



Targeting Class I Histone Deacetylases in Human Uterine Leiomyosarcoma

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Uterine Leiomyosarcoma Samples
2.2. Immunohistochemistry
2.3. Cells and Reagents
2.4. Proliferation Assay
2.5. Protein Extraction and Western Blot
2.6. RNA-Sequencing
2.7. Transcriptome Profiles Analysis
2.7.1. Transcriptome Data Analysis
2.7.2. Differential Gene Expression Analysis
2.7.3. Gene List Enrichment Analysis
2.7.4. Drug Similarity Analysis
2.7.5. Visualization of Aggregates and Intersection DEGs on UpSet Plot
2.7.6. Epithelial–Mesenchymal Transition Score Calculation
2.7.7. Co-Expression Network Analysis
2.8. Identification of the Potential Drugs
2.8.1. Drug-Gene Interaction Network
2.8.2. Module-Based Drug Prediction
2.9. Statistical Analysis
3. Results
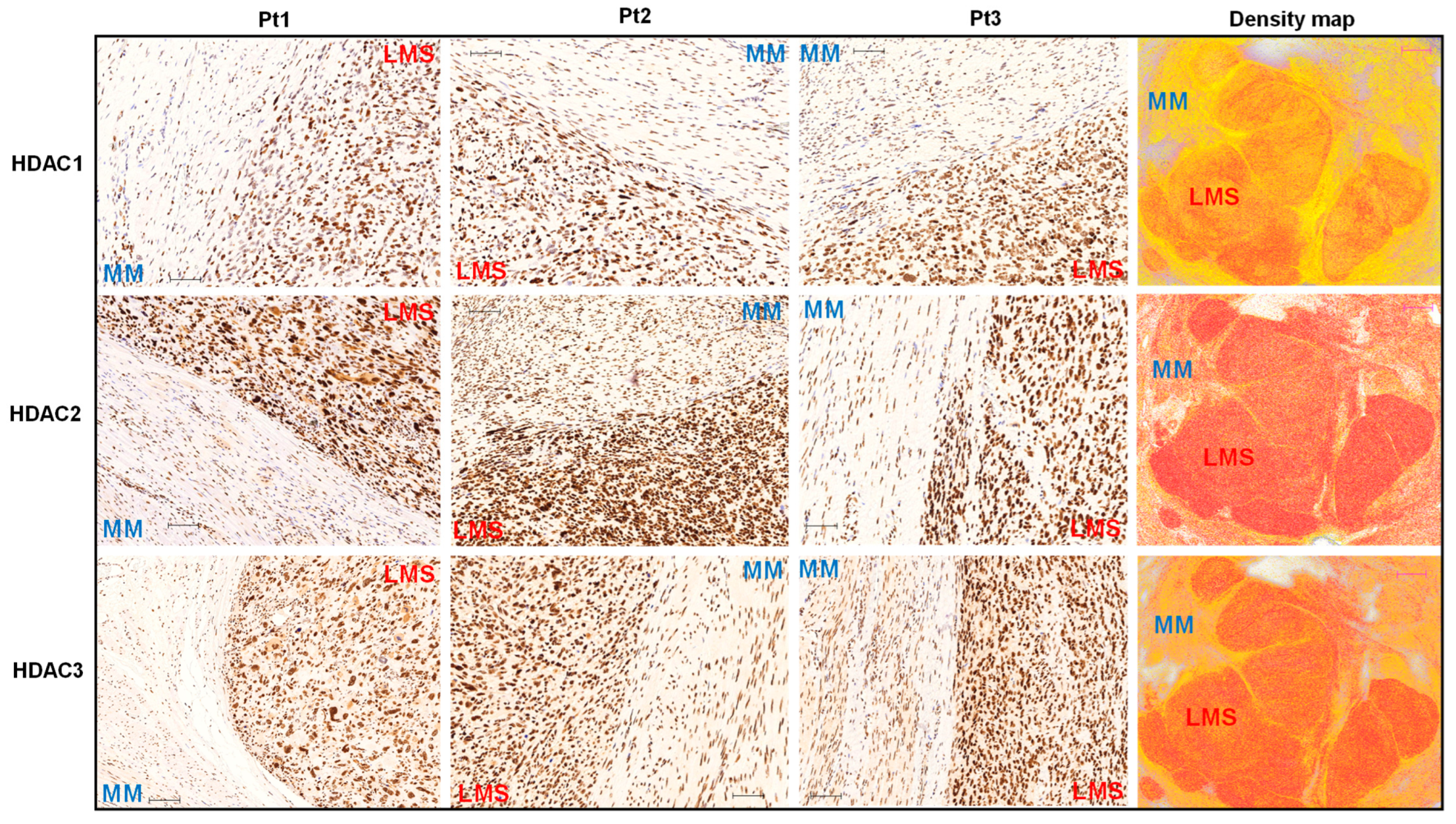
3.1. The Expression Levels of Class I HDACs Members Are Upregulated in uLMS Tissues Compared to Adjacent Myometrium from Women with uLMS
3.2. Class I HDAC Protein Levels Are Upregulated in uLMS Cell Lines
3.3. Inhibition of HDACs Decreased the Cell Proliferation in uLMS Cells
3.4. Tucidinostat and DL-Sulforaphane Sculpt the Transcriptome of uLMS Cells
3.4.1. Pathway Analysis of DEGs upon Tucidinostat and DL-Sulforaphane Treatment
3.4.2. The Expression of Cell Cycle- and Apoptosis-Related Genes Is Altered upon Tucidinostat and DL-Sulforaphane Treatment
3.4.3. Tucidinostat and DL-Sulforaphane Altered the Gene Expression Correlating to Histone Modifications
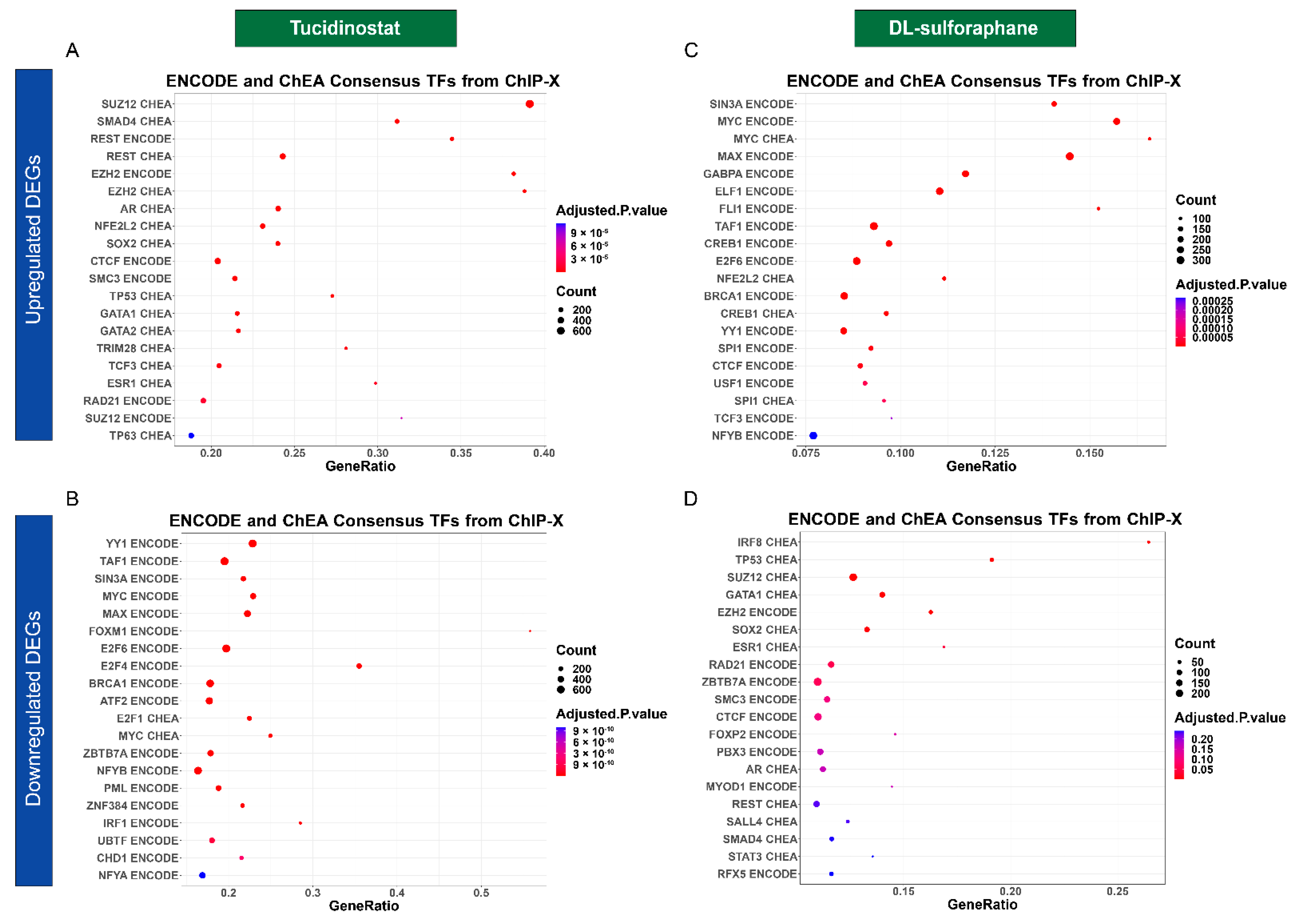
3.4.4. Tucidinostat and DL-Sulforaphane Altered the Gene Expression Associated with Transcriptional Factors
3.4.5. Tucidinostat and DL-Sulforaphane Altered the Gene Expression Correlating to the miRNA Regulation
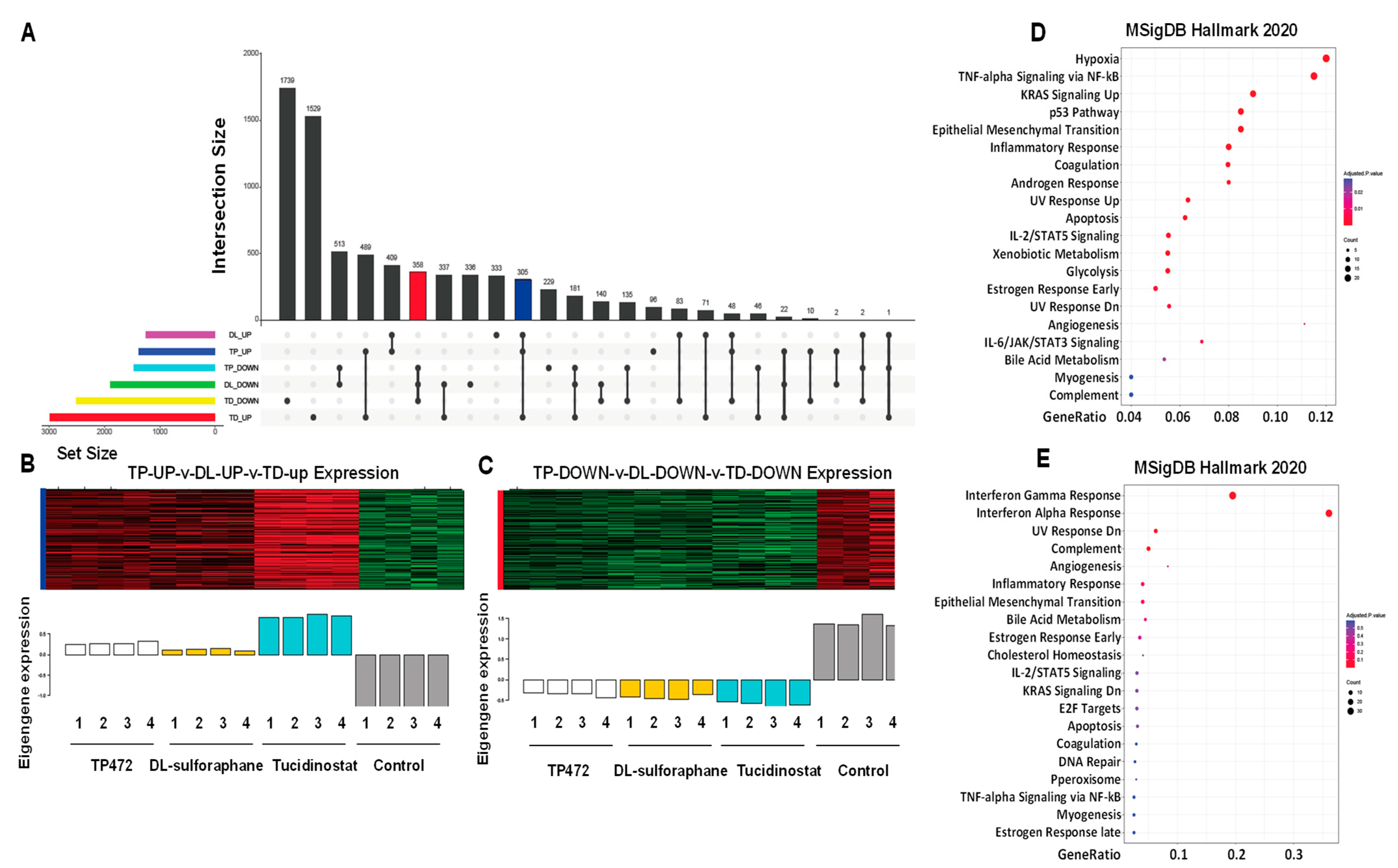
3.4.6. UpSet Plot Visualization
3.4.7. Apoptosis and EMT
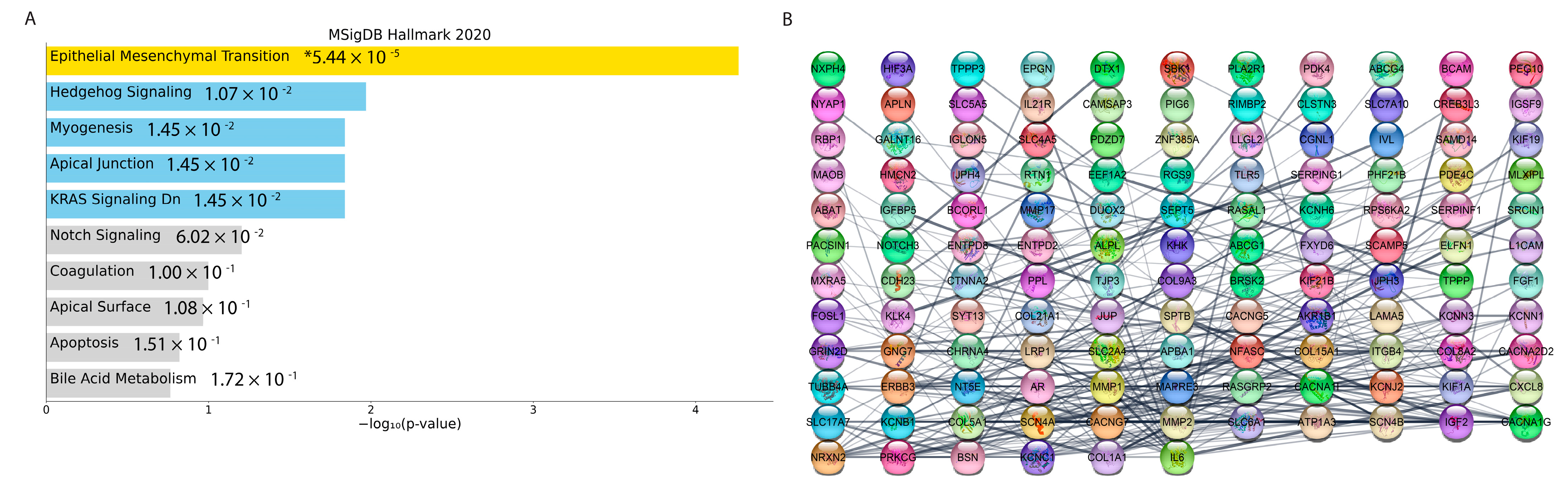
3.4.8. Co-Expression Network Analysis
3.5. Potential Drugs Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Angelo, E.; Prat, J. Uterine sarcomas: A review. Gynecol. Oncol. 2010, 116, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Skorstad, M.; Kent, A.; Lieng, M. Uterine leiomyosarcoma—Incidence, treatment, and the impact of morcellation: A nationwide cohort study. Acta Obstet. Gynecol Scand. 2016, 95, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Seagle, B.L.; Sobecki-Rausch, J.; Strohl, A.E.; Shilpi, A.; Grace, A.; Shahabi, S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecol. Oncol. 2017, 145, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hensley, M.L.; Blessing, J.A.; Mannel, R.; Rose, P.G. Fixed-dose rate gemcitabine plus docetaxel as first-line therapy for metastatic uterine leiomyosarcoma: A Gynecologic Oncology Group phase II trial. Gynecol. Oncol. 