
Higher Circulating Trimethylamine N-Oxide Aggravates Cognitive Impairment Probably via Downregulating Hippocampal SIRT1 in Vascular Dementia Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. TMAO Treatment and Experimental Model
2.3. Determination of TMAO in Plasma and Hippocampal Samples via High-Performance Liquid Chromatography–Tandem Mass Spectrometry (HPLC–MS/MS)
2.4. Open Field Test (OFT)
2.5. Morris Water Maze Test (MWM)
2.6. Nissl Staining
2.7. Immunofluorescence
2.8. Immunohistochemistry (IHC)
2.9. Golgi Staining
2.10. Oxidative Stress Parameters Measurement
2.11. Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
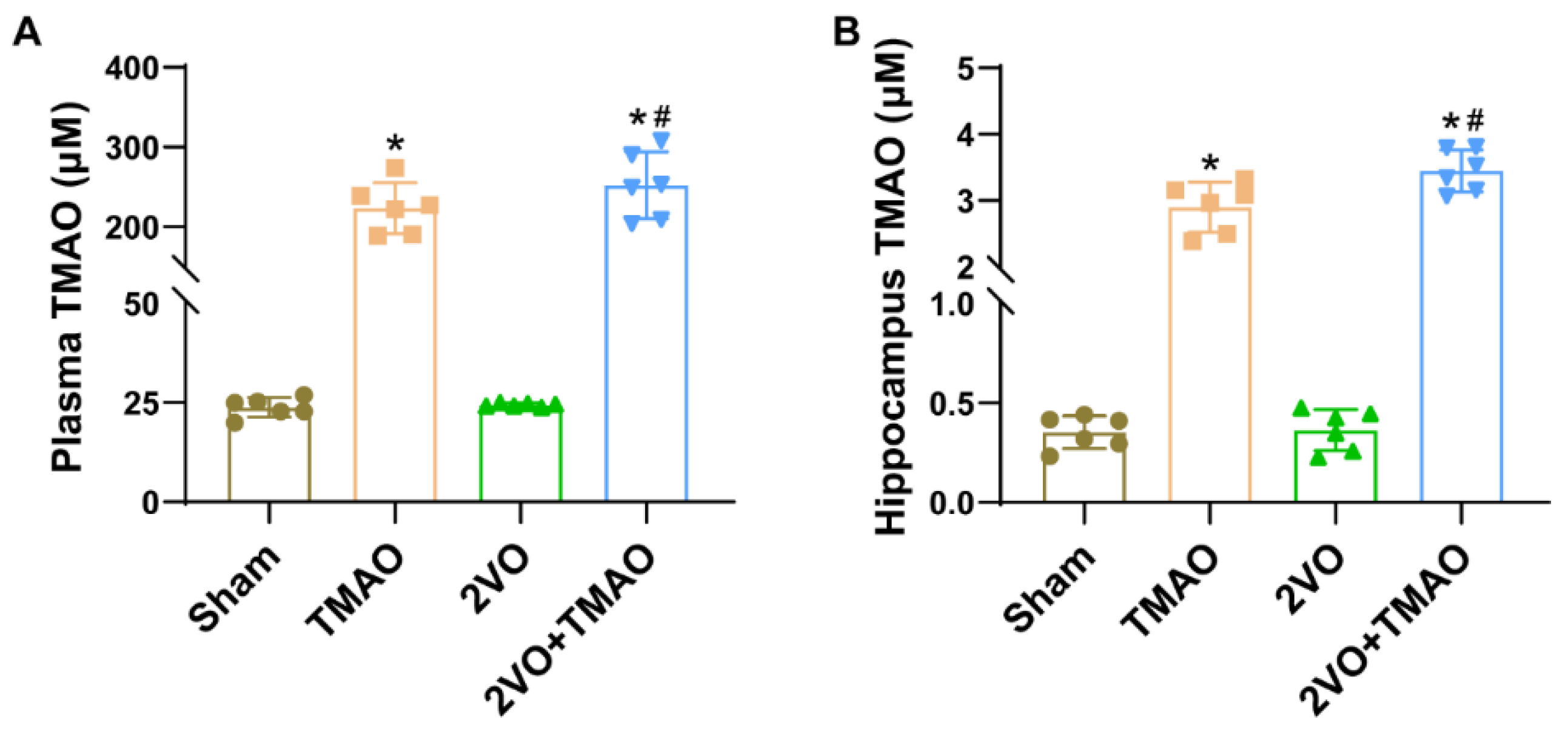
3.1. The Levels of TMAO Increased in TMAO-Treated Rats after 2VO Surgery
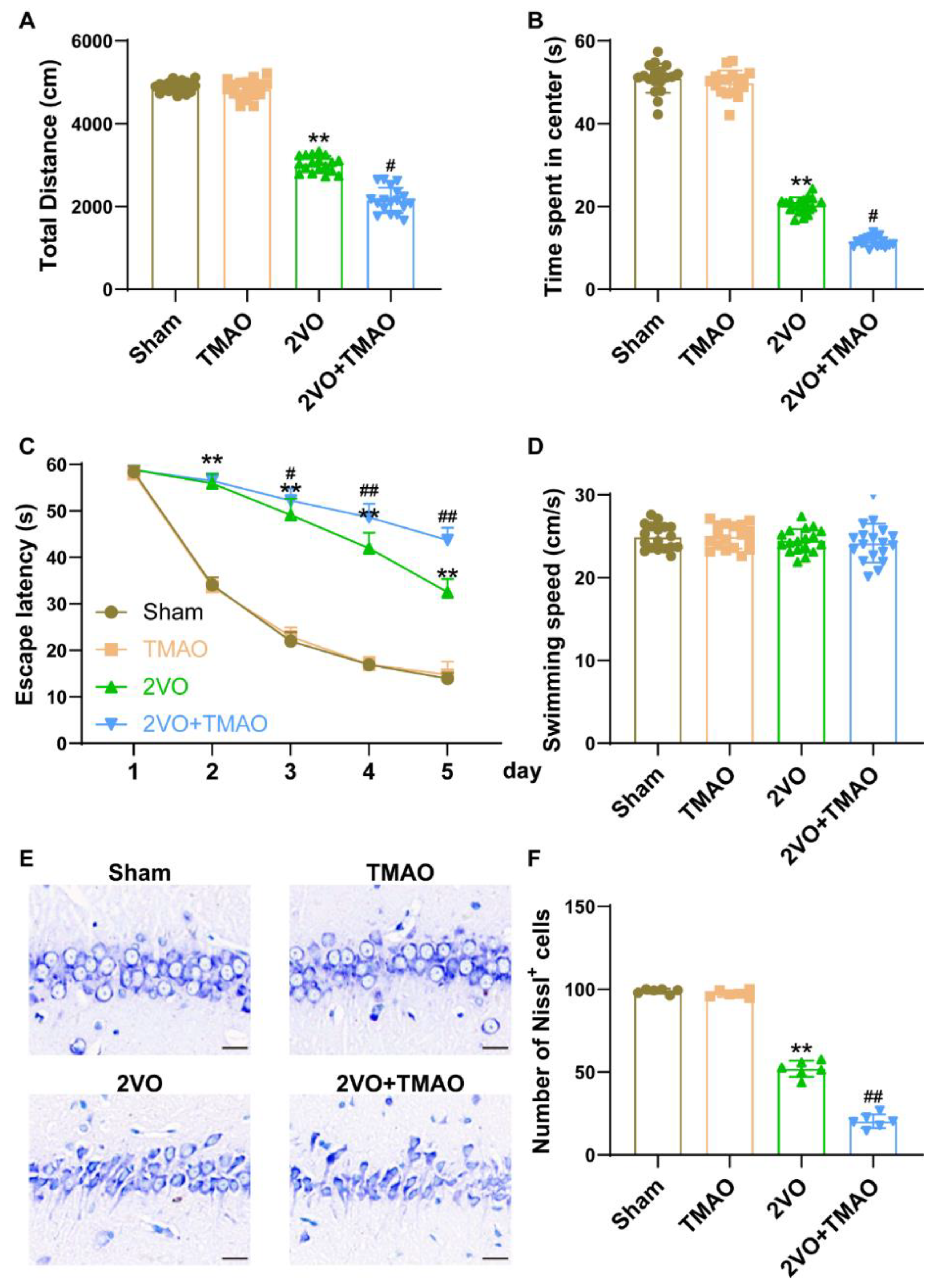
3.2. TMAO Aggravated Cognitive Function Deficits in 2VO Rats
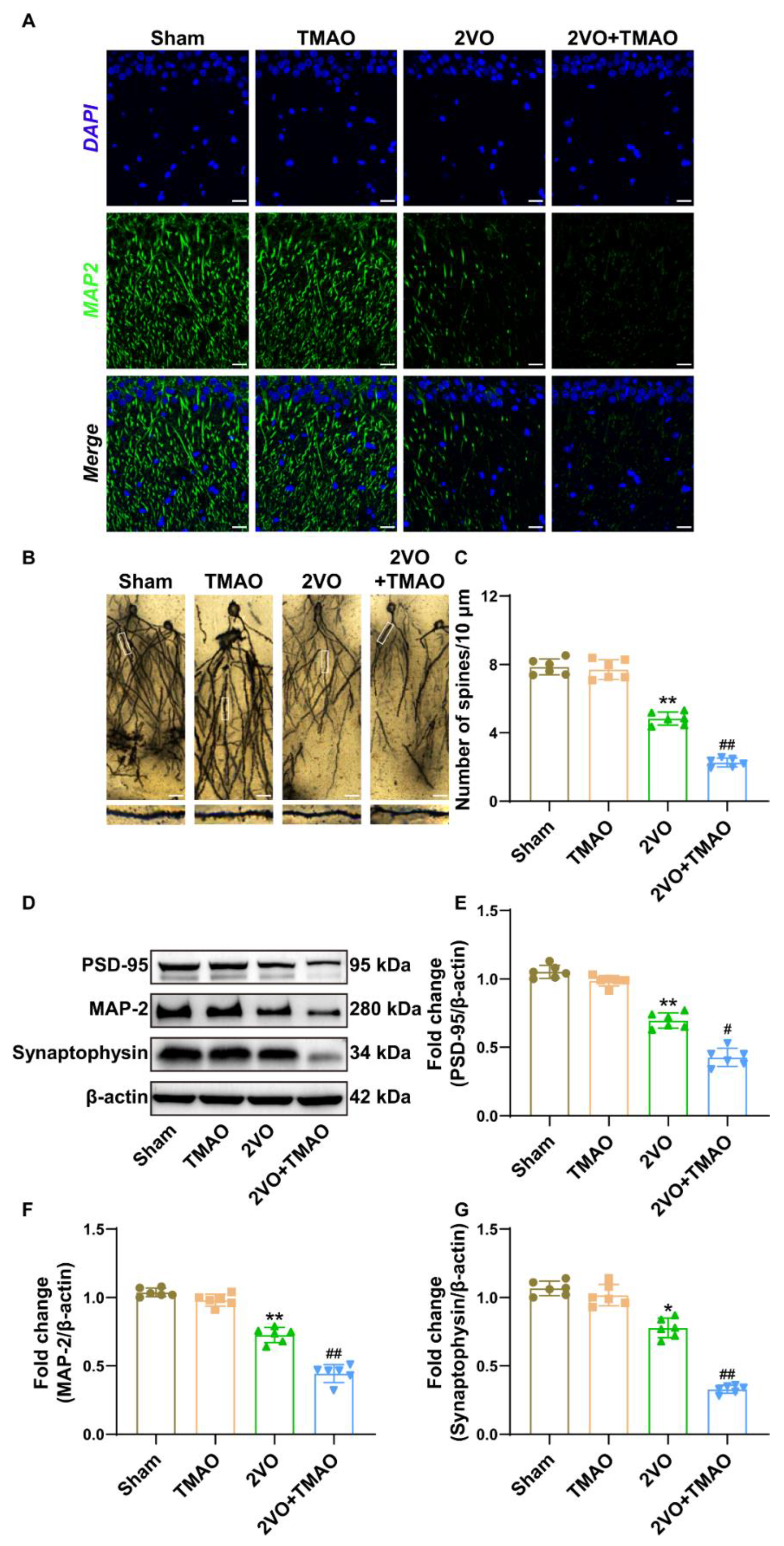
3.3. TMAO Attenuated Hippocampal Synaptic Plasticity within 2VO Rats
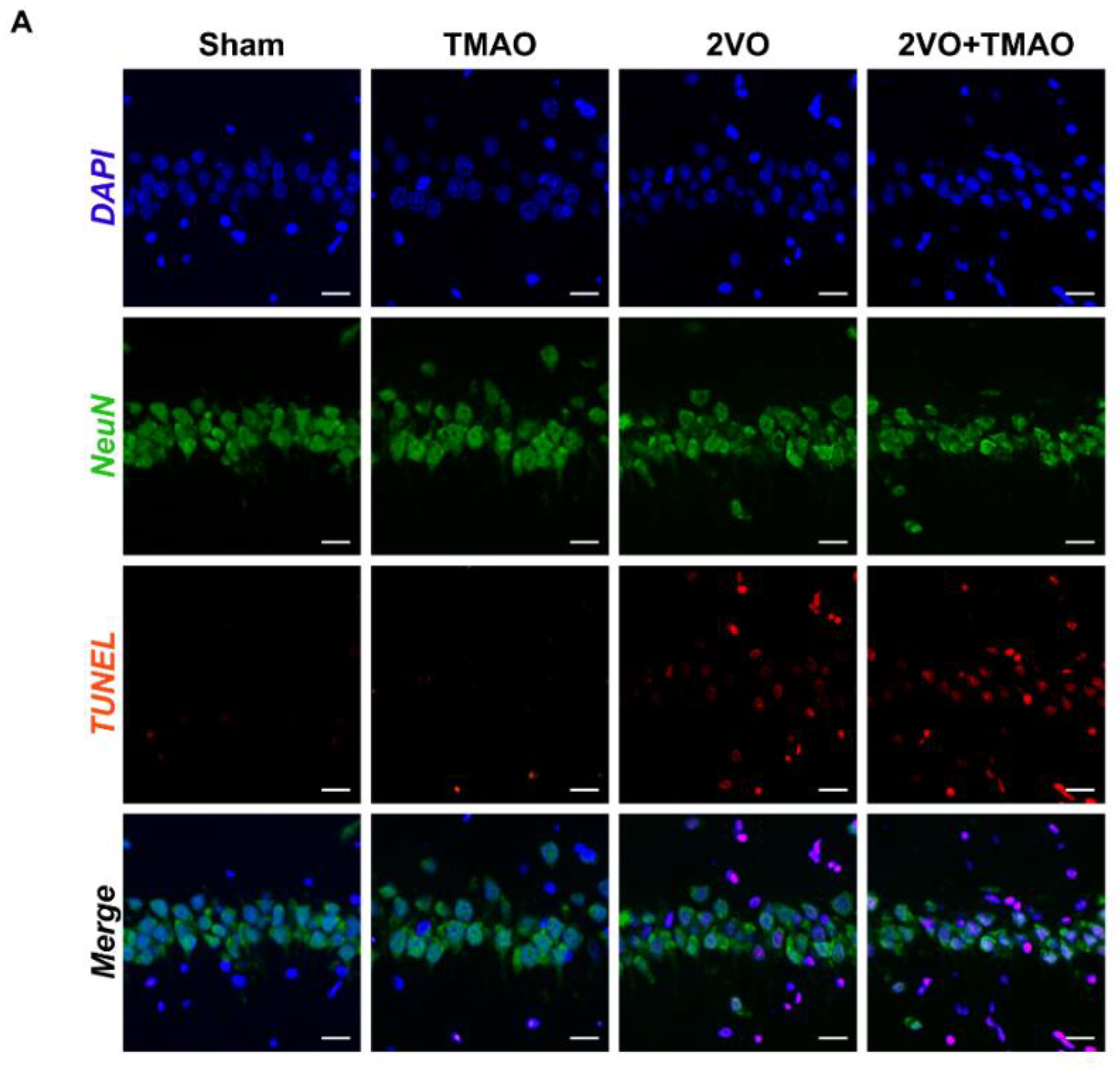
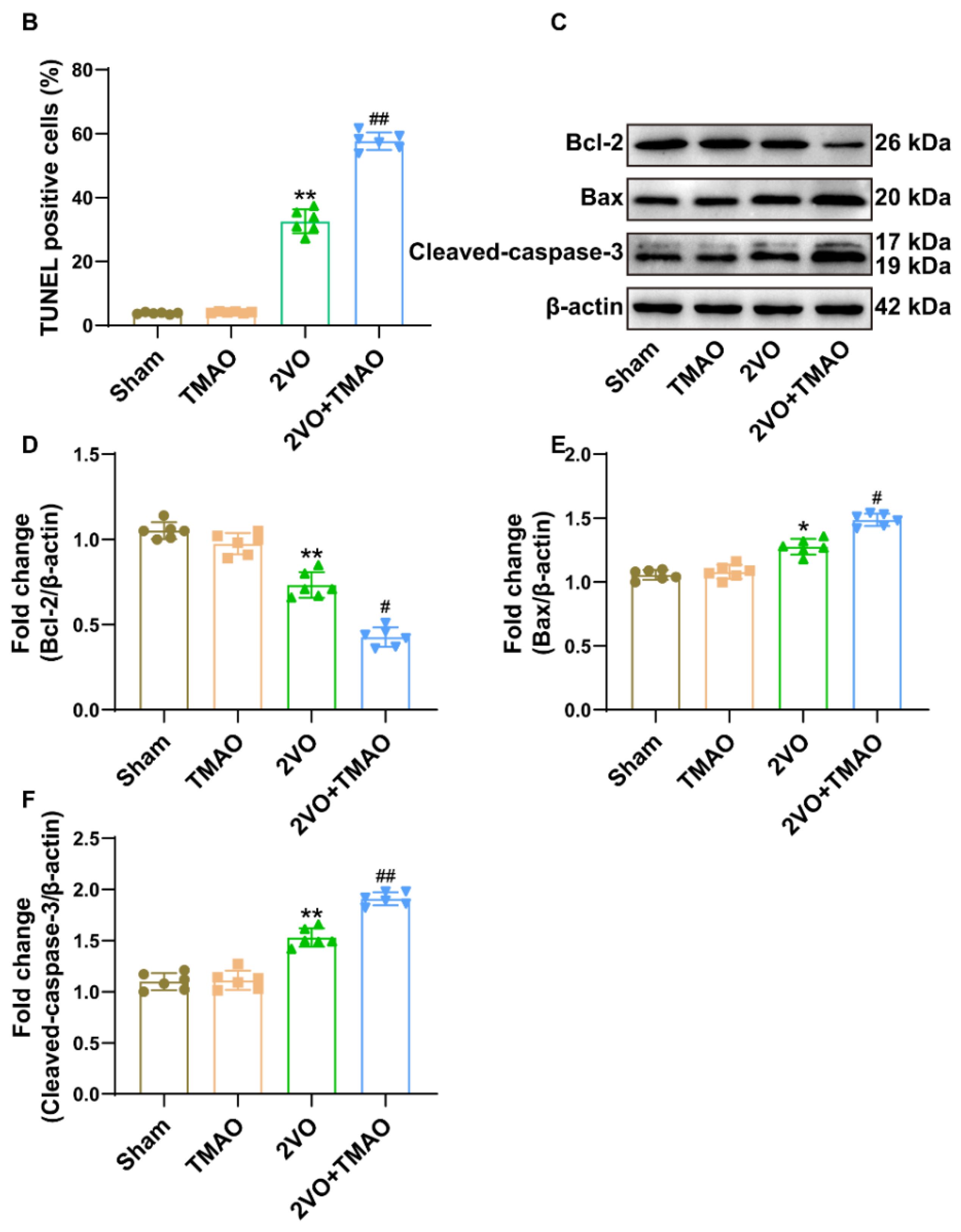
3.4. TMAO Induced Neuronal Apoptosis within 2VO Rats
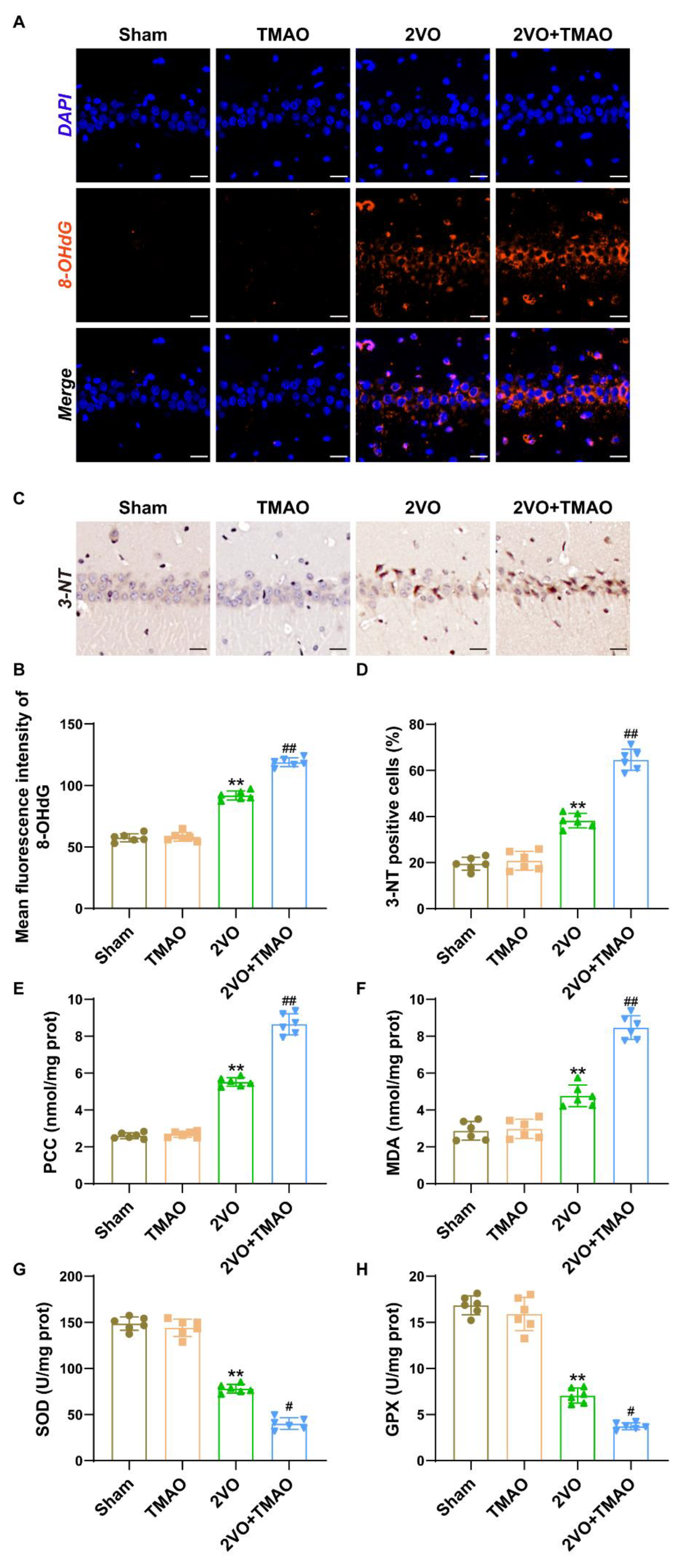
3.5. TMAO Promoted Hippocampal Oxidative Stress within 2VO Rats
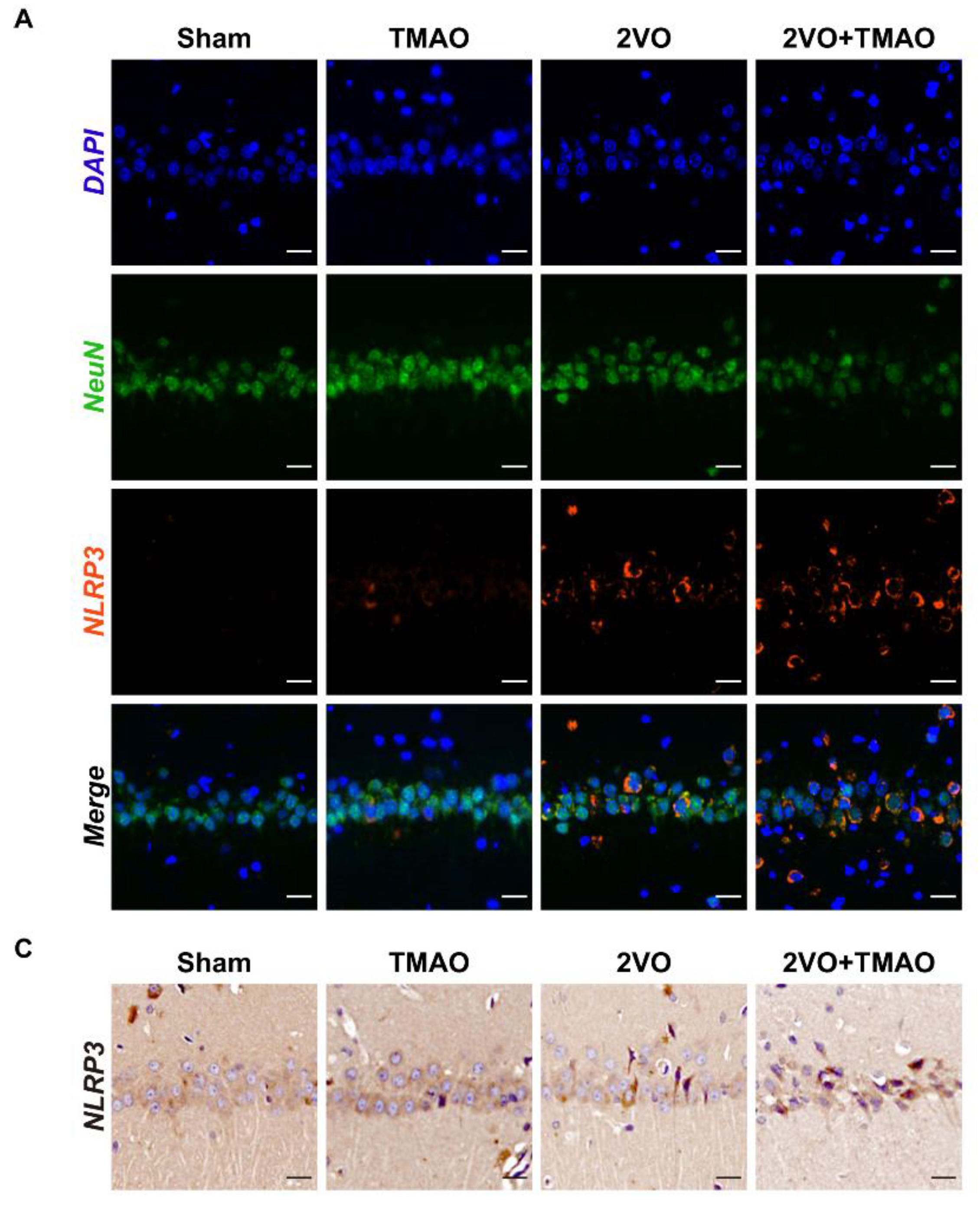
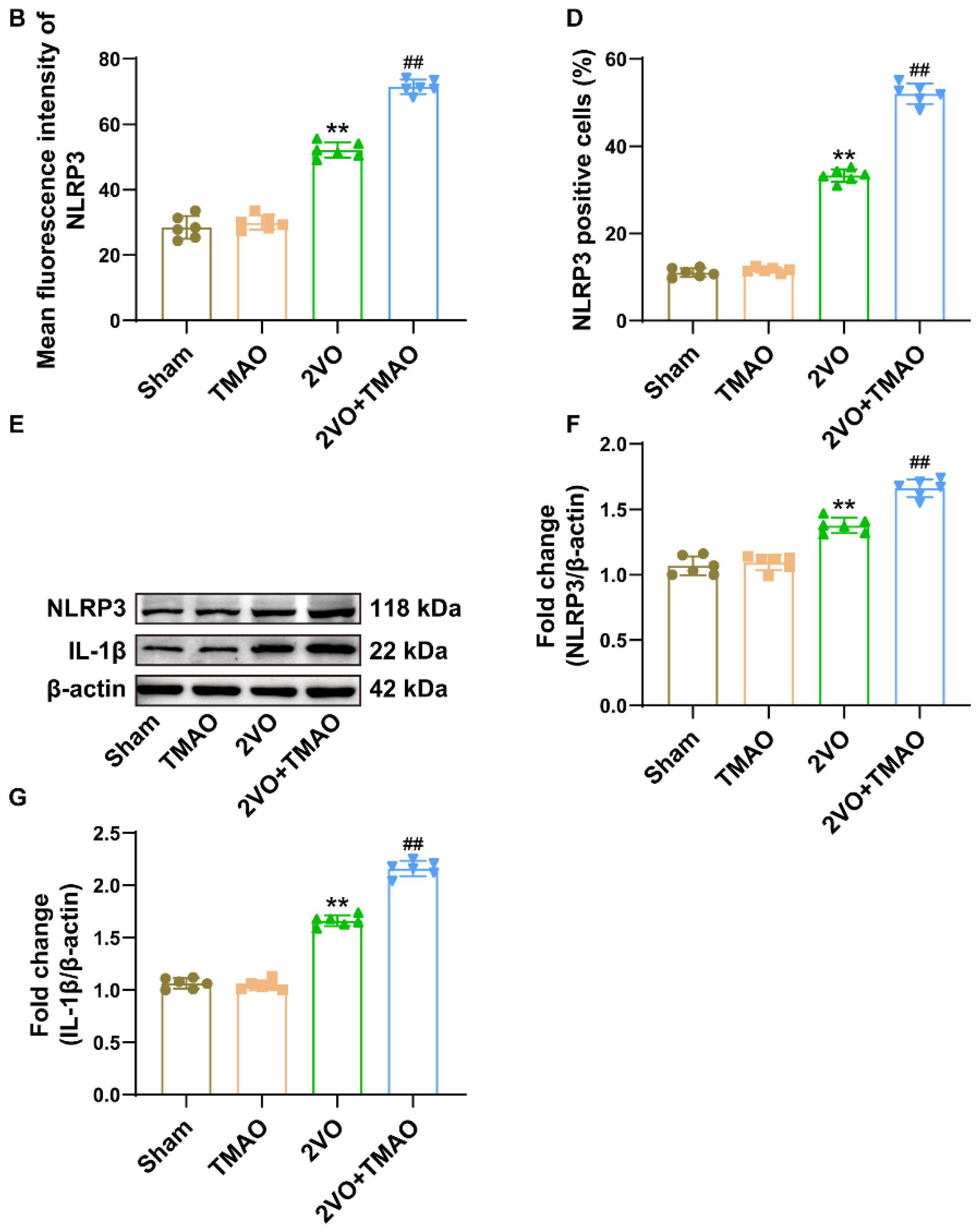
3.6. TMAO Drove Hippocampal NLRP3 Inflammasome Activation within 2VO Rats
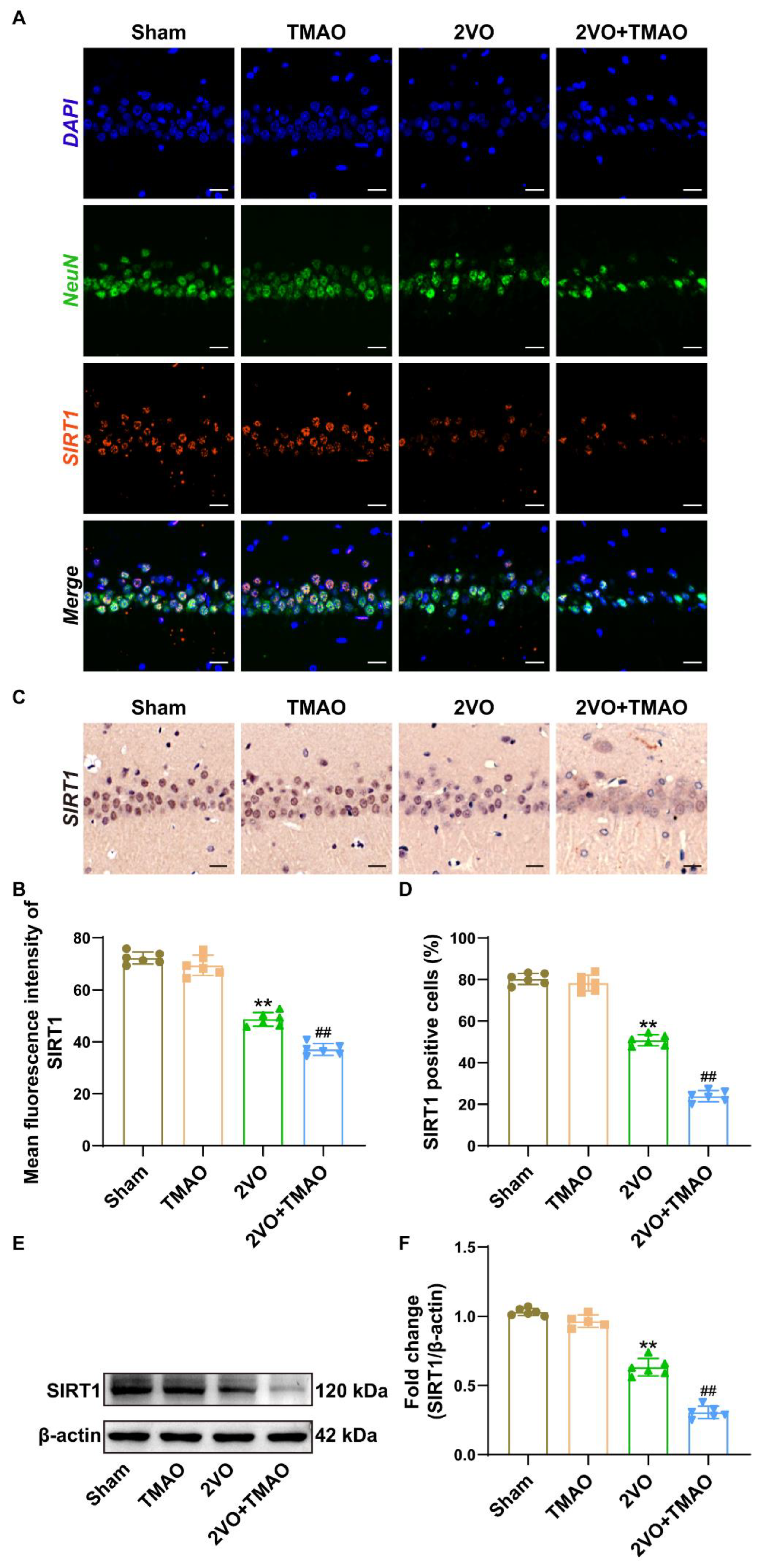
3.7. TMAO Decreased Hippocampal SIRT1 Expression within 2VO Rats
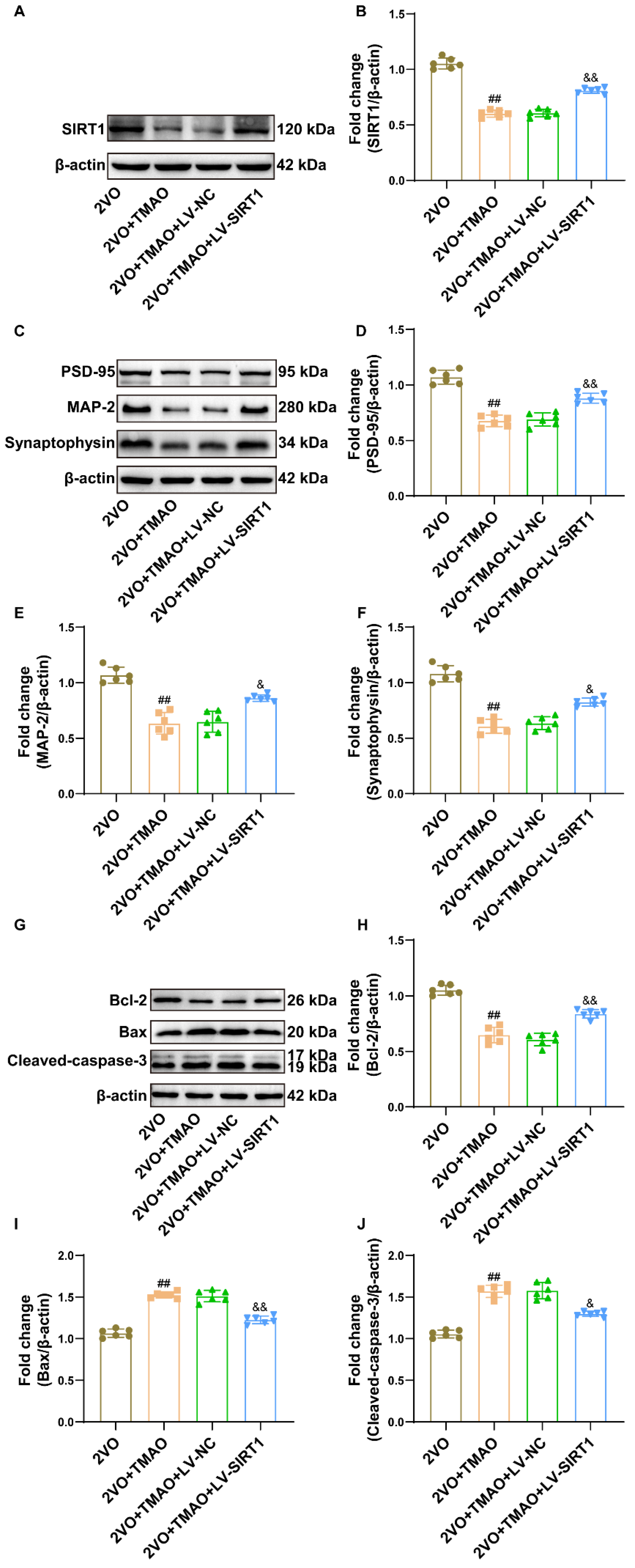
