

Neuroprotective Effects of Neuropeptide Y on Human Neuroblastoma SH-SY5Y Cells in Glutamate Excitotoxicity and ER Stress Conditions

 ,
,  ,
,
Abstract

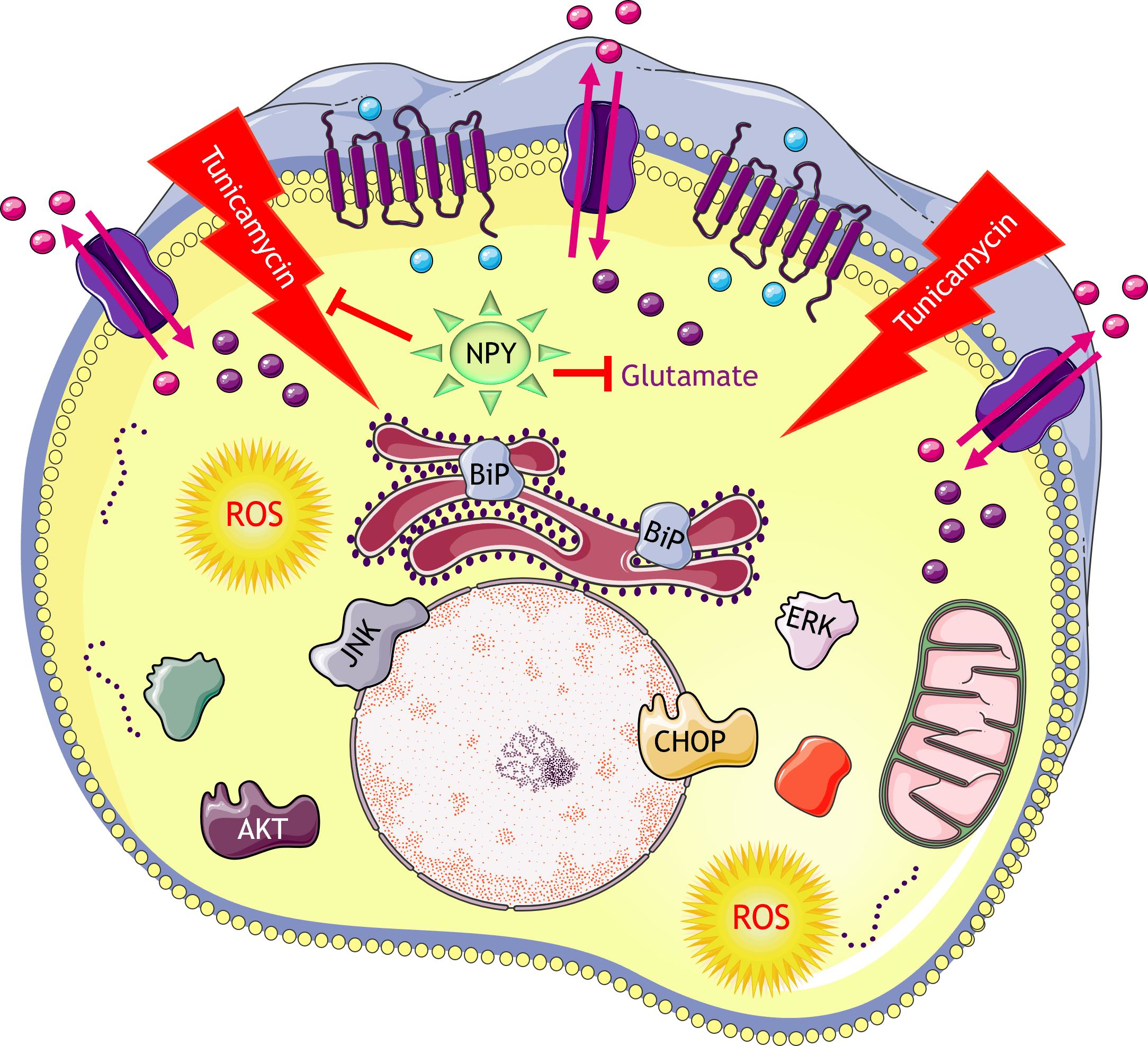{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
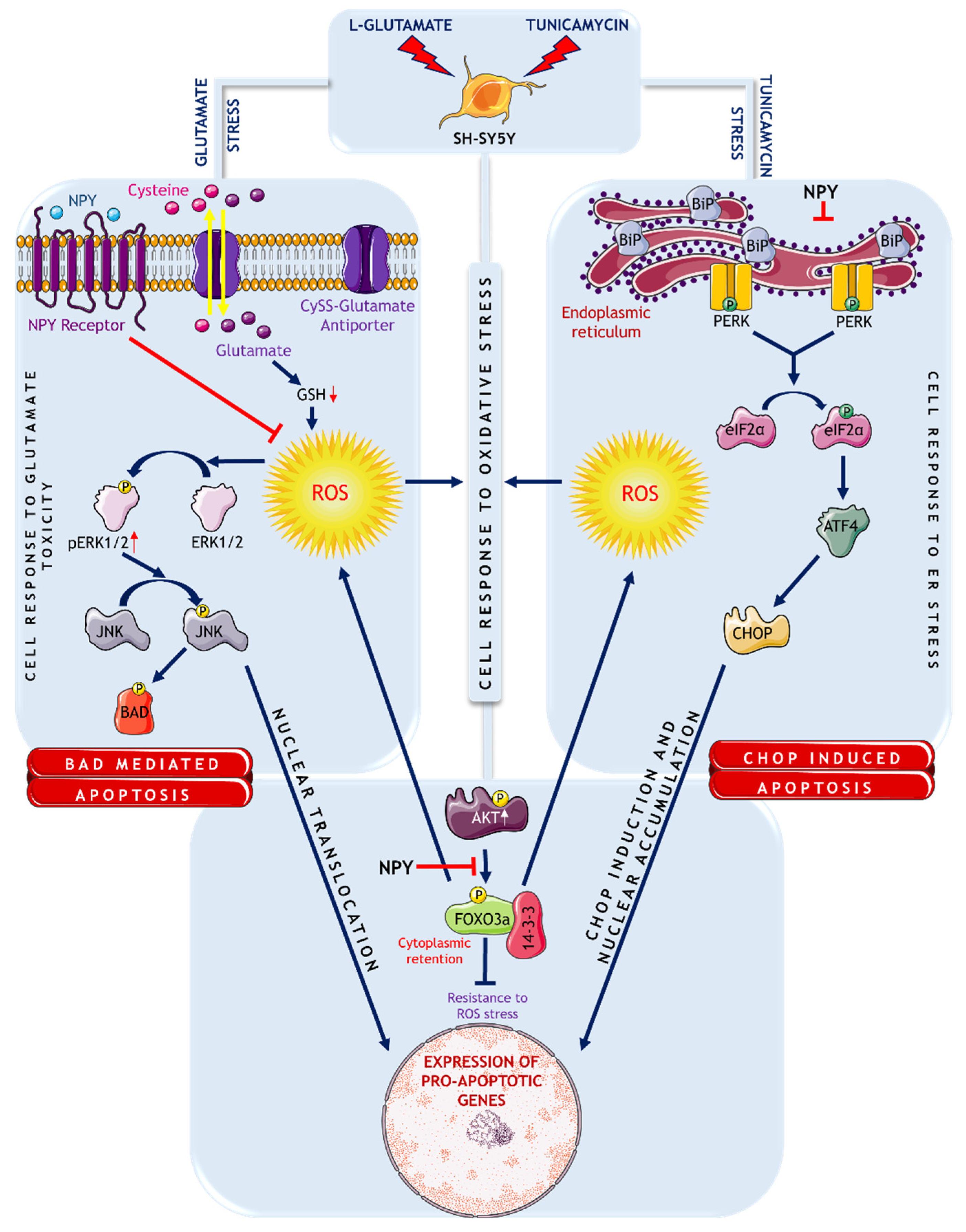{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatments with Glutamate, Tunicamycin, and Neuropeptide Y
2.3. Cell Viability Test
2.4. SDS-PAGE and Western Blotting
2.5. Immunocytochemistry
2.6. Statistical Analysis
3. Results
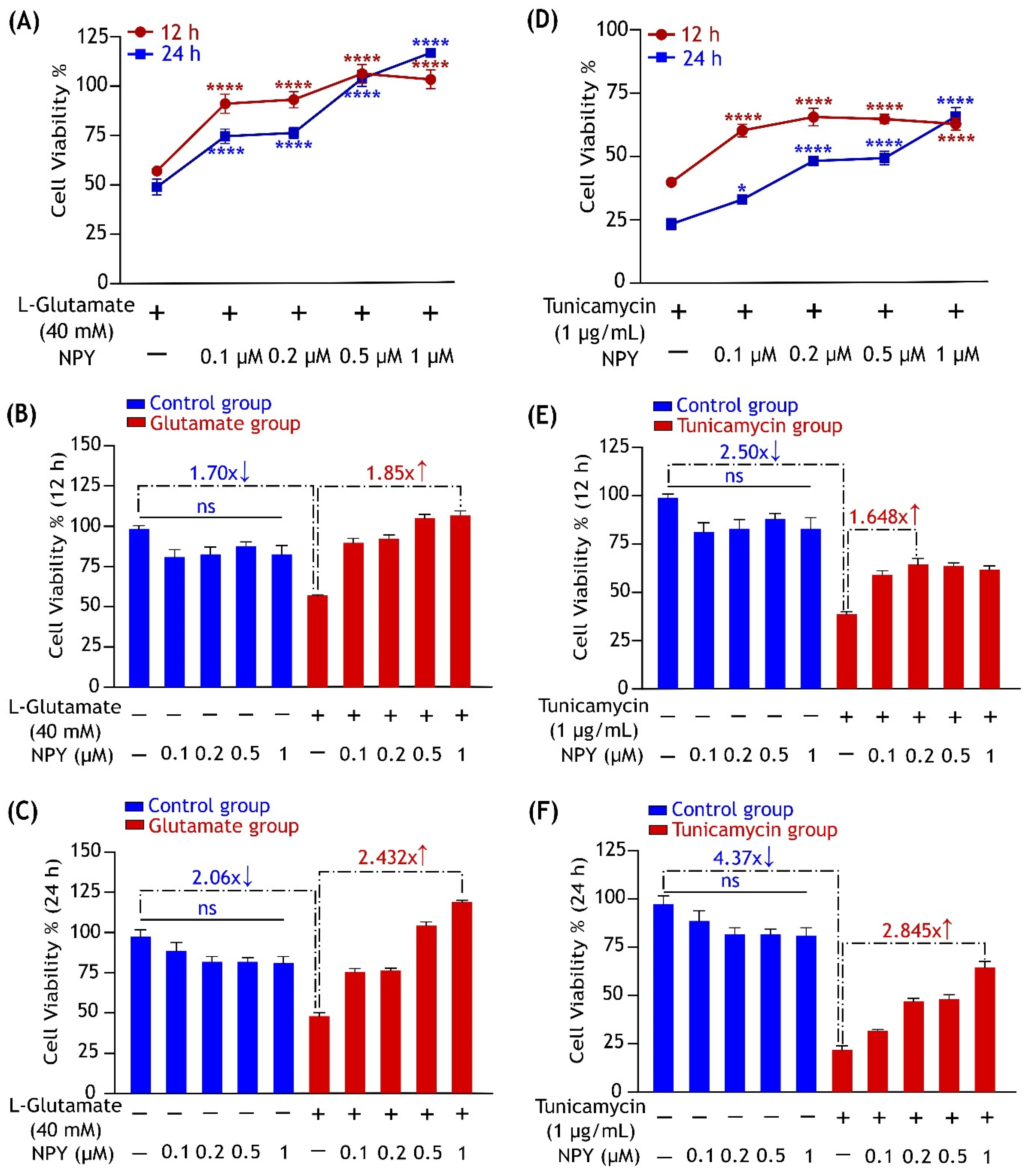
3.1. NPY Protects SH-SY5Y Cells from Glutamate and Tunicamycin-Induced Cell Death
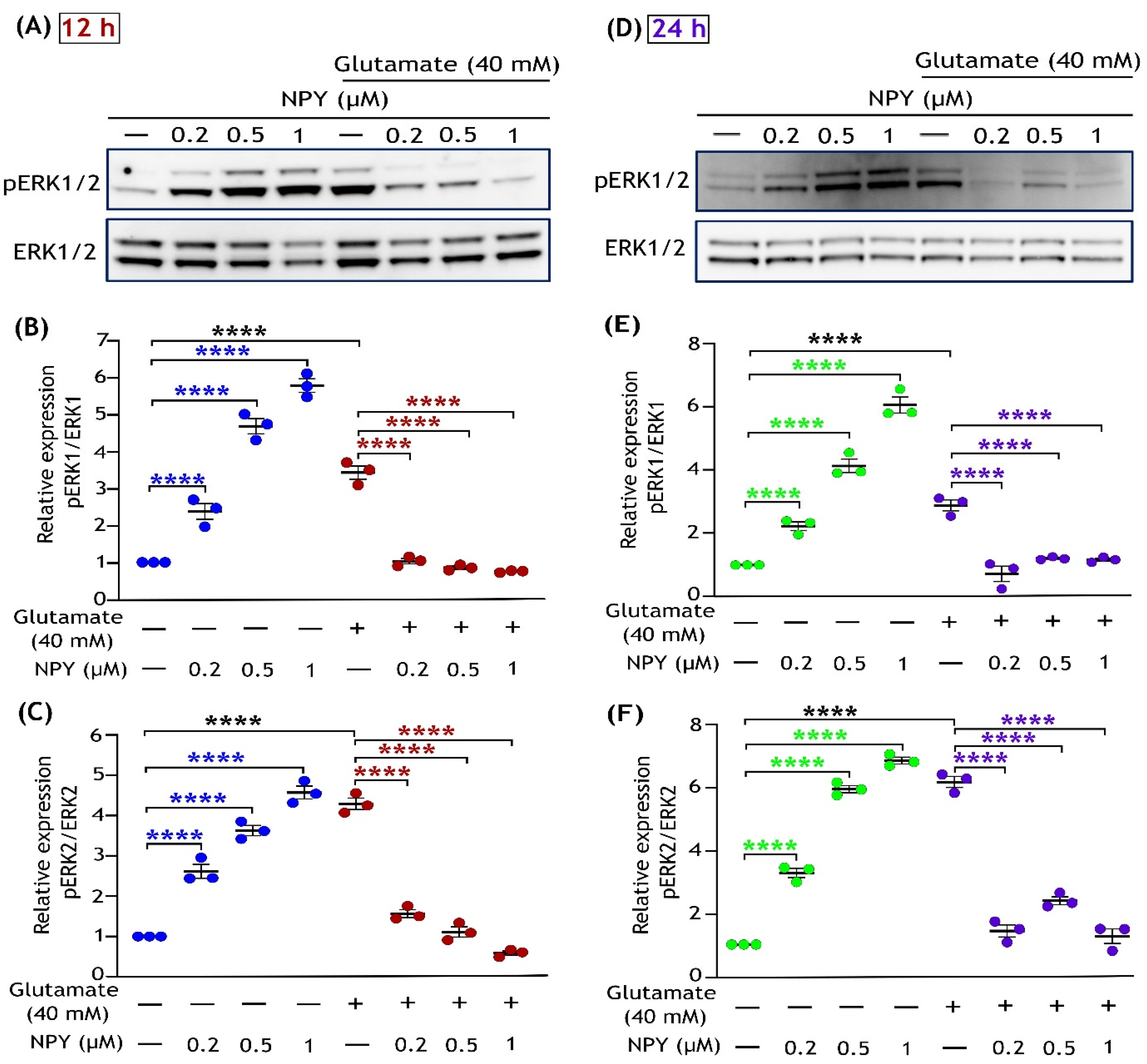
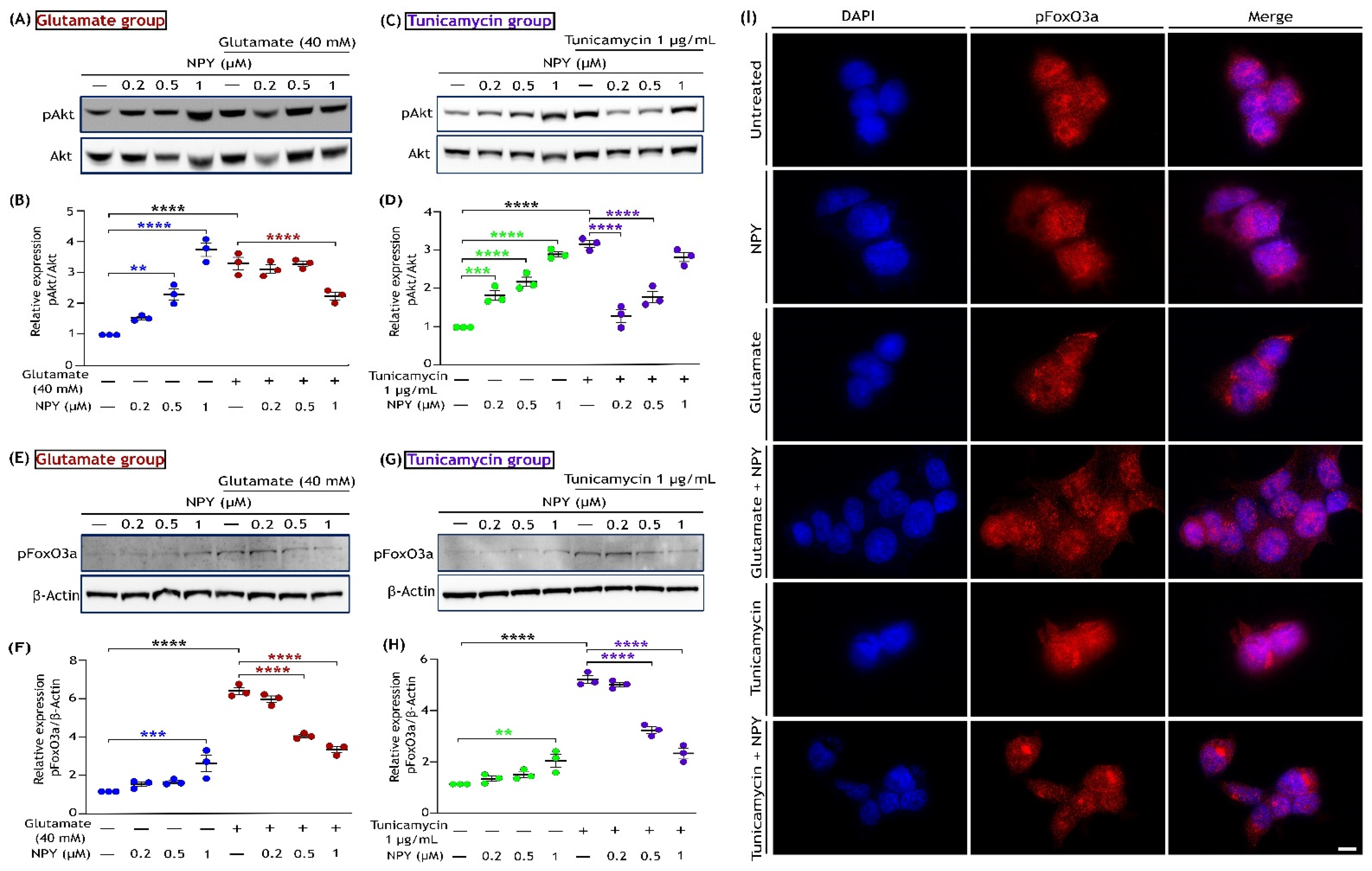
3.2. NPY Suppresses the Glutamate-Induced Apoptotic Activation of ERK1/2
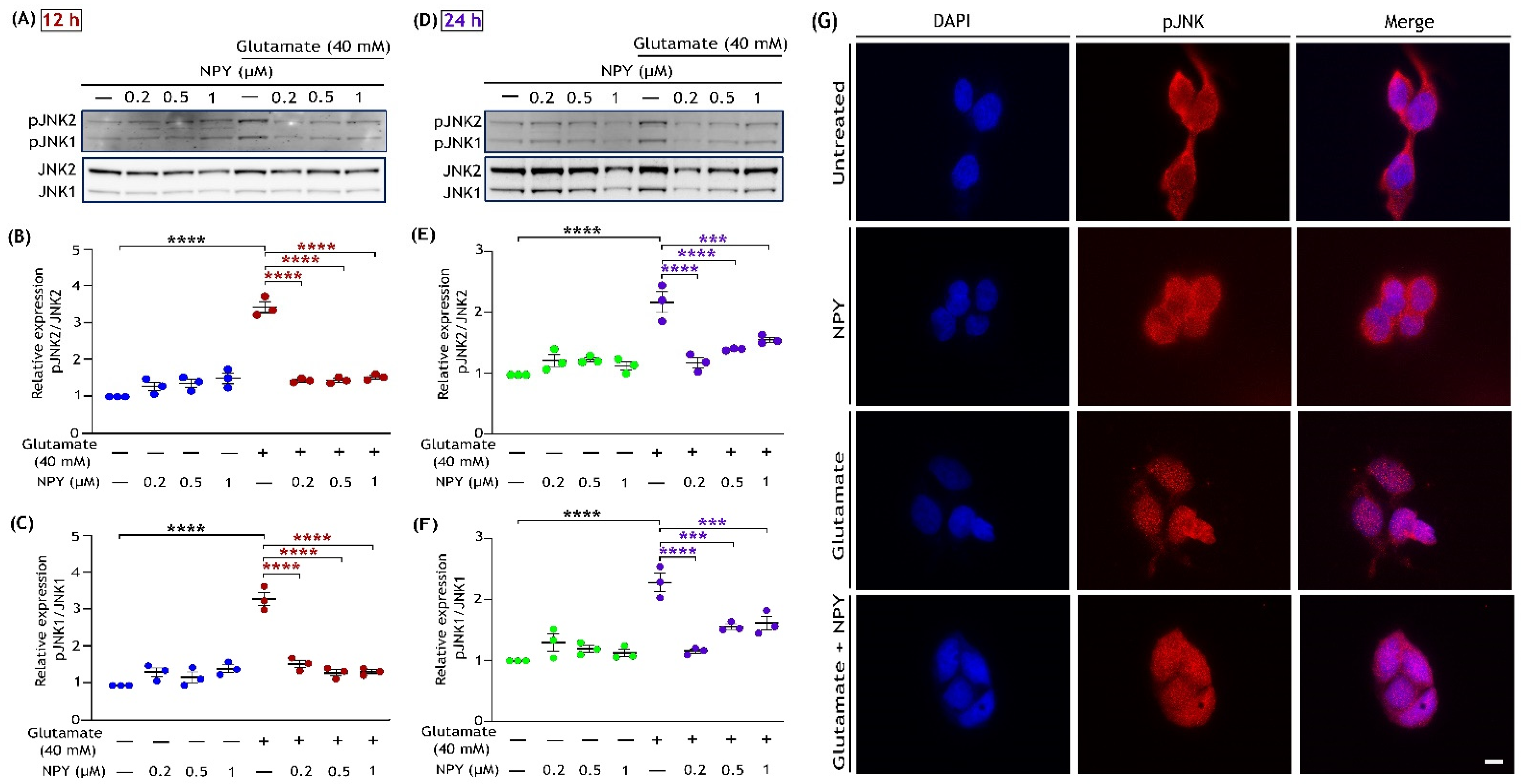
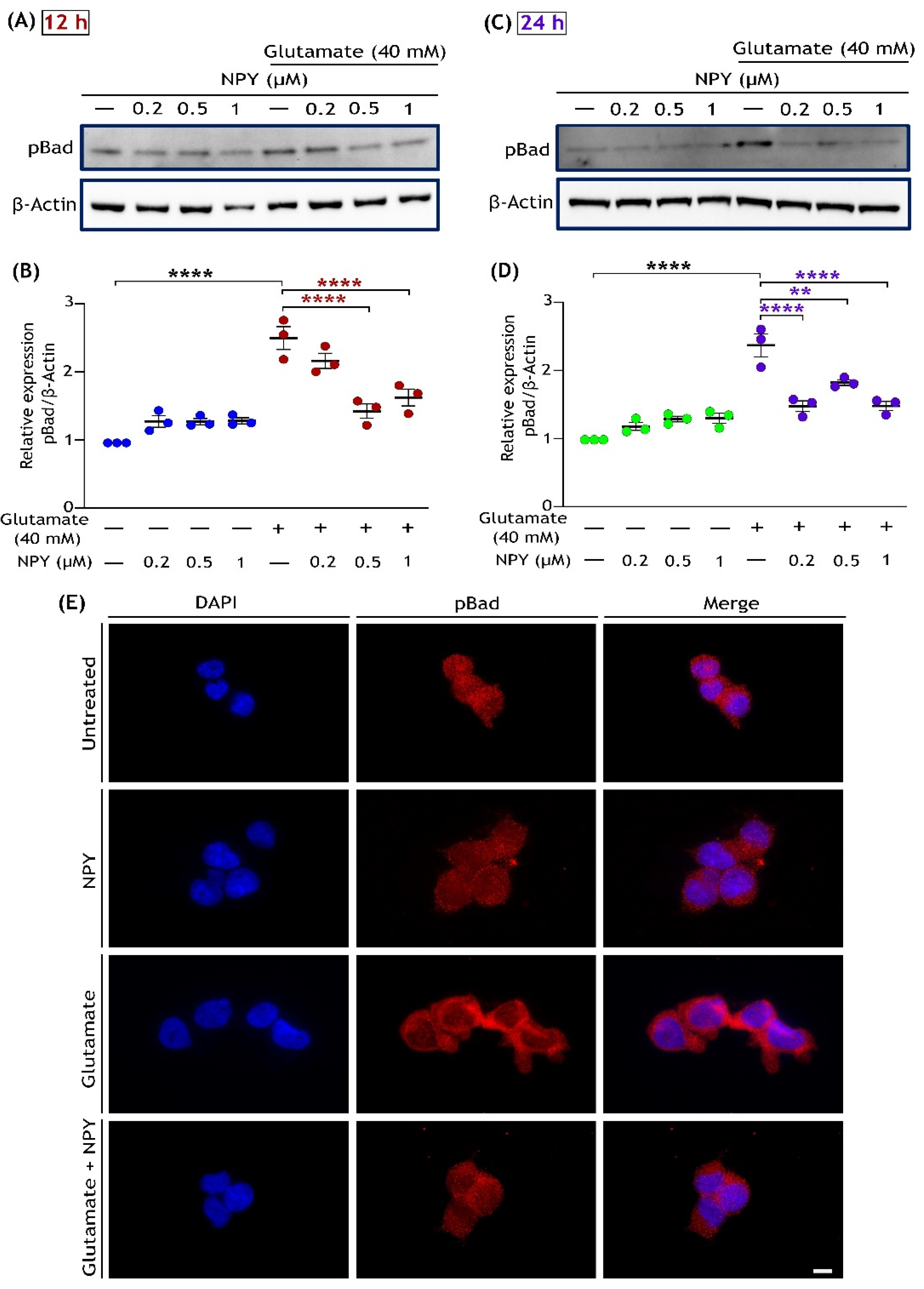
3.3. NPY Downregulates the ERK1/2 Mediated Activation of the JNK/BAD Apoptotic Networks in Glutamate Toxicity
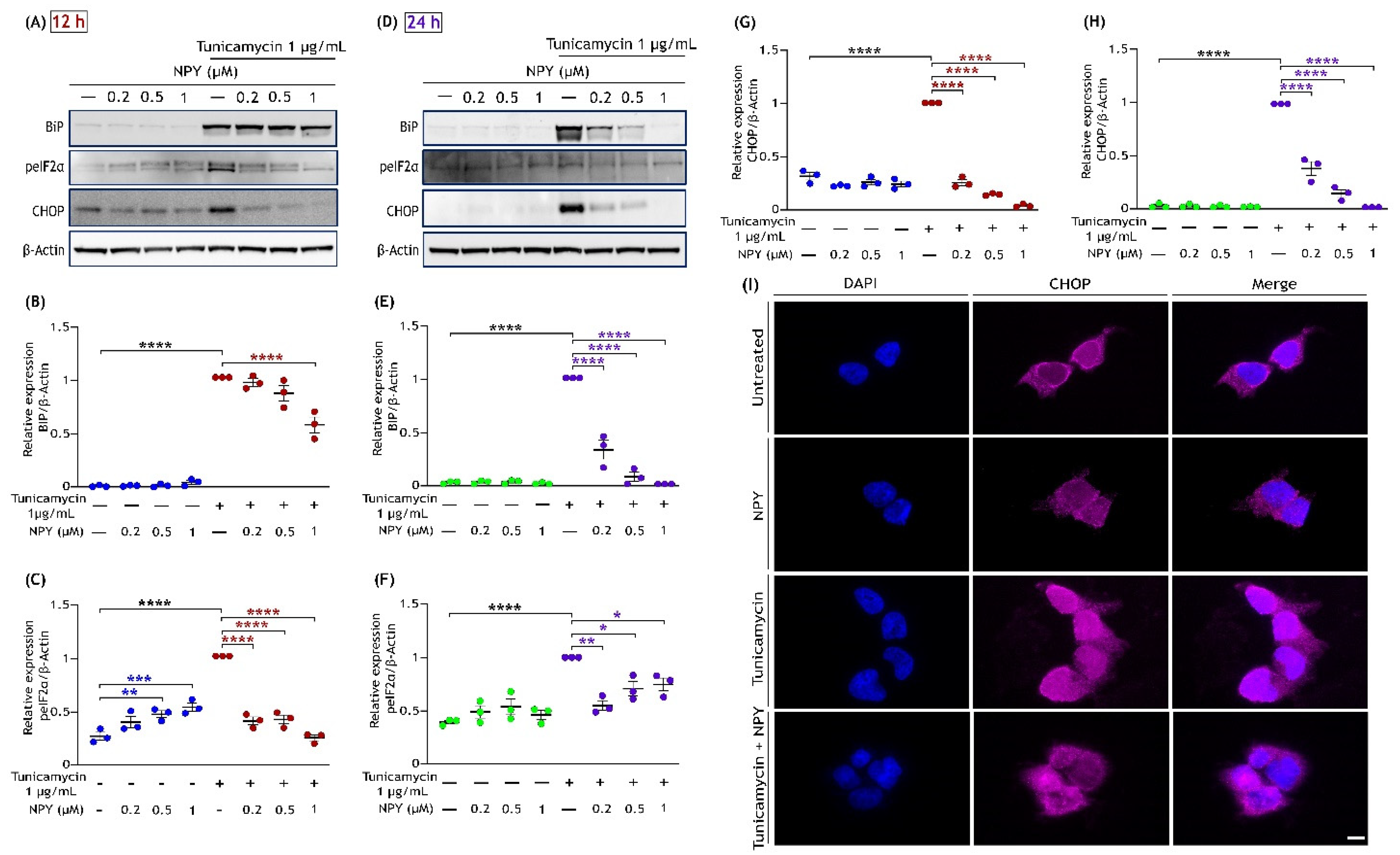
3.4. NPY Attenuates the ER Stress-Mediated Cell Death Induced by Tunicamycin
3.5. NPY Alleviates the Oxidative Damage Caused by Glutamate and Tunicamycin in SH-SY5Y Cells
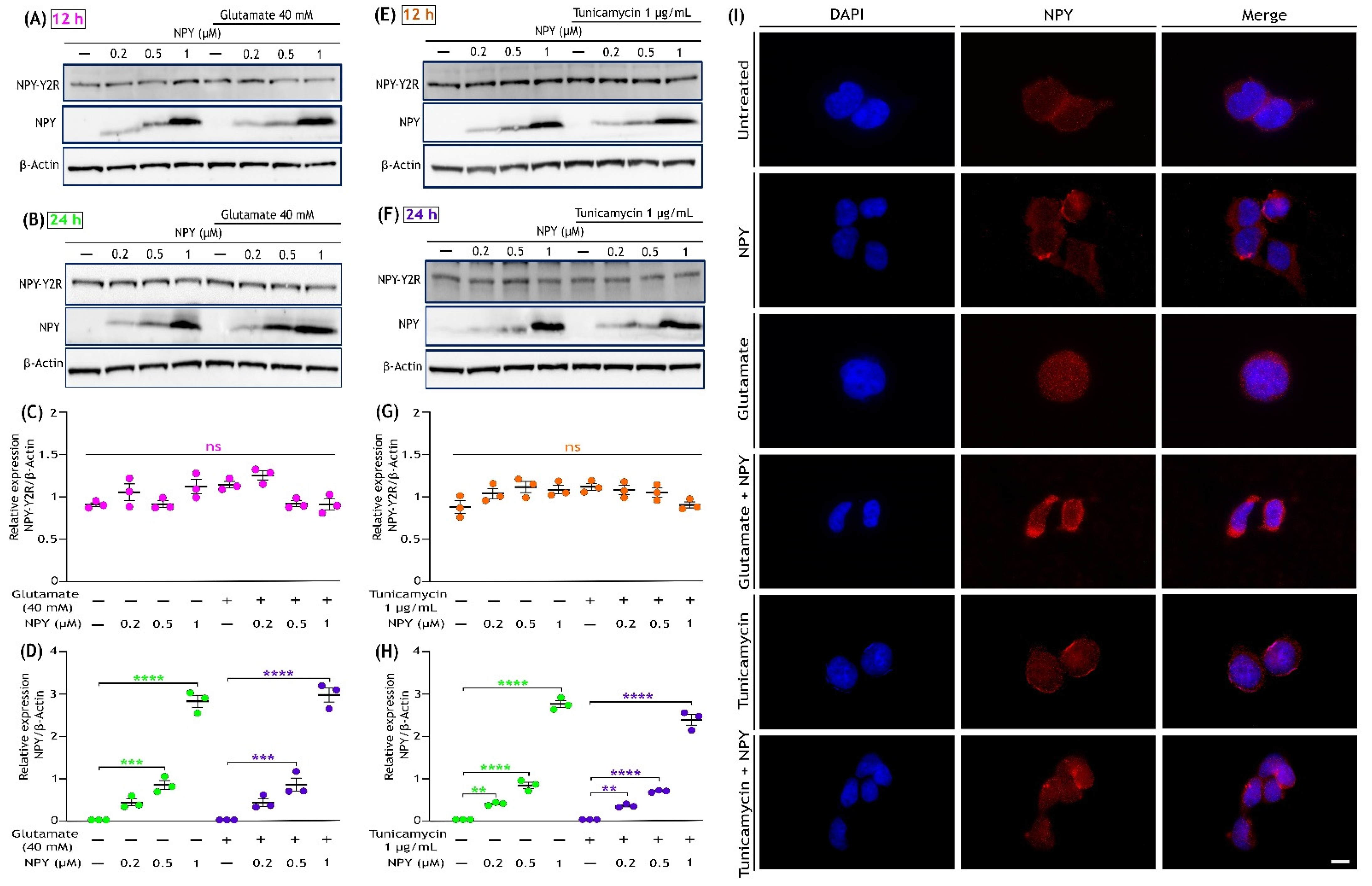
3.6. Glutamate Toxicity or Tunicamycin Does Not Affect NPY Receptor (NPY-Y2R) Expression, and NPY Treatment Increases Its Expression in SH-SY5Y Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schubert, D.; Piasecki, D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J. Neurosci. 2001, 21, 7455–7462. [Google Scholar] [CrossRef]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target Against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef]
- Morris, G.; Puri, B.K.; Walder, K.; Berk, M.; Stubbs, B.; Maes, M.; Carvalho, A.F. The Endoplasmic Reticulum Stress Response in Neuroprogressive Diseases: Emerging Pathophysiological Role and Translational Implications. Mol. Neurobiol. 2018, 55, 8765–8787. [Google Scholar] [CrossRef]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic reticulum-mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci 2022, 135, jcs248534. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.K.; Jeong, S.; Lee, J. The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2022, 23, 5894. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Park, C.; Kim, J.; Ko, S.B.; Choi, Y.K.; Jeong, H.; Woo, H.; Kang, H.; Bang, I.; Kim, S.A.; Yoon, T.Y.; et al. Structural basis of neuropeptide Y signaling through Y1 receptor. Nat. Commun. 2022, 13, 853. [Google Scholar] [CrossRef]