2008, 109, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Landoni, F.; Sartori, E.; Zola, P.; Maggino, T.; Lissoni, A.; Bazzurini, L.; Arisio, R.; Romagnolo, C.; Cristofani, R. Uterine leiomyosarcoma: Analysis of treatment failures and survival. Gynecol. Oncol. 1996, 62, 25–32. [Google Scholar] [CrossRef]
- de Almeida, B.C.; Dos Anjos, L.G.; Dobroff, A.S.; Baracat, E.C.; Yang, Q.; Al-Hendy, A.; Carvalho, K.C. Epigenetic Features in Uterine Leiomyosarcoma and Endometrial Stromal Sarcomas: An Overview of the Literature. Biomedicines 2022, 10, 2567. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkuhler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef]
- Ali, A.; Ali, A.; Khan, S.; Ibrahim, M.; Alshehri, M.A.; Thirupathi, A. Inhibition of HDACs Suppresses Cell Proliferation and Cell Migration of Gastric Cancer by Regulating E2F5 Targeting BCL2. Life 2021, 11, 1425. [Google Scholar] [CrossRef]
- Chen, L.; Fei, Y.; Zhao, Y.; Chen, Q.; Chen, P.; Pan, L. Expression and prognostic analyses of HDACs in human gastric cancer based on bioinformatic analysis. Medicine 2021, 100, e26554. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.S.; Abdelsalam, M.; Zeyn, Y.; Zessin, M.; Mustafa, A.M.; Fischer, M.A.; Zeyen, P.; Sun, P.; Bulbul, E.F.; Vecchio, A.; et al. Synthesis, Molecular Docking and Biological Characterization of Pyrazine Linked 2-Aminobenzamides as New Class I Selective Histone Deacetylase (HDAC) Inhibitors with Anti-Leukemic Activity. Int. J. Mol. Sci. 2021, 23, 369. [Google Scholar] [CrossRef] [PubMed]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Tian, Y.; Salwen, H.R.; Chlenski, A.; Godley, L.A.; Raj, J.U.; Yang, Q. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs 2013, 24, 484–493. [Google Scholar] [CrossRef]
- Ali, M.; Shahin, S.M.; Sabri, N.A.; Al-Hendy, A.; Yang, Q. Activation of beta-Catenin Signaling and its Crosstalk With Estrogen and Histone Deacetylases in Human Uterine Fibroids. J. Clin. Endocrinol. Metab. 2020, 105, e1517–e1535. [Google Scholar] [CrossRef]
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Sivaraj, D.; Green, M.M.; Gasparetto, C. Panobinostat for the management of multiple myeloma. Future Oncol. 2017, 13, 477–488. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Bose, P.; Verstovsek, S. Givinostat: An emerging treatment for polycythemia vera. Expert Opin. Investig. Drugs 2020, 29, 525–536. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.S.; El-Khoueiry, A.; Yin, J.; Oberg, A.L.; Flynn, P.; Adkins, D.; Sharma, A.; Weisenberger, D.J.; Brown, T.; Medvari, P.; et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat: A phase 2 consortium/stand up 2 cancer study. Oncotarget 2017, 8, 35326–35338. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, C.L.; Kasamon, Y.; Bociek, R.G.; Lee, P.; Gore, L.; Copeland, A.; Sorensen, R.; Ordentlich, P.; Cruickshank, S.; Kunkel, L.; et al. ENGAGE- 501: Phase II study of entinostat (SNDX-275) in relapsed and refractory Hodgkin lymphoma. Haematologica 2016, 101, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, C.L.; Crump, M.; Andreadis, C.; Rizzieri, D.; Assouline, S.E.; Fox, S.; van der Jagt, R.H.C.; Copeland, A.; Potvin, D.; Chao, R.; et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 2017, 178, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Arrighetti, N.; Corno, C.; Gatti, L. Drug Combinations with HDAC Inhibitors in Antitumor Therapy. Crit. Rev. Oncog. 2015, 20, 83–117. [Google Scholar] [CrossRef]
- Pathil, A.; Armeanu, S.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Gregor, M.; Lauer, U.M.; Bitzer, M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology 2006, 43, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Bariani, M.V.; Falahati, A.; Khosh, A.; Lastra, R.R.; Siblini, H.; Boyer, T.G.; Al-Hendy, A. The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma. Cells 2022, 11, 2160. [Google Scholar] [CrossRef] [PubMed]
- Ram, S.; Vizcarra, P.; Whalen, P.; Deng, S.; Painter, C.L.; Jackson-Fisher, A.; Pirie-Shepherd, S.; Xia, X.; Powell, E.L. Pixelwise H-score: A novel digital image analysis-based metric to quantify membrane biomarker expression from immunohistochemistry images. PLoS ONE 2021, 16, e0245638. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Duan, Q.; Reid, S.P.; Clark, N.R.; Wang, Z.; Fernandez, N.F.; Rouillard, A.D.; Readhead, B.; Tritsch, S.R.; Hodos, R.; Hafner, M.; et al. L1000CDS(2): LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2016, 2, 16015. [Google Scholar] [CrossRef]
- Yoshimitsu, M.; Ando, K.; Ishida, T.; Yoshida, S.; Choi, I.; Hidaka, M.; Takamatsu, Y.; Gillings, M.; Lee, G.T.; Onogi, H.; et al. Oral histone deacetylase inhibitor HBI-8000 (tucidinostat) in Japanese patients with relapsed or refractory non-Hodgkin’s lymphoma: Phase I safety and efficacy. JPN J. Clin. Oncol. 2022, 52, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, A.; Izutsu, K.; Jo, T.; Yoshida, S.; Tsukasaki, K.; Ando, K.; Choi, I.; Imaizumi, Y.; Kato, K.; Kurosawa, M.; et al. Oral histone deacetylase inhibitor tucidinostat (HBI-8000) in patients with relapsed or refractory adult T-cell leukemia/lymphoma: Phase IIb results. Cancer Sci. 2022, 113, 2778. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Chen, B.; Qi, F.; Fang, H.; Qi, S.N.; Guo, R.Y.; Li, N.; Yang, Y.; Wang, S.L.; Song, Y.W.; et al. First-Line Chemoradiation With or Without Chidamide (Tucidinostat) in Patients With Intermediate- and High-Risk Early-Stage Extranodal Nasal-Type Natural Killer/T-Cell Lymphoma: A Randomized Phase 2 Study in China. Int. J. Radiat. Oncol. Biol. Phys. 2022, 113, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xu, S.; Li, S. Selinexor improves the anti-cancer effect of tucidinostat on TP53 wild-type breast cancer. Mol. Cell Endocrinol. 2022, 545, 111558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.C.; Fang, Y.; Wang, L.; Cheng, S.; Fu, D.; He, Y.; Zhao, Y.; Wang, C.F.; Jiang, X.F.; Song, Q.; et al. Clinical efficacy and molecular biomarkers in a phase II study of tucidinostat plus R-CHOP in elderly patients with newly diagnosed diffuse large B-cell lymphoma. Clin. Epigenetics 2020, 12, 160. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Wang, F.; Liu, P.; An, H.; Zhang, Y. [Corrigendum] Sulforaphane suppresses the viability and metastasis, and promotes the apoptosis of bladder cancer cells by inhibiting the expression of FAT1. Int. J. Mol. Med. 2022, 50, 93. [Google Scholar] [CrossRef]
- Li, S.; Khoi, P.N.; Yin, H.; Sah, D.K.; Kim, N.H.; Lian, S.; Jung, Y.D. Sulforaphane Suppresses the Nicotine-Induced Expression of the Matrix Metalloproteinase-9 via Inhibiting ROS-Mediated AP-1 and NF-kappaB Signaling in Human Gastric Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5172. [Google Scholar] [CrossRef]
- Huang, B.; Lei, S.; Wang, D.; Sun, Y.; Yin, J. Sulforaphane exerts anticancer effects on human liver cancer cells via induction of apoptosis and inhibition of migration and invasion by targeting MAPK7 signalling pathway. J. BUON 2021, 26, 642. [Google Scholar]
- Iida, Y.; Okamoto-Katsuyama, M.; Maruoka, S.; Mizumura, K.; Shimizu, T.; Shikano, S.; Hikichi, M.; Takahashi, M.; Tsuya, K.; Okamoto, S.; et al. Effective ferroptotic small-cell lung cancer cell death from SLC7A11 inhibition by sulforaphane. Oncol. Lett. 2021, 21, 71. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Qorri, B.; Baluch, N.; Sparaneo, A.; Fabrizio, F.P.; Muscarella, L.A.; Tyker, A.; Kumar, S.; Cheng, H.M.; Szewczuk, M.R.; et al. Next-generation multimodality of nutrigenomic cancer therapy: Sulforaphane in combination with acetazolamide actively target bronchial carcinoid cancer in disabling the PI3K/Akt/mTOR survival pathway and inducing apoptosis. Oncotarget 2021, 12, 1470–1489. [Google Scholar] [CrossRef] [PubMed]