3.8. TMAO Reduced Synaptic Plasticity and Promoted Neuronal Apoptosis by Inhibiting SIRT1 within 2VO Rats
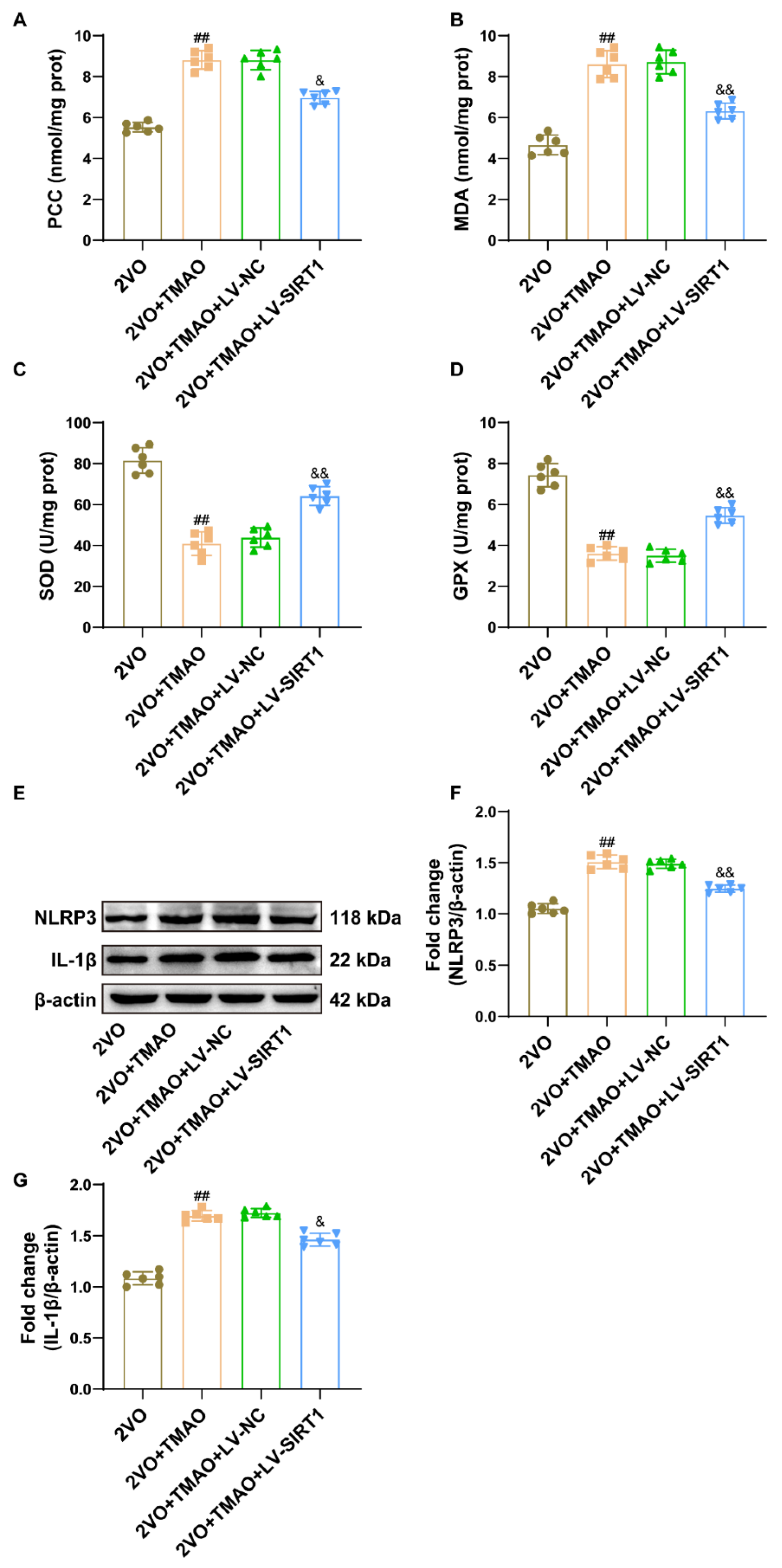
3.9. TMAO Promoted Neuronal Oxidative Stress and NLRP3 Inflammasome Signaling through a SIRT1-Based Pathway within 2VO Rats
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, A.; Kalaria, R.N.; Corbett, A.; Ballard, C. Update on Vascular Dementia. J. Geriatr. Psychiatry Neurol. 2016, 29, 281–301. [Google Scholar]
- Kalaria, R.N.; Akinyemi, R.; Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta 2016, 1862, 915–925. [Google Scholar]
- Wolters, F.J.; Ikram, M.A. Epidemiology of Vascular Dementia. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1542–1549. [Google Scholar]
- Rizzi, L.; Rosset, I.; Roriz-Cruz, M. Global epidemiology of dementia: Alzheimer’s and vascular types. BioMed Res. Int. 2014, 2014, 908915. [Google Scholar]
- Choi, D.H.; Lee, K.H.; Kim, J.H.; Seo, J.H.; Kim, H.Y.; Shin, C.Y.; Han, J.S.; Han, S.H.; Kim, Y.S.; Lee, J. NADPH oxidase 1, a novel molecular source of ROS in hippocampal neuronal death in vascular dementia. Antioxid. Redox Signal. 2014, 21, 533–550. [Google Scholar]
- Rosenberg, G.A.; Bjerke, M.; Wallin, A. Multimodal markers of inflammation in the subcortical ischemic vascular disease type of vascular cognitive impairment. Stroke 2014, 45, 1531–1538. [Google Scholar]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. TEM 2014, 25, 138–145. [Google Scholar]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar]