- Shende, P.; Desai, D. Physiological and Therapeutic Roles of Neuropeptide Y on Biological Functions. Adv. Exp. Med. Biol. 2020, 1237, 37–47. [Google Scholar] [CrossRef]
- Decressac, M.; Barker, R.A. Neuropeptide Y and its role in CNS disease and repair. Exp. Neurol. 2012, 238, 265–272. [Google Scholar] [CrossRef]
- Brothers, S.P.; Wahlestedt, C. Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Mol. Med. 2010, 2, 429–439. [Google Scholar] [CrossRef]
- Yi, M.; Li, H.; Wu, Z.; Yan, J.; Liu, Q.; Ou, C.; Chen, M. A Promising Therapeutic Target for Metabolic Diseases: Neuropeptide Y Receptors in Humans. Cell. Physiol. Biochem. 2018, 45, 88–107. [Google Scholar] [CrossRef]
- Silva, A.P.; Xapelli, S.; Grouzmann, E.; Cavadas, C. The putative neuroprotective role of neuropeptide Y in the central nervous system. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 331–347. [Google Scholar] [CrossRef]
- Malva, J.O.; Xapelli, S.; Baptista, S.; Valero, J.; Agasse, F.; Ferreira, R.; Silva, A.P. Multifaces of neuropeptide Y in the brain--neuroprotection, neurogenesis and neuroinflammation. Neuropeptides 2012, 46, 299–308. [Google Scholar] [CrossRef]
- Hansel, D.E.; Eipper, B.A.; Ronnett, G.V. Neuropeptide Y functions as a neuroproliferative factor. Nature 2001, 410, 940–944. [Google Scholar] [CrossRef]
- Aveleira, C.A.; Botelho, M.; Carmo-Silva, S.; Pascoal, J.F.; Ferreira-Marques, M.; Nóbrega, C.; Cortes, L.; Valero, J.; Sousa-Ferreira, L.; Álvaro, A.R.; et al. Neuropeptide Y stimulates autophagy in hypothalamic neurons. Proc. Natl. Acad. Sci. USA 2015, 112, E1642–E1651. [Google Scholar] [CrossRef]
- Silva, A.P.; Pinheiro, P.S.; Carvalho, A.P.; Carvalho, C.M.; Jakobsen, B.; Zimmer, J.; Malva, J.O. Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB J. 2003, 17, 1118–1120. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 2019, 13, 869. [Google Scholar] [CrossRef]
- Croce, N.; Ciotti, M.T.; Gelfo, F.; Cortelli, S.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. Neuropeptide Y protects rat cortical neurons against β-amyloid toxicity and re-establishes synthesis and release of nerve growth factor. ACS Chem. Neurosci. 2012, 3, 312–318. [Google Scholar] [CrossRef]