- Rutz, J.; Thaler, S.; Maxeiner, S.; Chun, F.K.; Blaheta, R.A. Sulforaphane Reduces Prostate Cancer Cell Growth and Proliferation In Vitro by Modulating the Cdk-Cyclin Axis and Expression of the CD44 Variants 4, 5, and 7. Int. J. Mol. Sci. 2020, 21, 8724. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, M.; Sun, N.X.; Zhu, C.; Lin, Y.M.; Li, C.; Liu, F.; Zhu, W.W. Sulforaphane suppresses carcinogenesis of colorectal cancer through the ERK/Nrf2UDP glucuronosyltransferase 1A metabolic axis activation. Oncol. Rep. 2020, 43, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.R.; Singh, S.V. Functional relevance of D,L-sulforaphane-mediated induction of vimentin and plasminogen activator inhibitor-1 in human prostate cancer cells. Eur. J. Nutr. 2014, 53, 843–852. [Google Scholar] [CrossRef]
- Qin, Y.; Liang, Y.; Jiang, G.; Peng, Y.; Feng, W. ACY-1215 suppresses the proliferation and induces apoptosis of chronic myeloid leukemia cells via the ROS/PTEN/Akt pathway. Cell Stress Chaperones 2022, 27, 383–396. [Google Scholar] [CrossRef]
- Lee, J.; Kim, K.; Ryu, T.Y.; Jung, C.R.; Lee, M.S.; Lim, J.H.; Park, K.; Kim, D.S.; Son, M.Y.; Hamamoto, R.; et al. EHMT1 knockdown induces apoptosis and cell cycle arrest in lung cancer cells by increasing CDKN1A expression. Mol. Oncol. 2021, 15, 2989–3002. [Google Scholar] [CrossRef]
- Li, B.; Li, A.; You, Z.; Xu, J.; Zhu, S. Epigenetic silencing of CDKN1A and CDKN2B by SNHG1 promotes the cell cycle, migration and epithelial-mesenchymal transition progression of hepatocellular carcinoma. Cell Death Dis. 2020, 11, 823. [Google Scholar] [CrossRef]
- He, C.; Chen, H.; Liu, Y.; Li, X.; Zhang, C.; Qin, Q.; Pang, Q. miR-106b-5p promotes cell proliferation and cell cycle progression by directly targeting CDKN1A in osteosarcoma. Exp. Ther. Med. 2020, 19, 3203–3210. [Google Scholar] [CrossRef]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [CrossRef]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006, 27, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Neuhoff, C.; Yang, Q.; Cinar, M.U.; Uddin, M.J.; Tholen, E.; Schellander, K.; Tesfaye, D. Sulforaphane Enhanced Proliferation of Porcine Satellite Cells via Epigenetic Augmentation of SMAD7. Animals 2022, 12, 1365. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, S.E.; Rusche, J.J.; Bec, S.L.; Horn, D.J.; Janda, J.; Rim, S.H.; Smith, C.L.; Bowden, G.T. The effect of sulforaphane on histone deacetylase activity in keratinocytes: Differences between in vitro and in vivo analyses. Mol. Carcinog. 2015, 54, 1513–1520. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, L.; Lin, R.Y.; Muschen, M.; Koeffler, H.P. Core transcriptional regulatory circuitries in cancer. Oncogene 2020, 39, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Kusunoki, S.; Hirayama, T.; Fujino, K.; Terao, Y.; Itakura, A. Case of leiomyosarcoma arising from subserosal leiomyoma. J. Obstet. Gynaecol. Res. 2019, 45, 1944–1947. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ciebiera, M.; Victoria Bariani, M.; Ali, M.; Elkafas, H.; Boyer, T.G.; Al-Hendy, A. Comprehensive Review of Uterine Fibroids: Developmental Origin, Pathogenesis, and Treatment. Endocr. Rev. 2021, 42, 678–719. [Google Scholar] [CrossRef]
