


Exploring the Characteristics of Monkeypox-Related Genes in Pan-Cancer


Abstract
1. Introduction
2. Materials and Methods
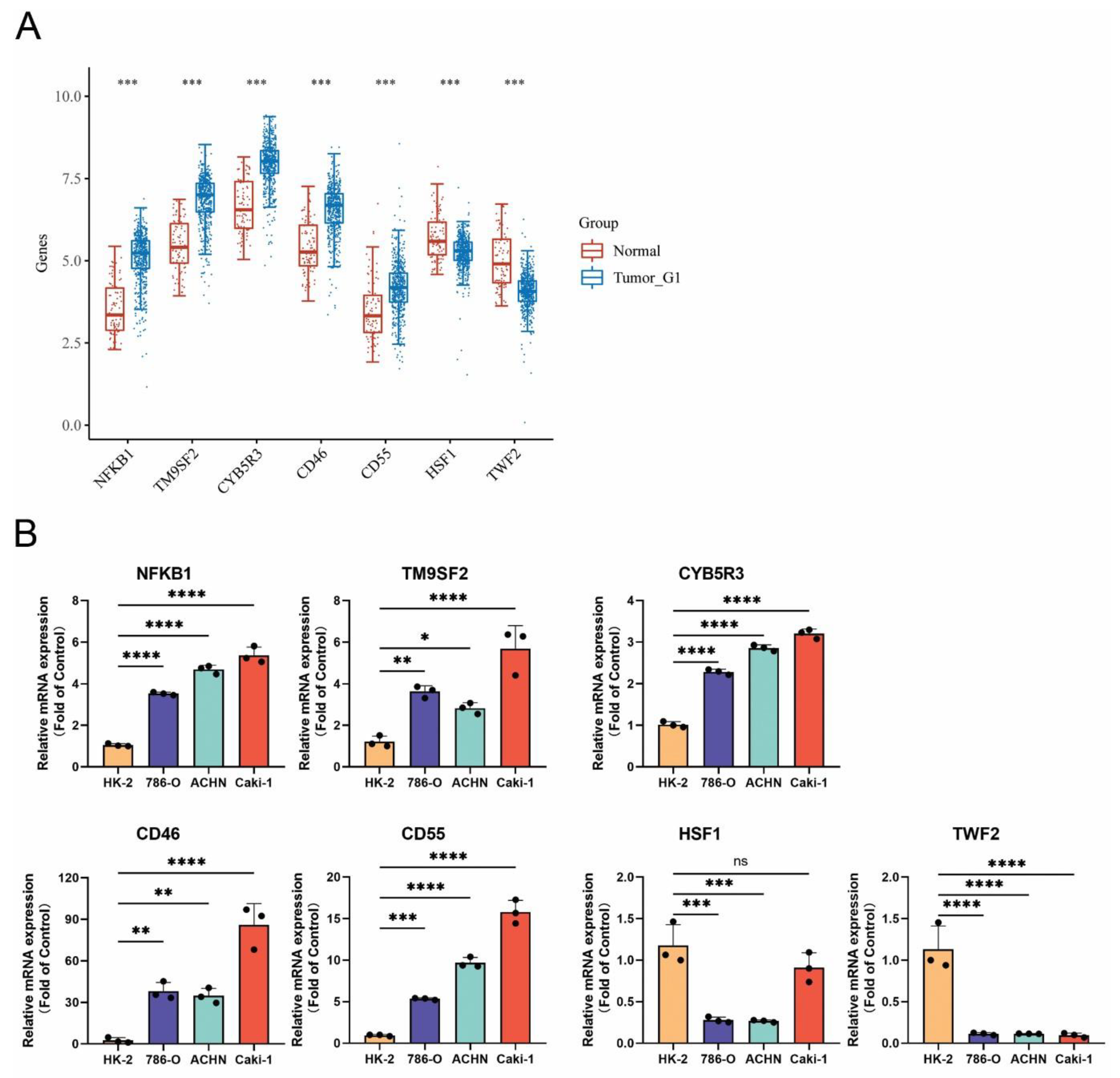
2.1. Cell Lines and qRT-PCR
2.2. Tumor Types
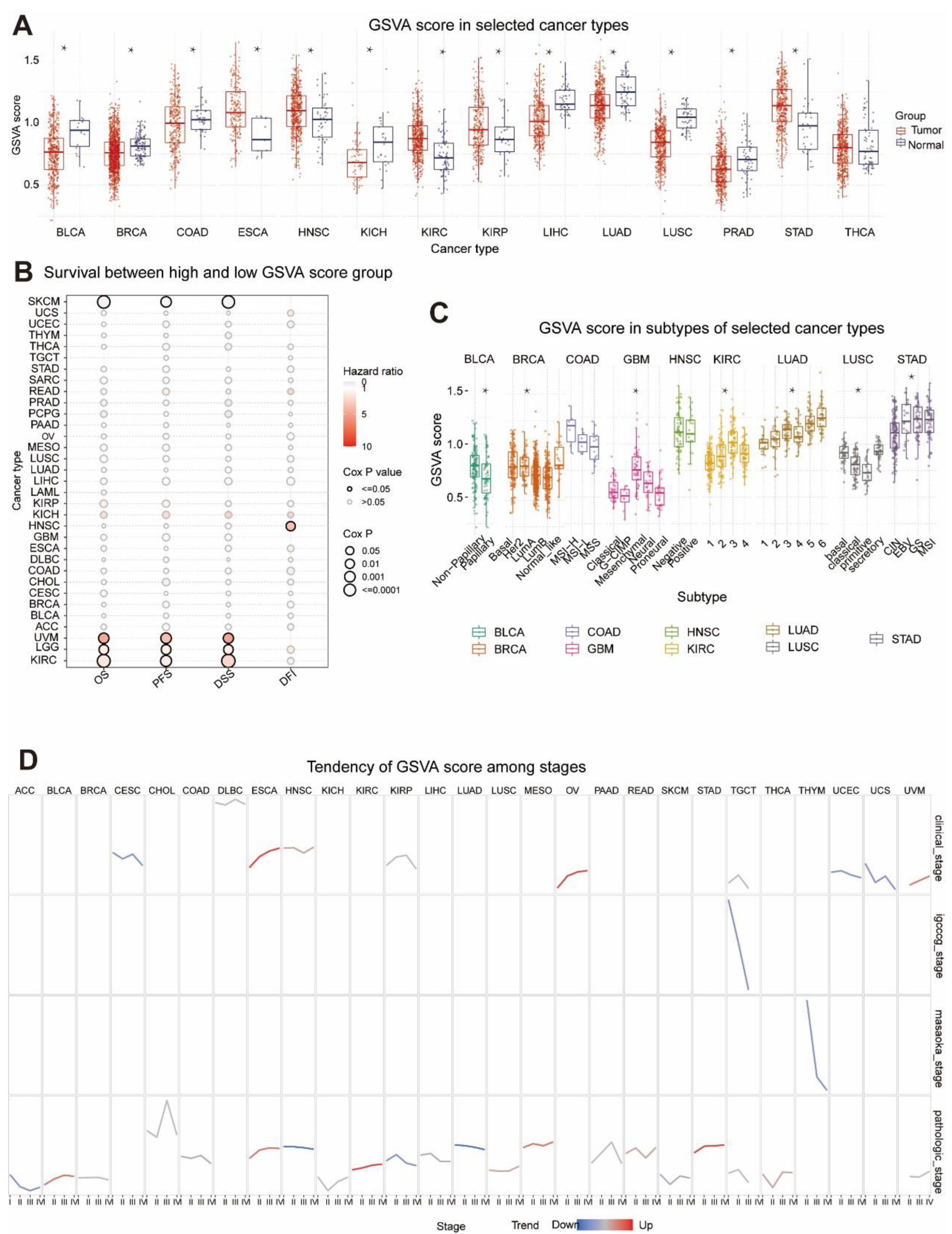
2.3. Comparison of GSVA Scores between Tumor and Normal Samples
2.4. GSVA Scores and Survival
2.5. GSVA Scores and Subtypes
2.6. Relationship between GSVA and Tumor Stage Staging
2.7. Correlation between GSVA Scores and Pathway Activity
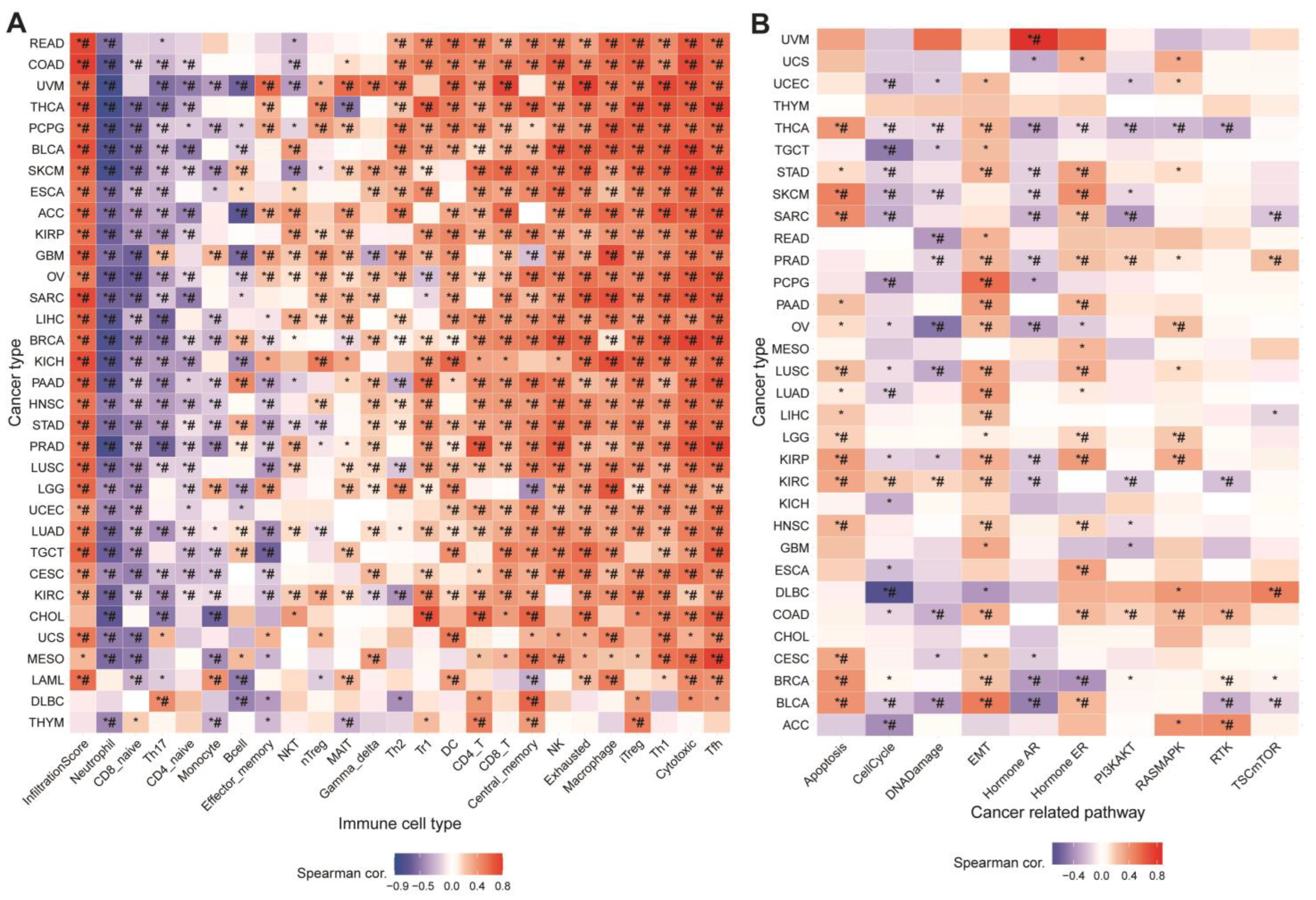
2.8. Relationship between GSVA Score and Immunization
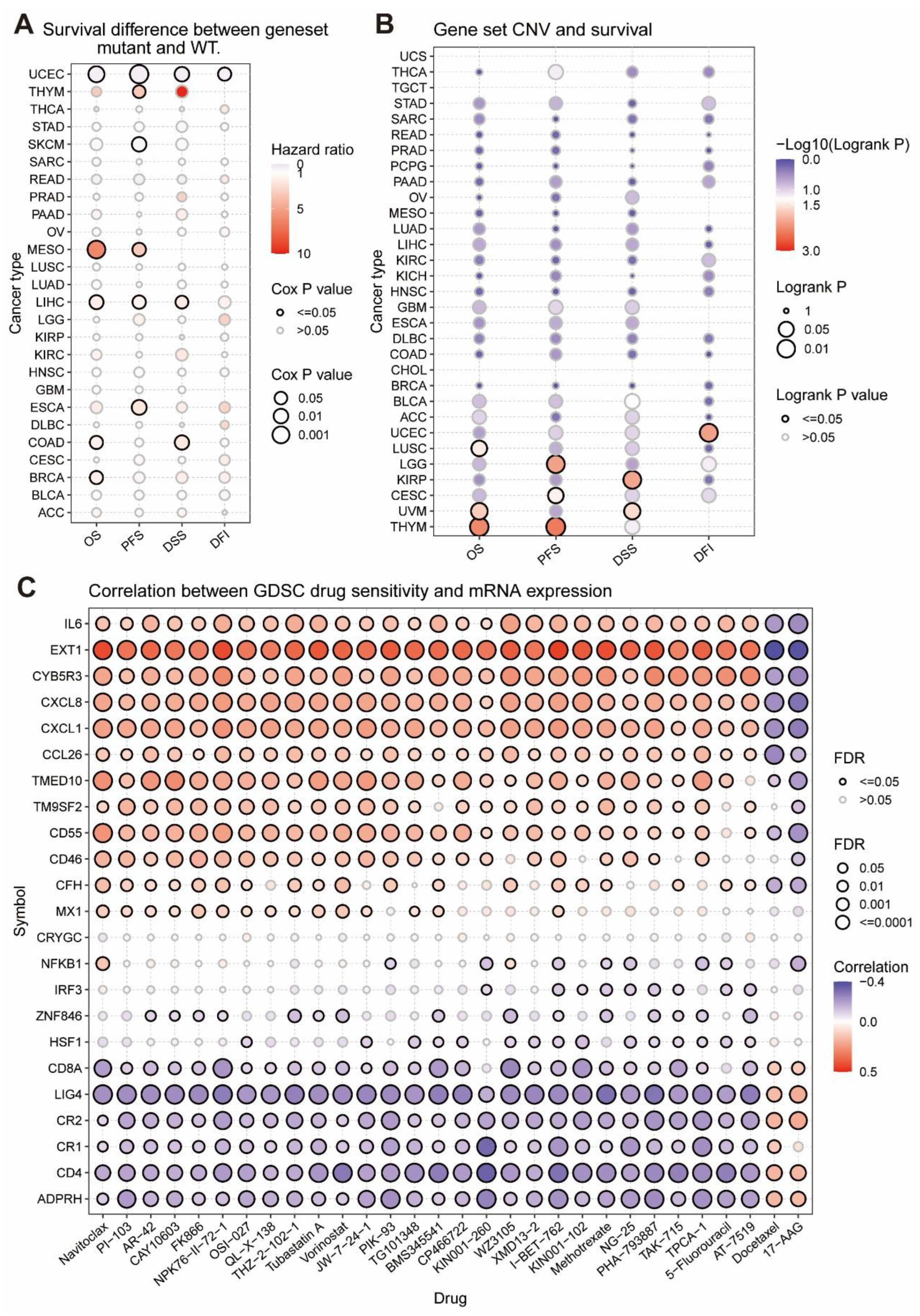
2.9. The Relationship between SNV and Survival
2.10. The Relationship between CNV and Survival
2.11. The Relationship between SNV and Immune Infiltration
2.12. Drug Sensitivity Analysis
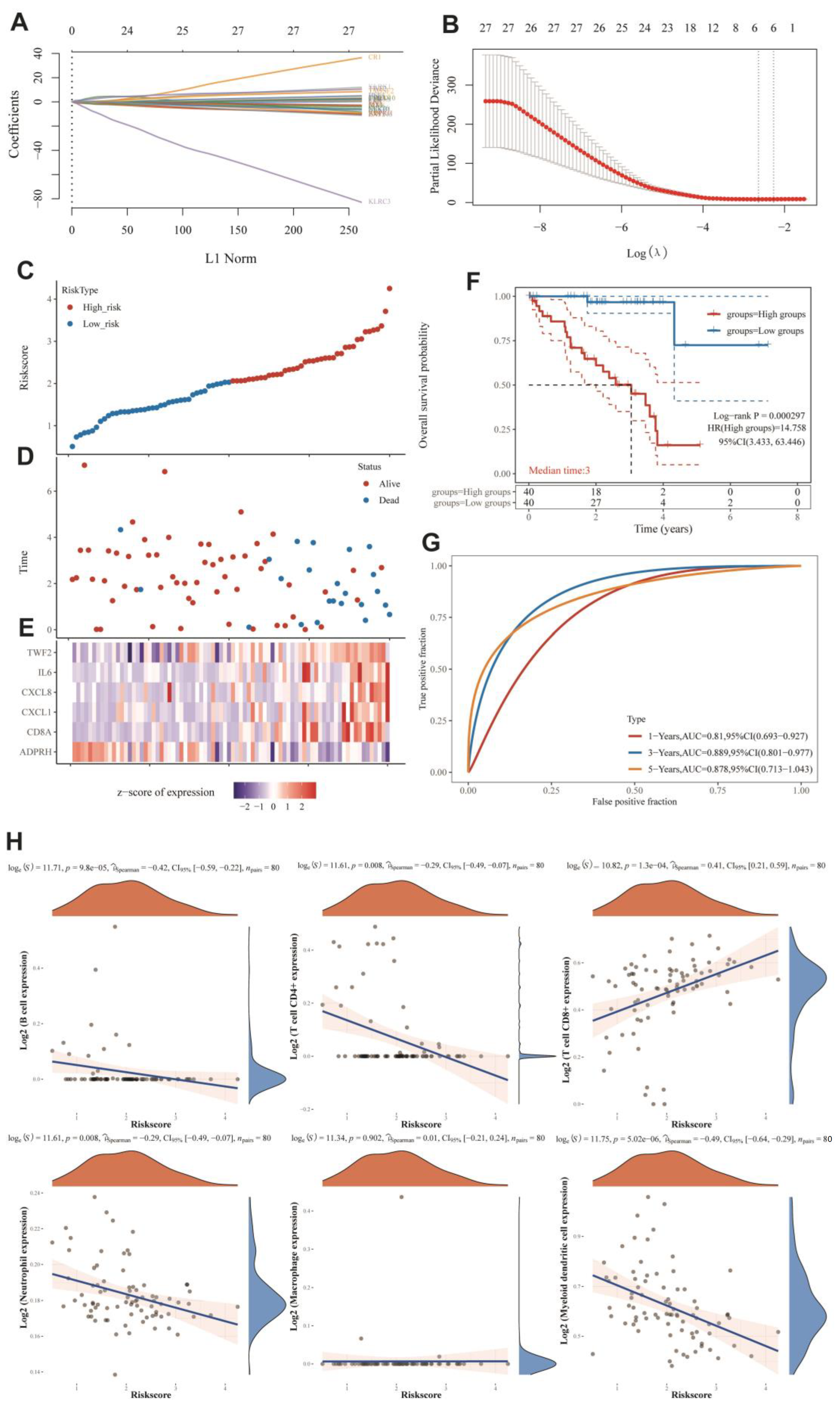
2.13. MRG-Related Prognostic Prediction Feature Construction in UVM
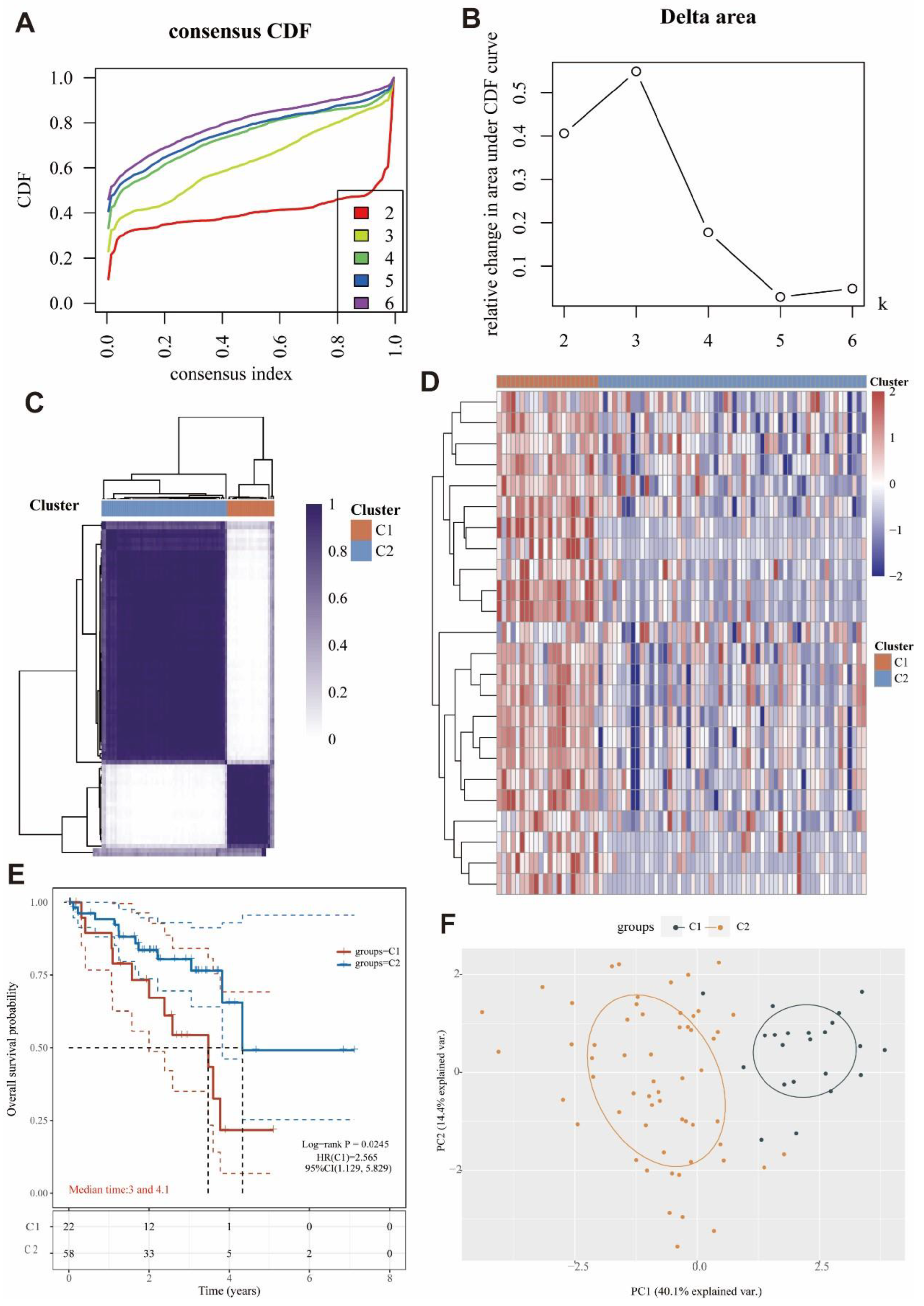
2.14. Identification and Survival Assessment of Molecular Subpopulations
2.15. Functional Analysis
2.16. Immunity Correlation Analysis of Subgroups
2.17. Statistical Analysis
3. Results
3.1. Difference of MRGs between Tumor and Normal Tissues
3.2. Relationship between GSVA Score and Immune Infiltration
3.3. Association between MRGs and Cancer-Related Pathways
3.4. The Relationship between Tumor Survival and MRGs’ SNV and CNV