- Saklani, P.; Khan, H.; Gupta, S.; Kaur, A.; Singh, T.G. Neuropeptides: Potential neuroprotective agents in ischemic injury. Life Sci. 2022, 288, 120186. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.Y.; Chen, W.C.; Shi, Y.C.; Wang, C.M.; Lin, S.; He, H.F. Regulation of neuropeptide Y in body microenvironments and its potential application in therapies: A review. Cell Biosci. 2021, 11, 151. [Google Scholar] [CrossRef]
- Pain, S.; Brot, S.; Gaillard, A. Neuroprotective Effects of Neuropeptide Y against Neurodegenerative Disease. Curr. Neuropharmacol. 2022, 20, 1717–1725. [Google Scholar] [CrossRef]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling Acute ER Stress In Vivo and In Vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef]
- Oda, T.; Kosuge, Y.; Arakawa, M.; Ishige, K.; Ito, Y. Distinct mechanism of cell death is responsible for tunicamycin-induced ER stress in SK-N-SH and SH-SY5Y cells. Neurosci. Res. 2008, 60, 29–39. [Google Scholar] [CrossRef]
- Sun, Z.W.; Zhang, L.; Zhu, S.J.; Chen, W.C.; Mei, B. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull. 2010, 26, 8–16. [Google Scholar] [CrossRef]
- Santos-Carvalho, A.; Elvas, F.; Alvaro, A.R.; Ambrosio, A.F.; Cavadas, C. Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Dis. 2013, 4, e636. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hong, S.H.; Kim, B.; Lee, D.S.; Yu, K.; Lee, K.S. Neuropeptide Y mitigates ER stress-induced neuronal cell death by activating the PI3K-XBP1 pathway. Eur. J. Cell Biol. 2018, 97, 339–348. [Google Scholar] [CrossRef]
- Dheer, Y.; Chitranshi, N.; Gupta, V.; Abbasi, M.; Mirzaei, M.; You, Y.; Chung, R.; Graham, S.L.; Gupta, V. Bexarotene Modulates Retinoid-X-Receptor Expression and Is Protective Against Neurotoxic Endoplasmic Reticulum Stress Response and Apoptotic Pathway Activation. Mol. Neurobiol. 2018, 55, 9043–9056. [Google Scholar] [CrossRef]
- Chitranshi, N.; Dheer, Y.; Gupta, V.; Abbasi, M.; Mirzaei, M.; You, Y.; Chung, R.; Graham, S.L.; Gupta, V. PTPN11 induces endoplasmic stress and apoptosis in SH-SY5Y cells. Neuroscience 2017, 364, 175–189. [Google Scholar] [CrossRef]
- Gupta, V.K.; Gowda, L.R. Alpha-1-proteinase inhibitor is a heparin binding serpin: Molecular interactions with the Lys rich cluster of helix-F domain. Biochimie 2008, 90, 749–761. [Google Scholar] [CrossRef]