- García, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3863–3875. [Google Scholar]
- Yao, H.; Tao, X.; Xu, L.; Qi, Y.; Yin, L.; Han, X.; Xu, Y.; Zheng, L.; Peng, J. Dioscin alleviates non-alcoholic fatty liver disease through adjusting lipid metabolism via SIRT1/AMPK signaling pathway. Pharmacol. Res. 2018, 131, 51–60. [Google Scholar]
- Zhou, C.C.; Yang, X.; Hua, X.; Liu, J.; Fan, M.B.; Li, G.Q.; Song, J.; Xu, T.Y.; Li, Z.Y.; Guan, Y.F.; et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharmacol. 2016, 173, 2352–2368. [Google Scholar]
- Manor, O.; Zubair, N.; Conomos, M.P.; Xu, X.; Rohwer, J.E.; Krafft, C.E.; Lovejoy, J.C.; Magis, A.T. A Multi-omic Association Study of Trimethylamine N-Oxide. Cell Rep. 2018, 24, 935–946. [Google Scholar]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar]
- Subramaniam, S.; Fletcher, C. Trimethylamine N-oxide: Breathe new life. Br. J. Pharmacol. 2018, 175, 1344–1353. [Google Scholar]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar]
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 124. [Google Scholar]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar]
- Li, D.; Ke, Y.; Zhan, R.; Liu, C.; Zhao, M.; Zeng, A.; Shi, X.; Ji, L.; Cheng, S.; Pan, B.; et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell 2018, 17, e12768. [Google Scholar]
- Ke, Y.; Li, D.; Zhao, M.; Liu, C.; Liu, J.; Zeng, A.; Shi, X.; Cheng, S.; Pan, B.; Zheng, L.; et al. Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic. Biol. Med. 2018, 116, 88–100. [Google Scholar]