3.5. The Considerable Impact of MRGs on the Drug Sensitivity of Tumors
3.6. MRGs-Related Prognostic Prediction Feature Construction in UVM
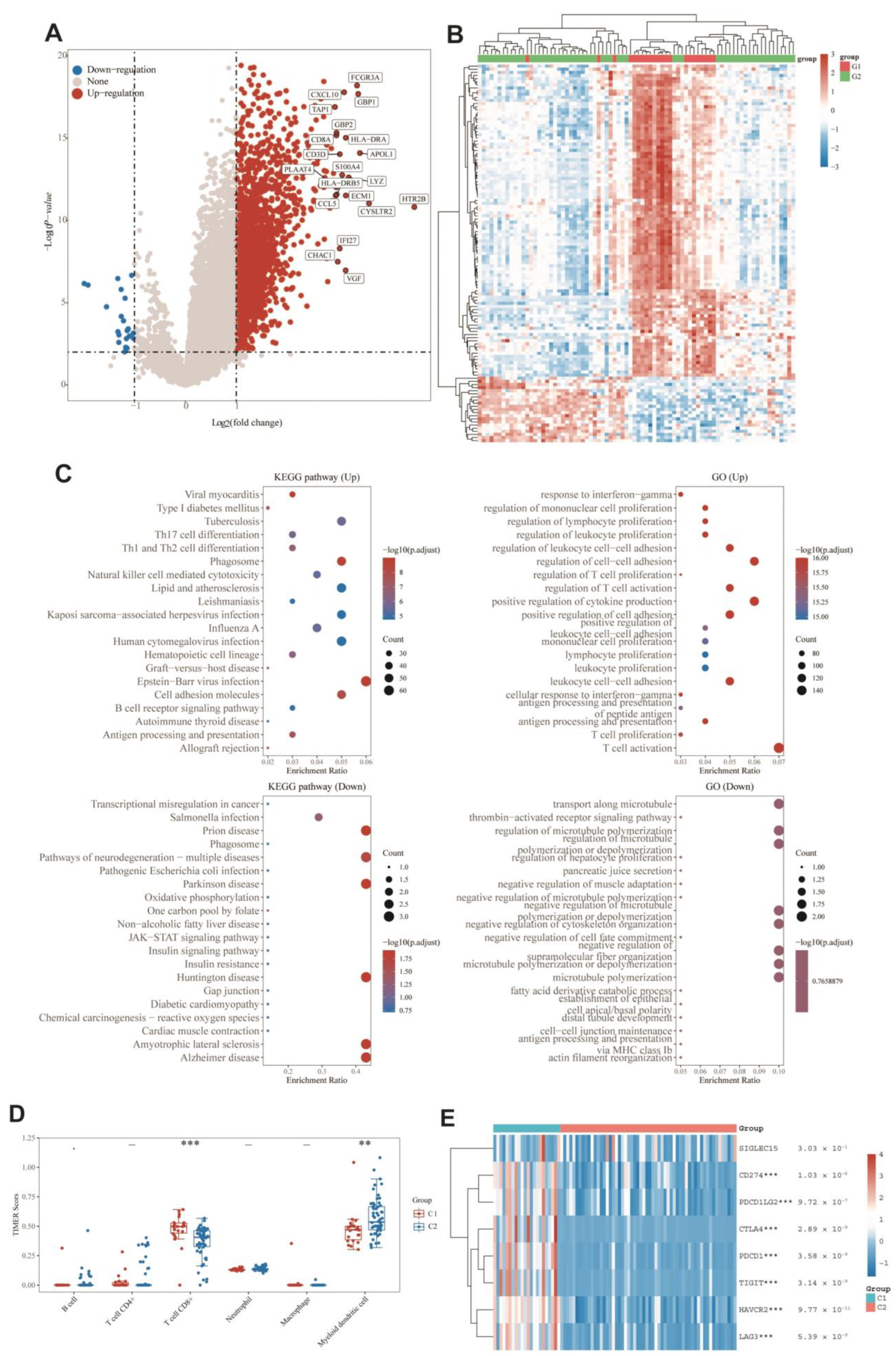
3.7. Immuno-Correlation Analysis of Prognostic Features of MRGs
3.8. Identification of Two Molecular Subtypes Based on MRGs
3.9. DEGs and Functional Analysis
3.10. Confirmation of MRGs Expression in KIRC
4. Discussion
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hopcraft, S.E.; Damania, B. Tumour viruses and innate immunity. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160267. [Google Scholar] [CrossRef] [PubMed]
- Valyi-Nagy, T.; Fredericks, B.; Ravindra, A.; Hopkins, J.; Shukla, D.; Valyi-Nagy, K. Herpes Simplex Virus 1 Infection Promotes the Growth of a Subpopulation of Tumor Cells in Three-Dimensional Uveal Melanoma Cultures. J. Virol. 2018, 92, e00700-18. [Google Scholar] [CrossRef] [PubMed]
- Ablashi, D.V.; Chatlynne, L.G.; Whitman, J.E., Jr.; Cesarman, E. Spectrum of Kaposi’s sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin. Microbiol. Rev. 2002, 15, 439–464. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- Peter, O.J.; Kumar, S.; Kumari, N.; Oguntolu, F.A.; Oshinubi, K.; Musa, R. Transmission dynamics of Monkeypox virus: A mathematical modelling approach. Model. Earth Syst. Environ. 2021, 8, 3423–3434. [Google Scholar] [CrossRef]