- Han, F.; Cheng, C.; Xu, Q.; Chen, J.; Yang, Z.; Liu, J. DEPDC1B promotes colorectal cancer via facilitating cell proliferation and migration while inhibiting apoptosis. Cell Cycle 2022, 21, 1–13. [Google Scholar] [CrossRef]
- Chen, Z.; Tang, W.J.; Zhou, Y.H.; Chen, Z.M.; Liu, K. Andrographolide inhibits non-small cell lung cancer cell proliferation through the activation of the mitochondrial apoptosis pathway and by reprogramming host glucose metabolism. Ann. Transl. Med. 2021, 9, 1701. [Google Scholar] [CrossRef]
- Luo, D.; Fan, H.; Ma, X.; Yang, C.; He, Y.; Ge, Y.; Jiang, M.; Xu, Z.; Yang, L. miR-1301-3p Promotes Cell Proliferation and Facilitates Cell Cycle Progression via Targeting SIRT1 in Gastric Cancer. Front. Oncol. 2021, 11, 664242. [Google Scholar] [CrossRef]
- Peng, Y.; Feng, H.; Wang, C.; Song, Z.; Zhang, Y.; Liu, K.; Cheng, X.; Zhao, R. The role of E26 transformation-specific variant transcription factor 5 in colorectal cancer cell proliferation and cell cycle progression. Cell Death Dis. 2021, 12, 427. [Google Scholar] [CrossRef]
- Qiao, Z.; Ren, S.; Li, W.; Wang, X.; He, M.; Guo, Y.; Sun, L.; He, Y.; Ge, Y.; Yu, Q. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, J.; Xu, Z.; Xu, J.; Shi, M.; Liu, P. Chidamide, a novel histone deacetylase inhibitor, inhibits multiple myeloma cells proliferation through succinate dehydrogenase subunit A. Am. J. Cancer Res. 2019, 9, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, Y.; Fu, M.; Liang, X.; Zhang, X.; Wang, R.; Lin, C.; Qian, H. Antitumor activity of Chidamide in hepatocellular carcinoma cell lines. Mol. Med. Rep. 2012, 5, 1503–1508. [Google Scholar] [CrossRef]
- Li, Y.; Chen, K.; Zhou, Y.; Xiao, Y.; Deng, M.; Jiang, Z.; Ye, W.; Wang, X.; Wei, X.; Li, J.; et al. A New Strategy to Target Acute Myeloid Leukemia Stem and Progenitor Cells Using Chidamide, a Histone Deacetylase Inhibitor. Curr. Cancer Drug Targets 2015, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; George, J.T.; Tripathi, S.; Levine, H.; Jolly, M.K. Comparative Study of Transcriptomics-Based Scoring Metrics for the Epithelial-Hybrid-Mesenchymal Spectrum. Front. Bioeng. Biotechnol. 2020, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Hu, C.; Yu, S.; Liu, L.; Zhao, J.; Zhao, Y.; Lin, F.; Du, X.; Yu, Q.; Xiao, Q. Identification of EMT-Related Gene Signatures to Predict the Prognosis of Patients With Endometrial Cancer. Front. Genet. 2020, 11, 582274. [Google Scholar] [CrossRef]