- Basavarajappa, D.K.; Gupta, V.K.; Dighe, R.; Rajala, A.; Rajala, R.V. Phosphorylated Grb14 is an endogenous inhibitor of retinal protein tyrosine phosphatase, 1B, and light-dependent activation of Src phosphorylates Grb14. Mol. Cell Biol. 2011, 31, 3975–3987. [Google Scholar] [CrossRef]
- Gupta, V.K.; Rajala, A.; Rajala, R.V. Insulin receptor regulates photoreceptor CNG channel activity. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1363–E1372. [Google Scholar] [CrossRef]
- Jiang, Q.; Gu, Z.; Zhang, G.; Jing, G. Diphosphorylation and involvement of extracellular signal-regulated kinases (ERK1/2) in glutamate-induced apoptotic-like death in cultured rat cortical neurons. Brain Res. 2000, 857, 71–77. [Google Scholar] [CrossRef]
- Vanhoutte, P.; Barnier, J.V.; Guibert, B.; Pages, C.; Besson, M.J.; Hipskind, R.A.; Caboche, J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol. Cell Biol. 1999, 19, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Szydlowska, K.; Gozdz, A.; Dabrowski, M.; Zawadzka, M.; Kaminska, B. Prolonged activation of ERK triggers glutamate-induced apoptosis of astrocytes: Neuroprotective effect of FK506. J. Neurochem. 2010, 113, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.V.; Rzhaninova, A.A.; Goldshtein, D.V.; Boldyrev, A.A. Expression of NMDA receptors in multipotent stromal cells of human adipose tissue under conditions of retinoic acid-induced differentiation. Bull. Exp. Biol. Med. 2007, 144, 626–629. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.R.; Hu, L.S.; Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. (Engl.) 2010, 123, 1086–1092. [Google Scholar]
- Centeno, C.; Repici, M.; Chatton, J.Y.; Riederer, B.M.; Bonny, C.; Nicod, P.; Price, M.; Clarke, P.G.; Papa, S.; Franzoso, G.; et al. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007, 14, 240–253. [Google Scholar] [CrossRef]
- Donovan, N.; Becker, E.B.; Konishi, Y.; Bonni, A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 2002, 277, 40944–40949. [Google Scholar] [CrossRef]
- Konishi, Y.; Lehtinen, M.; Donovan, N.; Bonni, A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol. Cell 2002, 9, 1005–1016. [Google Scholar] [CrossRef]