- Saxena, S.K.; Ansari, S.; Maurya, V.K.; Kumar, S.; Jain, A.; Paweska, J.T.; Tripathi, A.K.; Abdel-Moneim, A.S. Re-emerging human monkeypox: A major public-health debacle. J. Med. Virol. 2022, 95, e27902. [Google Scholar] [CrossRef]
- Silva, N.I.O.; de Oliveira, J.S.; Kroon, E.G.; Trindade, G.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2020, 13, 43. [Google Scholar] [CrossRef]
- Alkhalil, A.; Hammamieh, R.; Hardick, J.; Ichou, M.A.; Jett, M.; Ibrahim, S. Gene expression profiling of monkeypox virus-infected cells reveals novel interfaces for host-virus interactions. Virol. J. 2010, 7, 173. [Google Scholar] [CrossRef]
- Lipsick, J. A History of Cancer Research: Tumor Viruses. Cold Spring Harb. Perspect. Biol. 2021, 13, a035774. [Google Scholar] [CrossRef]
- Wang, L.; Chard Dunmall, L.S.; Cheng, Z.; Wang, Y. Remodeling the tumor microenvironment by oncolytic viruses: Beyond oncolysis of tumor cells for cancer treatment. J. Immunother. Cancer 2022, 10, e004167. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.R.; Wang, J.; Yates, T.J.; Hasanali, S.L.; Lokeshwar, S.D.; Morera, D.S.; Shamaladevi, N.; Li, C.S.; Klaassen, Z.; Terris, M.K.; et al. Molecular targeting of renal cell carcinoma by an oral combination. Oncogenesis 2020, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yan, S.; Wang, J.; Xu, Y.; Wang, Y.; Zhang, S.; Xu, X.; Yang, Q.; Zeng, X.; Zhou, Y.; et al. Endothelial adenosine A2a receptor-mediated glycolysis is essential for pathological retinal angiogenesis. Nat. Commun. 