.; Li, X.; Li, B.; Yang, X. Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma. Cells 2022, 11, 3924. https://doi.org/10.3390/cells11233924
Chen J, Wang R, Liu Z, Fan J, Liu S, Tan S, Li X, Li B, Yang X. Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma. Cells. 2022; 11(23):3924. https://doi.org/10.3390/cells11233924
Chicago/Turabian StyleChen, Jianfei, Rui Wang, Zhongliang Liu, Jun Fan, Shenglu Liu, Shunde Tan, Xinkai Li, Bo Li, and Xiaoli Yang. 2022. "Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma" Cells 11, no. 23: 3924. https://doi.org/10.3390/cells11233924
APA StyleChen, J., Wang, R., Liu, Z., Fan, J., Liu, S., Tan, S., Li, X., Li, B., & Yang, X. (2022). Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma. Cells, 11(23), 3924. https://doi.org/10.3390/cells11233924