- Kamada, H.; Nito, C.; Endo, H.; Chan, P.H. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2007, 27, 521–533. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130 Pt 12, 3111–3123. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Kobayashi, Y.; Chen, C.; Motoyama, N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid. Redox Signal. 2005, 7, 752–760. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.L.; Vaughan, P.F.; Beck-Sickinger, A.G.; Peers, C. Inhibition of Ca2+ channel currents in human neuroblastoma (SH-SY5Y) cells by neuropeptide Y and a novel cyclic neuropeptide Y analogue. Neuropharmacology 1995, 34, 1507–1514. [Google Scholar] [CrossRef]
- Hofliger, M.M.; Castejon, G.L.; Kiess, W.; Beck Sickinger, A.G. Novel cell line selectively expressing neuropeptide Y-Y2 receptors. J. Recept. Signal Transduct. Res. 2003, 23, 351–360. [Google Scholar] [CrossRef]
- Connor, M.; Yeo, A.; Henderson, G. Neuropeptide Y Y2 receptor and somatostatin sst2 receptor coupling to mobilization of intracellular calcium in SH-SY5Y human neuroblastoma cells. Br. J. Pharmacol. 1997, 120, 455–463. [Google Scholar] [CrossRef][Green Version]
- Chatenet, D.; Cescato, R.; Waser, B.; Erchegyi, J.; Rivier, J.E.; Reubi, J.C. Novel dimeric DOTA-coupled peptidic Y1-receptor antagonists for targeting of neuropeptide Y receptor-expressing cancers. EJNMMI Res. 2011, 1, 21. [Google Scholar] [CrossRef]
- Croce, N.; Dinallo, V.; Ricci, V.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. Neuroprotective effect of neuropeptide Y against beta-amyloid 25–35 toxicity in SH-SY5Y neuroblastoma cells is associated with increased neurotrophin production. Neurodegener. Dis. 2011, 8, 300–309. [Google Scholar] [CrossRef]
- Decressac, M.; Pain, S.; Chabeauti, P.Y.; Frangeul, L.; Thiriet, N.; Herzog, H.; Vergote, J.; Chalon, S.; Jaber, M.; Gaillard, A. Neuroprotection by neuropeptide Y in cell and animal models of Parkinson’s disease. Neurobiol. Aging 2012, 33, 2125–2137. [Google Scholar] [CrossRef]
- Decressac, M.; Wright, B.; Tyers, P.; Gaillard, A.; Barker, R.A. Neuropeptide Y modifies the disease course in the R6/2 transgenic model of Huntington’s disease. Exp. Neurol. 2010, 226, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Domanskyi, A.; Parlato, R. Oxidative Stress in Neurodegenerative Diseases. Antioxidants 2022, 11, 504. [Google Scholar] [CrossRef]