2017, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, H.; Xu, J.; Yang, Q.; Ma, Q.; Mao, X.; Xu, Z.; Zhou, Y.; Da, Q.; Cai, Y.; et al. Single-cell transcriptome analyses reveal unique microglia types associated with proliferative retinopathy. JCI Insight 2022. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.Y.; Broom, B.M.; Verhaak, R.G.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef]
- Miao, Y.R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.Y.; Wang, H.; Guo, A.Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The Genecards Suite. In Practical Guide to Life Science Databases; Springer: Singapore, 2021; pp. 27–56. [Google Scholar]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Akbani, R.; Ng, P.K.; Werner, H.M.; Shahmoradgoli, M.; Zhang, F.; Ju, Z.; Liu, W.; Yang, J.Y.; Yoshihara, K.; Li, J.; et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 2014, 5, 3887. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, E.; Zhuang, H.; Xie, L.; Feng, X.; Liu, J.; Yu, Y. Construction of a novel gene-based model for prognosis prediction of clear cell renal cell carcinoma. Cancer Cell Int. 2020, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control. 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Ma, J.; He, N.; Wang, J.; Wang, L.; Jin, G.; Lin, R.; Yang, Y. CD8A and HAPLN3 expression profiling to reveal an immunologic subtype of bladder cancer with favorable survival. J. Clin. Oncol. 