
Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington’s Disease and Other Neurodegenerative Disorders

 , , and
, , and 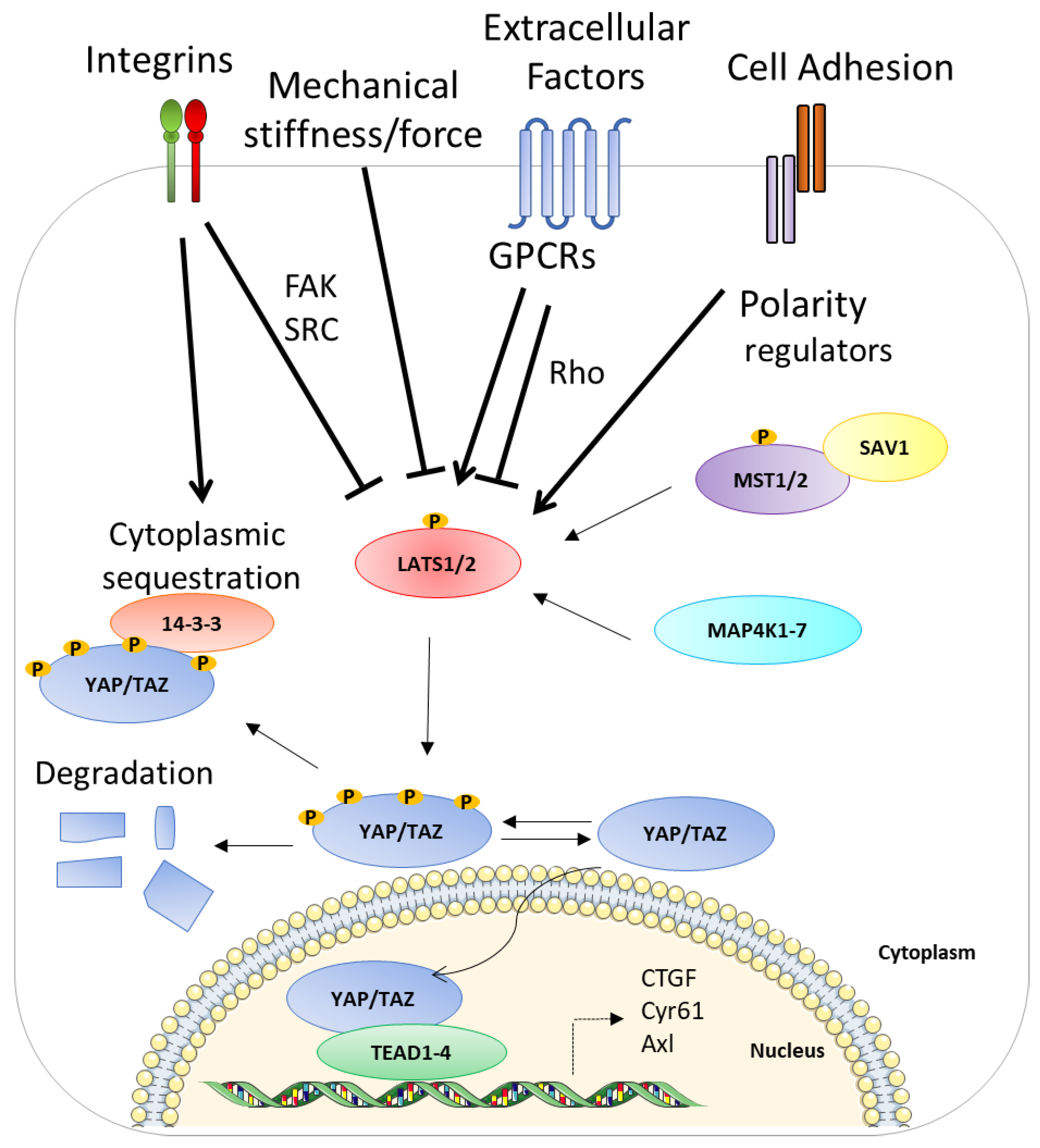{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hippo Pathway
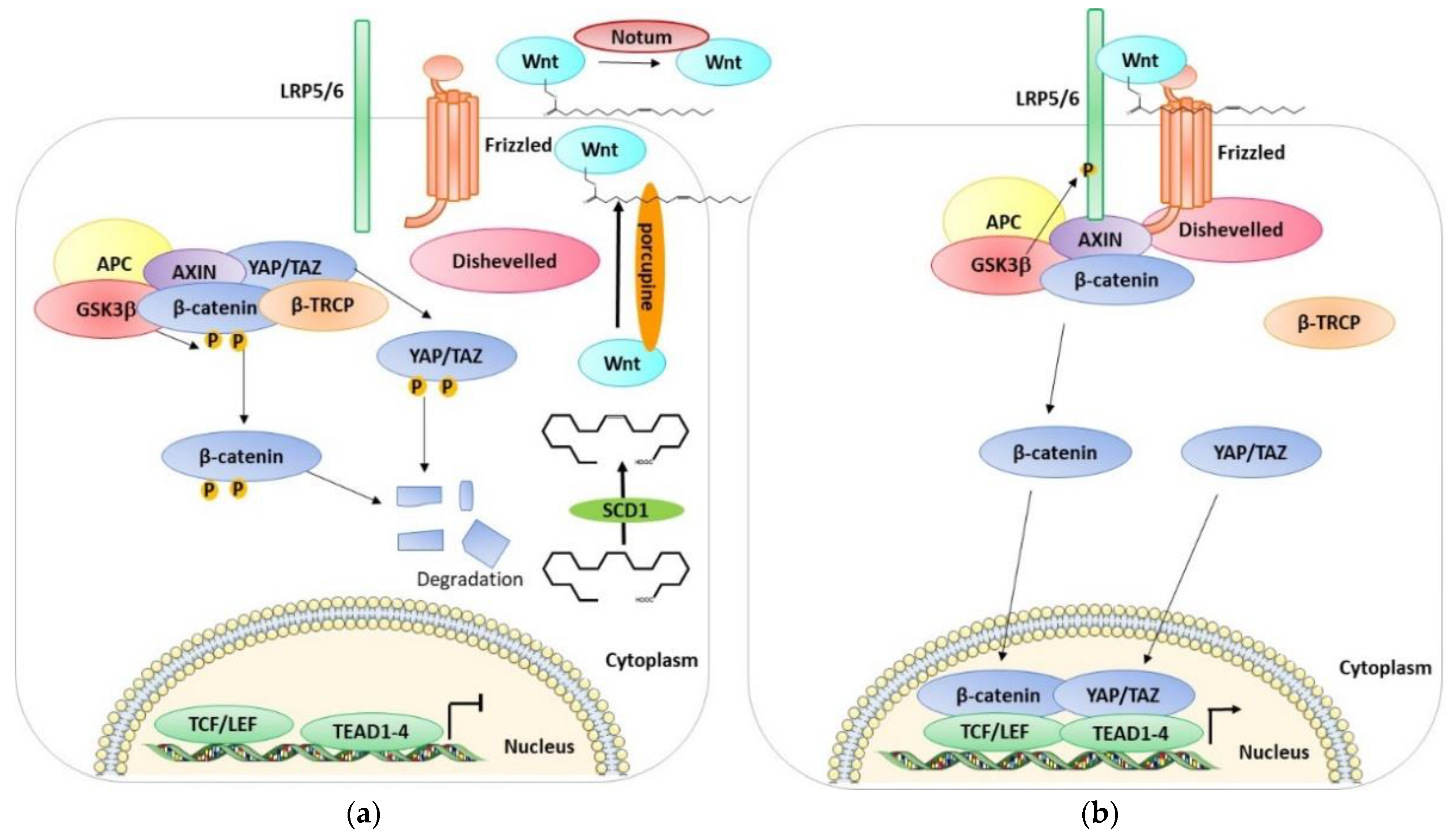
3. Wnt/β-Catenin Pathway
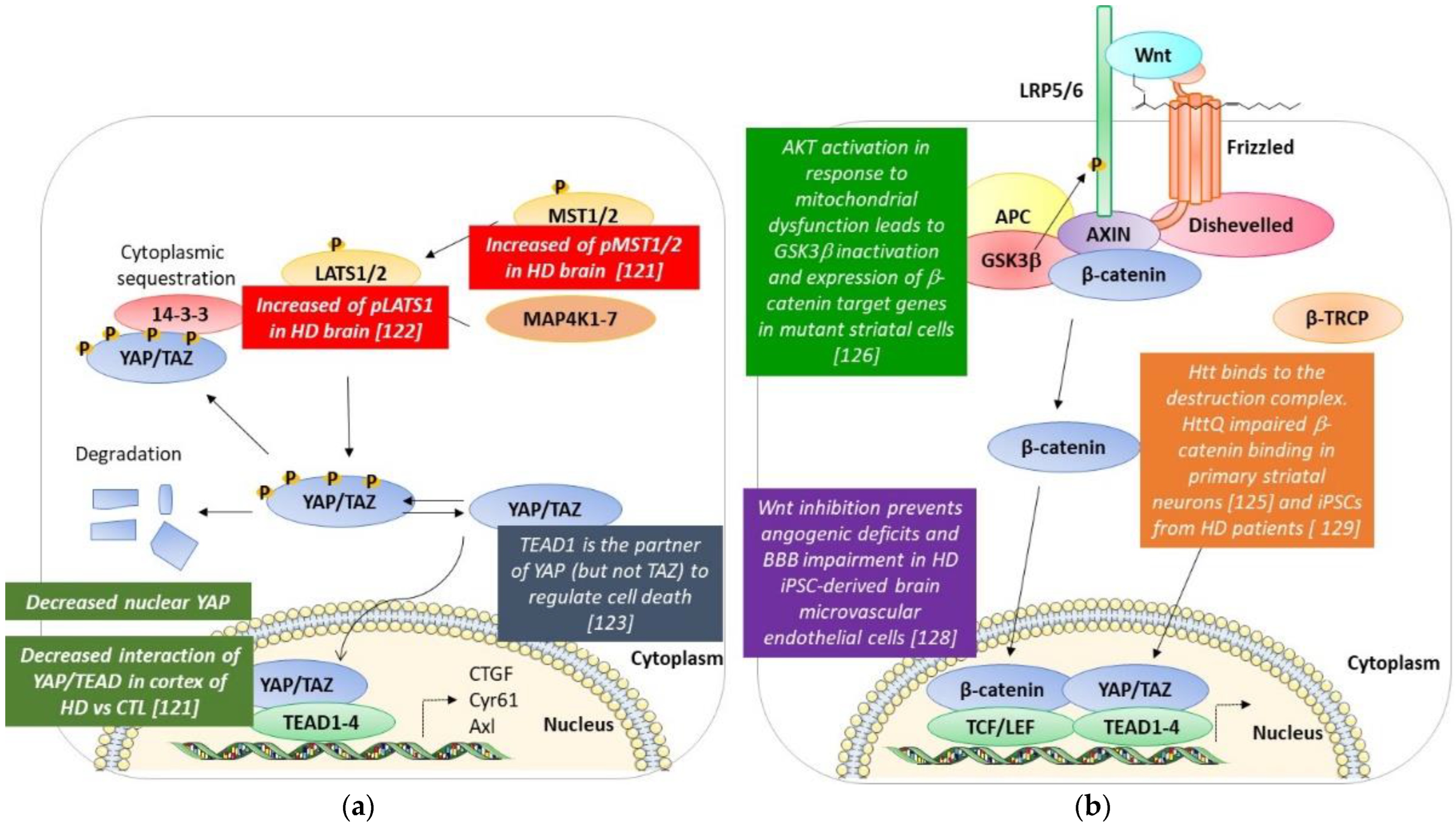
4. Hippo Pathway and Huntington’s Disease (Figure 3a)

5. Wnt/β-Catenin and Huntington’s Disease (Figure 3b)
6. Hippo Pathway and Wnt Pathway in Other Neurodegenerative Diseases
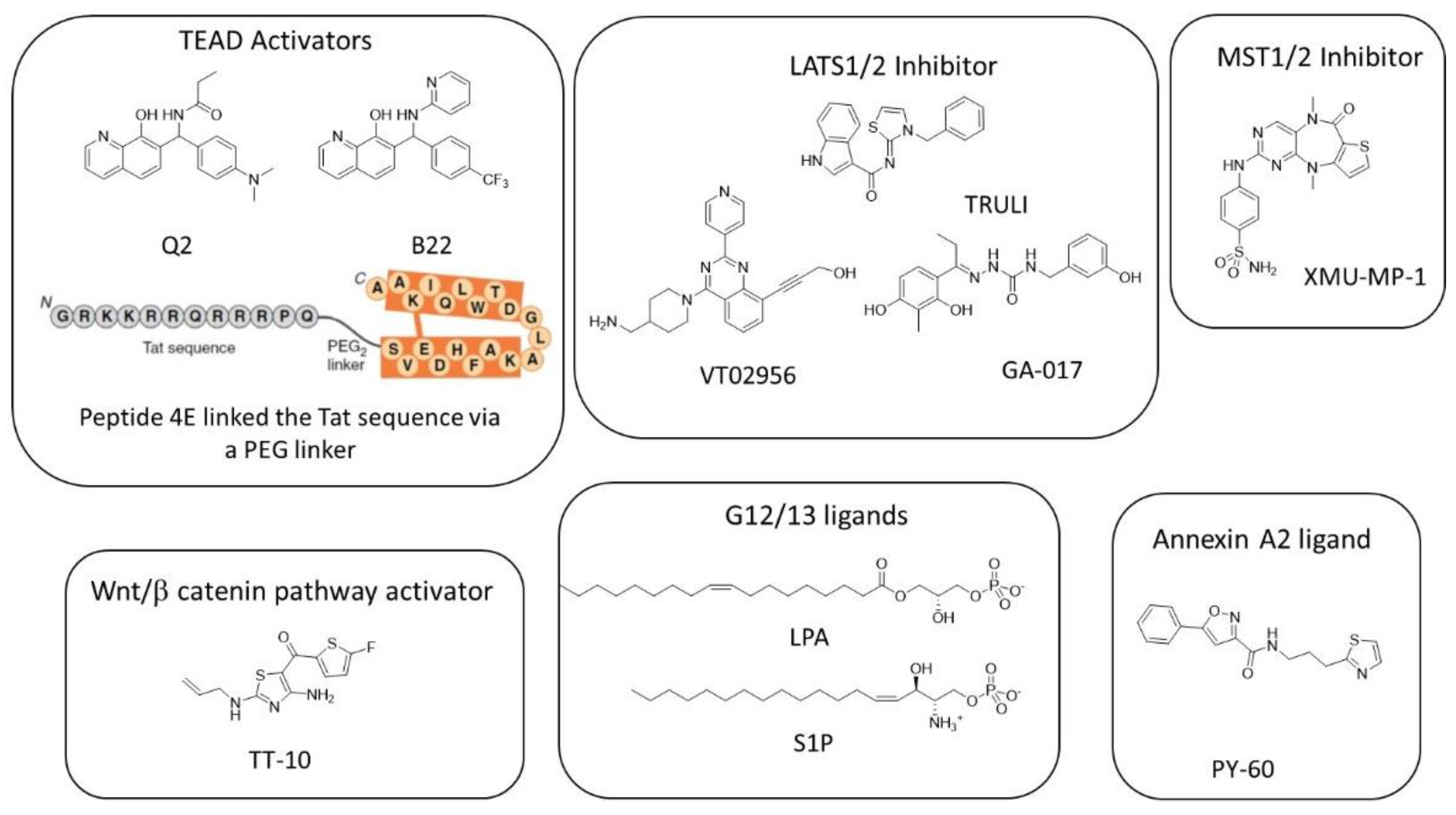
7. Favoring YAP/TAZ-TEAD Interaction
8. Targeting Wnt/β-Catenin Signaling Pathway
9. Discussion/Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo Pathway and Human Cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Zygulska, A.L.; Krzemieniecki, K.; Pierzchalski, P. Hippo Pathway—Brief Overview of Its Relevance in Cancer. J. Physiol. Pharmacol. 2017, 68, 311–335. [Google Scholar] [PubMed]