- Simoes, A.P.; Silva, C.G.; Marques, J.M.; Pochmann, D.; Porciuncula, L.O.; Ferreira, S.; Oses, J.P.; Beleza, R.O.; Real, J.I.; Kofalvi, A.; et al. Glutamate-induced and NMDA receptor-mediated neurodegeneration entails P2Y1 receptor activation. Cell Death Dis. 2018, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc(-) cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Klein, M.; Methner, A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity. J. Neurochem. 2006, 98, 916–925. [Google Scholar] [CrossRef]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Poddar, R.; Paul, S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009, 110, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Zhu, J.; Kulich, S.M.; Chu, C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: Implications for Parkinson’s disease. Autophagy 2008, 4, 770–782. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Cheung, A.; Newland, P.L.; Zaben, M.; Attard, G.S.; Gray, W.P. Intracellular nitric oxide mediates neuroproliferative effect of neuropeptide y on postnatal hippocampal precursor cells. J. Biol. Chem. 2012, 287, 20187–20196. [Google Scholar] [CrossRef]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 2000, 275, 35778–35785. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal. Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Fan, M.; Chambers, T.C. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist. Updates 2001, 4, 253–267. [Google Scholar] [CrossRef]
- Deng, Y.; Ren, X.; Yang, L.; Lin, Y.; Wu, X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003, 115, 61–70. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apop.ptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qian, Y.; Li, J.; Zhou, X.; Xu, H.; Yan, J.; Xiang, J.; Yuan, X.; Sun, B.; Sisodia, S.S.; et al. BAD-mediated neuronal apoptosis and neuroinflammation contribute to Alzheimer’s disease pathology. iScience 2021, 24, 102942. [Google Scholar] [CrossRef]
- Jiang, L.; Luo, M.; Liu, D.; Chen, B.; Zhang, W.; Mai, L.; Zeng, J.; Huang, N.; Huang, Y.; Mo, X.; et al. BAD overexpression inhibits cell growth and induces apoptosis via mitochondrial-dependent pathway in non-small cell lung cancer. Cancer Cell Int. 2013, 13, 53. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef]
- Chen, G.; Fan, Z.; Wang, X.; Ma, C.; Bower, K.A.; Shi, X.; Ke, Z.J.; Luo, J. Brain-derived neurotrophic factor suppresses tunicamycin-induced upregulation of CHOP in neurons. J. Neurosci. Res. 2007, 85, 1674–1684. [Google Scholar] [CrossRef]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006, 13, 363–373. [Google Scholar] [CrossRef]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