2020, 38, e15193. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Yi, M.; Peng, C.; Xia, B.; Gan, L. CXCL8 facilitates the survival and Paclitaxel-resistance of triple-negative breast cancers. Clin. Breast Cancer 2022, 22, e191–e198. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Kitamura, H.; Xiang, H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 Modulates the Immune Status of the Tumor Microenvironment to Facilitate Metastatic Colonization of Colorectal Cancer CellsIL6 Facilitates Metastatic Colonization of Colon Cancer. Cancer Immunol. Res. 2019, 7, 1944–1957. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K. High-Fat Diet-Induced Inflammation Accelerates Prostate Cancer Growth via IL6 SignalingHFD-Induced Inflammation and Prostate Cancer Growth. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef]
- Galon, J.; Dieu-Nosjean, M.; Tartour, E.; Sautes-Fridman, C.; Fridman, W. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar]
- Pehlivan, D.; Beck, C.R.; Okamoto, Y.; Harel, T.; Akdemir, Z.H.; Jhangiani, S.N.; Withers, M.A.; Goksungur, M.T.; Carvalho, C.; Czesnik, D. The role of combined SNV and CNV burden in patients with distal symmetric polyneuropathy. Genet. Med. 2016, 18, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, L.; Liu, H.; Deng, G.; Xu, P.; Tan, Y.; Xu, Y.; Liu, B.; Chen, Q.; Tian, D. ADPRH is a prognosis-related biomarker and correlates with immune infiltrates in low grade glioma. J. Cancer 2021, 12, 2912. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Fu, H.; Yin, J.; Zhao, Q. CHAF1B knockdown blocks migration in a hepatocellular carcinoma model. Oncol. Rep. 2018, 40, 405–413. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers |
|---|---|
| CD46 | Forward: AAGTGGTCAAATGTCGATTTCCA |
| Reverse: TCGAGGTAAAAACCCTTATCGC | |
| CD55 | Forward: TTCCAGTCGGTACTGTTGTGG |
| Reverse: CCCGGATTAGGGCATGATTTCT | |
| CYB5R3 | Forward: AAAGTCCAACCCTATCATCAGGA |
| Reverse: AAGCGTGCAGAATGTTTGTTC | |
| HSF1 | Forward: GCACATTCCATGCCCAAGTAT |
| Reverse: GGCCTCTCGTCTATGCTCC | |
| NFKB1 | Forward: AACAGAGAGGATTTCGTTTCCG |
| Reverse: TTTGACCTGAGGGTAAGACTTCT | |
| TM9SF2 | Forward: CGTCAACTTCTGCGACGAAGA |
| Reverse: GGCAAAAATCAAACGCTGTGTA | |
| TWF2 | Forward: AGAAACACCTGTCGTCCTGTG |
| Reverse: CACCTCGTTAATGCGGATCTG | |
| β-actin | Forward: CATGTACGTTGCTATCCAGGC |
| Reverse: CTCCTTAATGTCACGCACGAT |
| Cancer Type | Abbreviation |
|---|---|
| acute myeloid leukemia | LAML |
| adrenocortical carcinoma | ACC |
| uroepithelial carcinoma of the bladder | BLCA |
| invasive breast cancer | BRCA |
| squamous cell carcinoma of the cervix and endocervical adenocarcinoma | CESC |
| cholangiocarcinoma | CHOL |
| colonic adenocarcinoma | COAD |
| esophageal cancer | ESCA |
| glioblastoma multiforme | GBM |
| head and neck squamous cell carcinoma | HNSC |