- Song, J. EMT or apoptosis: A decision for TGF-beta. Cell Res. 2007, 17, 289–290. [Google Scholar] [CrossRef]
- Yang, Y.; Pan, X.; Lei, W.; Wang, J.; Song, J. Transforming growth factor-beta1 induces epithelial-to-mesenchymal transition and apoptosis via a cell cycle-dependent mechanism. Oncogene 2006, 25, 7235–7244. [Google Scholar] [CrossRef]
- Amin, S.; Walsh, M.; Wilson, C.; Parker, A.E.; Oscier, D.; Willmore, E.; Mann, D.; Mann, J. Cross-talk between DNA methylation and active histone modifications regulates aberrant expression of ZAP70 in CLL. J. Cell Mol. Med. 2012, 16, 2074–2084. [Google Scholar] [CrossRef]
- Szulwach, K.E.; Li, X.; Smrt, R.D.; Li, Y.; Luo, Y.; Lin, L.; Santistevan, N.J.; Li, W.; Zhao, X.; Jin, P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 2010, 189, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Fischle, W. Epigenetic markers and their cross-talk. Essays Biochem. 2010, 48, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Song, Y.; Lam, R.; Ruder, D.; Creighton, C.J.; Bid, H.K.; Bill, K.L.; Bolshakov, S.; Zhang, X.; Lev, D.; et al. HDAC Inhibition for the Treatment of Epithelioid Sarcoma: Novel Cross Talk Between Epigenetic Components. Mol. Cancer Res. 2016, 14, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Morris, C.D.; Williams, R.M.; Loring, J.F.; Thomas, E.A. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. USA 2015, 112, E56–E64. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Dalla, E.; Franforte, E.; Paluvai, H.; Minisini, M.; Trevisanut, M.; Picco, R.; Brancolini, C. Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 2020, 48, 646–664. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Ratan, R.R. The interplay between microRNAs and histone deacetylases in neurological diseases. Neurochem. Int. 2014, 77, 33–39. [Google Scholar] [CrossRef]
- Baltan, S.; Sandau, U.S.; Brunet, S.; Bastian, C.; Tripathi, A.; Nguyen, H.; Liu, H.; Saugstad, J.A.; Zarnegarnia, Y.; Dutta, R. Identification of miRNAs That Mediate Protective Functions of Anti-Cancer Drugs During White Matter Ischemic Injury. ASN Neuro 2021, 13, 17590914211042220. [Google Scholar] [CrossRef]
- Klieser, E.; Urbas, R.; Swierczynski, S.; Stattner, S.; Primavesi, F.; Jager, T.; Mayr, C.; Kiesslich, T.; Fazio, P.D.; Helm, K.; et al. HDAC-Linked “Proliferative” miRNA Expression Pattern in Pancreatic Neuroendocrine Tumors. Int. J. Mol. Sci. 2018, 19, 2781. [Google Scholar] [CrossRef]
- Wang, J.; Tian, K.Y.; Fang, Y.; Chang, H.M.; Han, Y.N.; Chen, F.Q. Sulforaphane attenuates cisplatin-induced hearing loss by inhibiting histone deacetylase expression. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211034086. [Google Scholar] [CrossRef]