- Jauhiainen, A.; Thomsen, C.; Strombom, L.; Grundevik, P.; Andersson, C.; Danielsson, A.; Andersson, M.K.; Nerman, O.; Rorkvist, L.; Stahlberg, A.; et al. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS ONE 2012, 7, e33208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 629–640. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Domin, H. Neuropeptide Y Y2 and Y5 receptors as potential targets for neuroprotective and antidepressant therapies: Evidence from preclinical studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110349. [Google Scholar] [CrossRef]
- Duarte-Neves, J.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 2016, 95, 210–224. [Google Scholar] [CrossRef]
- Ramos, B.; Baglietto-Vargas, D.; del Rio, J.C.; Moreno-Gonzalez, I.; Santa-Maria, C.; Jimenez, S.; Caballero, C.; Lopez-Tellez, J.F.; Khan, Z.U.; Ruano, D.; et al. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1658–1672. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef]
- Miyauchi, H.; Minamino, T.; Tateno, K.; Kunieda, T.; Toko, H.; Komuro, I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004, 23, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Dolado, I.; Nebreda, A.R. AKT and oxidative stress team up to kill cancer cells. Cancer Cell 2008, 14, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef]
- Los, M.; Maddika, S.; Erb, B.; Schulze-Osthoff, K. Switching Akt: From survival signaling to deadly response. Bioessays 2009, 31, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Shi, C.; Viccaro, K.; Lee, H.G.; Shah, K. Cdk5-Foxo3 axis: Initially neuroprotective, eventually neurodegenerative in Alzheimer’s disease models. J. Cell Sci. 2016, 129, 1815–1830. [Google Scholar] [CrossRef]
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. (Lausanne) 2013, 4, 5. [Google Scholar] [CrossRef]
- Vezzani, A.; Sperk, G. Overexpression of NPY and Y2 r.receptors in epileptic brain tissue: An endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides 2004, 38, 245–252. [Google Scholar] [CrossRef]
- Schwarzer, C.; Kofler, N.; Sperk, G. Up-regulation of neuropeptide Y-Y2 receptors in an animal model of temporal lobe epilepsy. Mol. Pharmacol. 1998, 53, 6–13. [Google Scholar] [CrossRef]