| renal suspicious cell carcinoma | KICH |
| renal clear cell carcinoma | KIRC |
| renal papillary cell carcinoma | KIRP |
| low grade glioma | LGG |
| hepatocellular carcinoma | LIHC |
| lung adenocarcinoma | LUAD |
| lung squamous cell carcinoma | LUSC |
| lymphoid neoplasm spreading large b-cell lymphoma | DLBC |
| mesothelioma | MESO |
| ovarian plasmacytoid cystic adenocarcinoma | OV |
| pancreatic adenocarcinoma | PAAD |
| pheochromocytoma and paraganglioma | PCPG |
| prostate adenocarcinoma | PRAD |
| rectal adenocarcinoma | READ |
| sarcoma | SARC |
| cutaneous melanoma | SKCM |
| gastric adenocarcinoma | STAD |
| testicular germ cell tumor | TGCT |
| thymoma | THYM |
| thyroid cancer | THCA |
| uterine carcinosarcoma | UCS |
| endometrial cancer of the uterine corpus | UCEC |
| uveal melanoma | UVM |
| Gene | Abbreviation |
|---|---|
| ADP-Ribosylarginine Hydrolase | ADPRH |
| BMS1 Pseudogene 20 | BMS1P20 |
| Complement C4A | C4A |
| Complement C4B | C4B |
| Chemokine (C-C motif) ligand 26 | CCL26 |
| Cluster of differentiation 4 | CD4 |
| CD46 molecule | CD46 |
| CD55 molecule | CD55 |
| CD8a Molecule | CD8A |
| Complement Factor H | CFH |
| Complement C3d Receptor 1 | CR1 |
| Complement C3d Receptor 2 | CR2 |
| Crystallin Gamma C | CRYGC |
| C-X-C Motif Chemokine Ligand 1 | CXCL1 |
| C-X-C Motif Chemokine Ligand 8 | CXCL8 |
| Cytochrome B5 Reductase 3 | CYB5R3 |
| Exostosin Glycosyltransferase 1 | EXT1 |