- Zhang, H.; Meng, J.; Yu, H. Trimethylamine N-oxide Supplementation Abolishes the Cardioprotective Effects of Voluntary Exercise in Mice Fed a Western Diet. Front. Physiol. 2017, 8, 944. [Google Scholar]
- Zhang, R.; Wu, Y.; Xie, F.; Zhong, Y.; Wang, Y.; Xu, M.; Feng, J.; Charish, J.; Monnier, P.P.; Qin, X. RGMa mediates reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling after stroke. Cell Death Differ. 2018, 25, 1503–1516. [Google Scholar]
- Feng, Z.; Lu, Y.; Wu, X.; Zhao, P.; Li, J.; Peng, B.; Qian, Z.; Zhu, L. Ligustilide alleviates brain damage and improves cognitive function in rats of chronic cerebral hypoperfusion. J. Ethnopharmacol. 2012, 144, 313–321. [Google Scholar]
- Li, M.; Meng, N.; Guo, X.; Niu, X.; Zhao, Z.; Wang, W.; Xie, X.; Lv, P. Dl-3-n-Butylphthalide Promotes Remyelination and Suppresses Inflammation by Regulating AMPK/SIRT1 and STAT3/NF-κB Signaling in Chronic Cerebral Hypoperfusion. Front. Aging Neurosci. 2020, 12, 137. [Google Scholar]
- Feng, X.; Degos, V.; Koch, L.G.; Britton, S.L.; Zhu, Y.; Vacas, S.; Terrando, N.; Nelson, J.; Su, X.; Maze, M. Surgery results in exaggerated and persistent cognitive decline in a rat model of the Metabolic Syndrome. Anesthesiology 2013, 118, 1098–1105. [Google Scholar]
- Liu, C.; Wu, Y.; Zha, S.; Liu, M.; Wang, Y.; Yang, G.; Ma, K.; Fei, Y.; Zhang, Y.; Hu, X.; et al. Treatment effects of tanshinone IIA against intracerebroventricular streptozotocin induced memory deficits in mice. Brain Res. 2016, 1631, 137–146. [Google Scholar]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Gu, L.Z.; Zhang, Y.D.; Tan, L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol. Aging 2014, 35, 1243–1251. [Google Scholar]