- Clevers, H. Wnt/Beta-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Liu, G.; Mong, P.; Mudbhary, R.; Biswas, R.; Arroyave, R.; Vijayakumar, S.; Economides, A.N.; Aaronson, S.A. Canonical and Noncanonical Wnts Use a Common Mechanism to Activate Completely Unrelated Coreceptors. Genes Dev. 2010, 24, 2517. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and Non-Canonical WNT Signaling in Cancer Stem Cells and Their Niches: Cellular Heterogeneity, Omics Reprogramming, Targeted Therapy and Tumor Plasticity (Review). Int. J. Oncol. 2017, 51, 1357. [Google Scholar] [CrossRef]
- Seifert, J.R.K.; Mlodzik, M. Frizzled/PCP Signaling: A Conserved Mechanism Regulating Cell Polarity and Directed Motility. Nat. Rev. Genet. 2007, 8, 126–138. [Google Scholar] [CrossRef]
- Wang, Y.; Nathans, J. Tissue/Planar Cell Polarity in Vertebrates: New Insights and New Questions. Development 2007, 134, 647–658. [Google Scholar] [CrossRef]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic Approaches to Huntington Disease: From the Bench to the Clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Kwon, D. Failure of Genetic Therapies for Huntington’s Devastates Community. Nature 2021, 593, 180. [Google Scholar] [CrossRef] [PubMed]