| Heat Shock Transcription Factor 1 | HSF1 |
| Interferon Alpha 1 | IFNA1 |
| Interleukin 6 | IL6 |
| Interferon Regulatory Factor 3 | IRF3 |
| Killer Cell Lectin-Like Receptor C3 | KLRC3 |
| Killer Cell Lectin-Like Receptor K1 | KLRK1 |
| DNA Ligase 4 | LIG4 |
| MX Dynamin-Like GTPase 1 | MX1 |
| Nuclear Factor Kappa B Subunit 1 | NFKB1 |
| Transmembrane 9 Superfamily Member 2 | TM9SF2 |
| Transmembrane P24 Trafficking Protein 10 | TMED10 |
| Twinfilin Actin-Binding Protein 2 | TWF2 |
| Zinc Finger Protein 846 | ZNF846 |
| Olfactory Receptor Family 10 Subfamily G Member 6 | OR10G6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Y.; Liu, Z.; Ye, W.; Huang, Z.; Wang, J. Exploring the Characteristics of Monkeypox-Related Genes in Pan-Cancer. Cells 2022, 11, 3909. https://doi.org/10.3390/cells11233909
Liao Y, Liu Z, Ye W, Huang Z, Wang J. Exploring the Characteristics of Monkeypox-Related Genes in Pan-Cancer. Cells. 2022; 11(23):3909. https://doi.org/10.3390/cells11233909
Chicago/Turabian StyleLiao, Yong, Zhiping Liu, Weile Ye, Zunnan Huang, and Jiaojiao Wang. 2022. "Exploring the Characteristics of Monkeypox-Related Genes in Pan-Cancer" Cells 11, no. 23: 3909. https://doi.org/10.3390/cells11233909
APA StyleLiao, Y., Liu, Z., Ye, W., Huang, Z., & Wang, J. (2022). Exploring the Characteristics of Monkeypox-Related Genes in Pan-Cancer. Cells, 11(23), 3909. https://doi.org/10.3390/cells11233909