- Chen, J.; Shu, S.; Chen, Y.; Liu, Z.; Yu, L.; Yang, L.; Xu, Y.; Zhang, M. AIM2 deletion promotes neuroplasticity and spatial memory of mice. Brain Res. Bull. 2019, 152, 85–94. [Google Scholar]
- Yin, Y.L.; Liu, Y.H.; Zhu, M.L.; Wang, H.H.; Qiu, Y.; Wan, G.R.; Li, P. Floralozone improves cognitive impairment in vascular dementia rats via regulation of TRPM2 and NMDAR signaling pathway. Physiol. Behav. 2022, 249, 113777. [Google Scholar]
- Kelleher, R.J., 3rd; Govindarajan, A.; Tonegawa, S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 2004, 44, 59–73. [Google Scholar]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar]
- Choi, S.A.; Chong, S.; Kwak, P.A.; Moon, Y.J.; Jangra, A.; Phi, J.H.; Lee, J.Y.; Park, S.H.; Kim, S.K. Impaired functional recovery of endothelial colony-forming cells from moyamoya disease in a chronic cerebral hypoperfusion rat model. J. Neurosurg. Pediatrics 2018, 23, 204–213. [Google Scholar]
- Dinan, T.G.; Cryan, J.F. Gut instincts: Microbiota as a key regulator of brain development, ageing and neurodegeneration. J. Physiol. 2017, 595, 489–503. [Google Scholar]
- Gao, Q.; Wang, Y.; Wang, X.; Fu, S.; Zhang, X.; Wang, R.T.; Zhang, X. Decreased levels of circulating trimethylamine N-oxide alleviate cognitive and pathological deterioration in transgenic mice: A potential therapeutic approach for Alzheimer’s disease. Aging 2019, 11, 8642–8663. [Google Scholar]