- Chen, X.; Westfall, T.C. Modulation of intracellular calcium transients and dopamine release by neuropeptide Y in PC-12 cells. Am. J. Physiol. 1994, 266 Pt 1, C784–C793. [Google Scholar] [CrossRef] [PubMed]
- Agasse, F.; Bernardino, L.; Kristiansen, H.; Christiansen, S.H.; Ferreira, R.; Silva, B.; Grade, S.; Woldbye, D.P.; Malva, J.O. Neuropeptide Y promotes neurogenesis in murine subventricular zone. Stem Cells 2008, 26, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Lecat, S.; Belemnaba, L.; Galzi, J.L.; Bucher, B. Neuropeptide Y receptor mediates activation of ERK1/2 via transactivation of the IGF receptor. Cell Signal. 2015, 27, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Santos-Carvalho, A.; Alvaro, A.R.; Martins, J.; Ambrosio, A.F.; Cavadas, C. Emerging novel roles of neuropeptide Y in the retina: From neuromodulation to neuroprotection. Prog. Neurobiol. 2014, 112, 70–79. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palanivel, V.; Gupta, V.; Mirshahvaladi, S.S.O.; Sharma, S.; Gupta, V.; Chitranshi, N.; Mirzaei, M.; Graham, S.L.; Basavarajappa, D. Neuroprotective Effects of Neuropeptide Y on Human Neuroblastoma SH-SY5Y Cells in Glutamate Excitotoxicity and ER Stress Conditions. Cells 2022, 11, 3665. https://doi.org/10.3390/cells11223665
Palanivel V, Gupta V, Mirshahvaladi SSO, Sharma S, Gupta V, Chitranshi N, Mirzaei M, Graham SL, Basavarajappa D. Neuroprotective Effects of Neuropeptide Y on Human Neuroblastoma SH-SY5Y Cells in Glutamate Excitotoxicity and ER Stress Conditions. Cells. 2022; 11(22):3665. https://doi.org/10.3390/cells11223665
Chicago/Turabian StylePalanivel, Viswanthram, Vivek Gupta, Seyed Shahab Oddin Mirshahvaladi, Samridhi Sharma, Veer Gupta, Nitin Chitranshi, Mehdi Mirzaei, Stuart L Graham, and Devaraj Basavarajappa. 2022. "Neuroprotective Effects of Neuropeptide Y on Human Neuroblastoma SH-SY5Y Cells in Glutamate Excitotoxicity and ER Stress Conditions" Cells 11, no. 22: 3665. https://doi.org/10.3390/cells11223665
APA StylePalanivel, V., Gupta, V., Mirshahvaladi, S. S. O., Sharma, S., Gupta, V., Chitranshi, N., Mirzaei, M., Graham, S. L., & Basavarajappa, D. (2022). Neuroprotective Effects of Neuropeptide Y on Human Neuroblastoma SH-SY5Y Cells in Glutamate Excitotoxicity and ER Stress Conditions. Cells, 11(22), 3665. https://doi.org/10.3390/cells11223665