- Alese, O.B.; Zakka, K.; Huo, X.; Jiang, R.; Shaib, W.L.; Akce, M.; Behera, M.; Sullivan, P.; Wu, C.; El-Rayes, B.F. Perioperative therapy in metastatic colorectal cancer: Pattern of use and survival outcomes. J. Surg. Oncol. 2021, 123, 596–605. [Google Scholar] [CrossRef]
- Soh, J.; Hamada, A.; Fujino, T.; Mitsudomi, T. Perioperative Therapy for Non-Small Cell Lung Cancer with Immune Checkpoint Inhibitors. Cancers 2021, 13, 4035. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.; Basu, A. Emerging perioperative therapeutic approaches in muscle invasive bladder cancer. Ther. Adv. Urol. 2022, 14, 17562872221134389. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Serrano, C.; Hensley, M.L.; Ray-Coquard, I. Soft Tissue and Uterine Leiomyosarcoma. J. Clin. Oncol. 2018, 36, 144–150. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Company | Catalog Number | Host | Application | Dilution | Size |
|---|---|---|---|---|---|---|
| HDAC1 | Cell signaling | 34589 | Rabbit | WB | 1:1000 | 62 kd |
| HDAC2 | Cell signaling | 57159 | Rabbit | WB | 1:1000 | 60 kd |
| HDAC3 | Cell signaling | 85057 | Rabbit | WB | 1:1000 | 49 kd |
| HDAC8 | Abcam | ab187139 | Rabbit | WB | 1:1000 | 42 kd |
| PCNA | Abcam | Ab18197 | Rabbit | WB | 1:1000 | 29 kd |
| β-actin | Sigma | A5316 | Mouse | WB | 1:8000 | 42 kd |
| HDAC1 | Abcam | Ab109411 | Rabbit | IHC | 1:50 | |
| HDAC2 | Abcam | Ab32117 | Rabbit | IHC | 1:500 | |
| HDAC3 | Abcam | Ab32369 | Rabbit | IHC | 1:4000 | |
| Ki67 | Abcam | ab15580 | Rabbit | IHC | 1: 3000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Falahati, A.; Khosh, A.; Mohammed, H.; Kang, W.; Corachán, A.; Bariani, M.V.; Boyer, T.G.; Al-Hendy, A. Targeting Class I Histone Deacetylases in Human Uterine Leiomyosarcoma. Cells 2022, 11, 3801. https://doi.org/10.3390/cells11233801
Yang Q, Falahati A, Khosh A, Mohammed H, Kang W, Corachán A, Bariani MV, Boyer TG, Al-Hendy A. Targeting Class I Histone Deacetylases in Human Uterine Leiomyosarcoma. Cells. 2022; 11(23):3801. https://doi.org/10.3390/cells11233801
Chicago/Turabian StyleYang, Qiwei, Ali Falahati, Azad Khosh, Hanaa Mohammed, Wenjun Kang, Ana Corachán, Maria Victoria Bariani, Thomas G. Boyer, and Ayman Al-Hendy. 2022. "Targeting Class I Histone Deacetylases in Human Uterine Leiomyosarcoma" Cells 11, no. 23: 3801. https://doi.org/10.3390/cells11233801
APA StyleYang, Q., Falahati, A., Khosh, A., Mohammed, H., Kang, W., Corachán, A., Bariani, M. V., Boyer, T. G., & Al-Hendy, A. (2022). Targeting Class I Histone Deacetylases in Human Uterine Leiomyosarcoma. Cells, 11(23), 3801. https://doi.org/10.3390/cells11233801