- Bennett, S.; Grant, M.M.; Aldred, S. Oxidative stress in vascular dementia and Alzheimer’s disease: A common pathology. J. Alzheimer’s Dis. JAD 2009, 17, 245–257. [Google Scholar]
- Raz, L.; Knoefel, J.; Bhaskar, K. The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2016, 36, 172–186. [Google Scholar]
- Maxwell, A.J. Mechanisms of dysfunction of the nitric oxide pathway in vascular diseases. Nitric Oxide Biol. Chem. 2002, 6, 101–124. [Google Scholar]
- Del Rio, D.; Zimetti, F.; Caffarra, P.; Tassotti, M.; Bernini, F.; Brighenti, F.; Zini, A.; Zanotti, I. The Gut Microbial Metabolite Trimethylamine-N-Oxide Is Present in Human Cerebrospinal Fluid. Nutrients 2017, 9, 1053. [Google Scholar]
- Zhao, L.; Zhang, C.; Cao, G.; Dong, X.; Li, D.; Jiang, L. Higher Circulating Trimethylamine N-oxide Sensitizes Sevoflurane-Induced Cognitive Dysfunction in Aged Rats Probably by Downregulating Hippocampal Methionine Sulfoxide Reductase A. Neurochem. Res. 2019, 44, 2506–2516. [Google Scholar]
- Meng, F.; Li, N.; Li, D.; Song, B.; Li, L. The presence of elevated circulating trimethylamine N-oxide exaggerates postoperative cognitive dysfunction in aged rats. Behav. Brain Res. 2019, 368, 111902. [Google Scholar]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 2017, 66, 693–702. [Google Scholar]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar]
- Takahashi, M. NLRP3 inflammasome as a novel player in myocardial infarction. Int. Heart J. 2014, 55, 101–105. [Google Scholar]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar]
- Yang, C.C.; Yang, C.M. Chinese Herbs and Repurposing Old Drugs as Therapeutic Agents in the Regulation of Oxidative Stress and Inflammation in Pulmonary Diseases. J. Inflamm. Res. 2021, 14, 657–687. [Google Scholar]
- Yang, G.; Zhang, X. TMAO promotes apoptosis and oxidative stress of pancreatic acinar cells by mediating IRE1α-XBP-1 pathway. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2021, 27, 361–369. [Google Scholar]
- Dong, F.; Jiang, S.; Tang, C.; Wang, X.; Ren, X.; Wei, Q.; Tian, J.; Hu, W.; Guo, J.; Fu, X.; et al. Trimethylamine N-oxide promotes hyperoxaluria-induced calcium oxalate deposition and kidney injury by activating autophagy. Free Radic. Biol. Med. 2022, 179, 288–300. [Google Scholar]
- Zhang, Q.; Fan, Z.; Xue, W.; Sun, F.; Zhu, H.; Huang, D.; Wang, Z.; Dong, L. Vitexin regulates Epac and NLRP3 and ameliorates chronic cerebral hypoperfusion injury. Can. J. Physiol. Pharmacol. 2021, 99, 1079–1087. [Google Scholar]
- Wang, R.; Wu, Z.; Bai, L.; Liu, R.; Ba, Y.; Zhang, H.; Cheng, X.; Zhou, G.; Huang, H. Resveratrol improved hippocampal neurogenesis following lead exposure in rats through activation of SIRT1 signaling. Environ. Toxicol. 2021, 36, 1664–1673. [Google Scholar]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Gräff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar]
- Yan, T.; Huang, J.; Nisar, M.F.; Wan, C.; Huang, W. The Beneficial Roles of SIRT1 in Drug-Induced Liver Injury. Oxidative Med. Cell. Longev. 2019, 2019, 8506195. [Google Scholar]
- Hattori, Y.; Okamoto, Y.; Maki, T.; Yamamoto, Y.; Oishi, N.; Yamahara, K.; Nagatsuka, K.; Takahashi, R.; Kalaria, R.N.; Fukuyama, H.; et al. Silent information regulator 2 homolog 1 counters cerebral hypoperfusion injury by deacetylating endothelial nitric oxide synthase. Stroke 2014, 45, 3403–3411. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Zou, J.; Hong, Y.; Peng, Q.; Fu, X.; Duan, R.; Chen, J.; Chen, X. Higher Circulating Trimethylamine N-Oxide Aggravates Cognitive Impairment Probably via Downregulating Hippocampal SIRT1 in Vascular Dementia Rats. Cells 2022, 11, 3650. https://doi.org/10.3390/cells11223650
Deng Y, Zou J, Hong Y, Peng Q, Fu X, Duan R, Chen J, Chen X. Higher Circulating Trimethylamine N-Oxide Aggravates Cognitive Impairment Probably via Downregulating Hippocampal SIRT1 in Vascular Dementia Rats. Cells. 2022; 11(22):3650. https://doi.org/10.3390/cells11223650
Chicago/Turabian StyleDeng, Yang, Junqing Zou, Ye Hong, Qiang Peng, Xinxin Fu, Rui Duan, Jie Chen, and Xiangliang Chen. 2022. "Higher Circulating Trimethylamine N-Oxide Aggravates Cognitive Impairment Probably via Downregulating Hippocampal SIRT1 in Vascular Dementia Rats" Cells 11, no. 22: 3650. https://doi.org/10.3390/cells11223650
APA StyleDeng, Y., Zou, J., Hong, Y., Peng, Q., Fu, X., Duan, R., Chen, J., & Chen, X. (2022). Higher Circulating Trimethylamine N-Oxide Aggravates Cognitive Impairment Probably via Downregulating Hippocampal SIRT1 in Vascular Dementia Rats. Cells, 11(22), 3650. https://doi.org/10.3390/cells11223650