- Serafino, A.; Giovannini, D.; Rossi, S.; Cozzolino, M. Targeting the Wnt/β-Catenin Pathway in Neurodegenerative Diseases: Recent Approaches and Current Challenges. Expert Opin Drug Discov. 2020, 15, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo Signaling Pathway Coordinately Regulates Cell Proliferation and Apoptosis by Inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo Pathway Regulation. Genes Dev. 2016, 30, 1. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Sasaki, H. Mammalian Tead Proteins Regulate Cell Proliferation and Contact Inhibition as Transcriptional Mediators of Hippo Signaling. Development 2008, 135, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, C.Y.; Zha, Z.Y.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. TEAD Transcription Factors Mediate the Function of TAZ in Cell Growth and Epithelial-Mesenchymal Transition. J. Biol. Chem. 2009, 284, 13355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD Mediates YAP-Dependent Gene Induction and Growth Control. Genes Dev. 2008, 22, 1962. [Google Scholar] [CrossRef]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP Induces Malignant Mesothelioma Cell Proliferation by Upregulating Transcription of Cell Cycle-Promoting Genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL Receptor Kinase Is a Mediator of YAP-Dependent Oncogenic Functions in Hepatocellular Carcinoma. Oncogene 2011, 30, 1229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-Dependent Induction of Amphiregulin Identifies a Non-Cell-Autonomous Component of the Hippo Pathway. Nat. Cell Biol. 2009, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.-Y.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway. Mol. Cell. Biol. 2008, 28, 2426. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The Hippo Tumor Pathway Promotes TAZ Degradation by Phosphorylating a Phosphodegron and Recruiting the SCFβ-TrCP E3 Ligase. J. Biol. Chem. 2010, 285, 37159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A Coordinated Phosphorylation by Lats and CK1 Regulates YAP Stability through SCFβ-TRCP. Genes Dev. 2010, 24, 72. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747. [Google Scholar] [CrossRef]
- Boggiano, J.C.; Vanderzalm, P.J.; Fehon, R.G. Tao-1 Phosphorylates Hippo/MST Kinases to Regulate the Hippo-Salvador-Warts Tumor Suppressor Pathway. Dev. Cell 2011, 21, 888. [Google Scholar] [CrossRef]
- Poon, C.L.C.; Lin, J.I.; Zhang, X.; Harvey, K.F. The Sterile 20-like Kinase Tao-1 Controls Tissue Growth by Regulating the Salvador-Warts-Hippo Pathway. Dev. Cell 2011, 21, 896–906. [Google Scholar] [CrossRef]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the MST1 Kinase by Autophosphorylation, by the Growth Inhibitory Proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004, 381, 453. [Google Scholar] [CrossRef]
- Glantschnig, H.; Rodan, G.A.; Reszka, A.A. Mapping of MST1 Kinase Sites of Phosphorylation: ACTIVATION AND AUTOPHOSPHORYLATION *. J. Biol. Chem. 2002, 277, 42987–42996. [Google Scholar] [CrossRef]
- Callus, B.A.; Verhagen, A.M.; Vaux, D.L. Association of Mammalian Sterile Twenty Kinases, Mst1 and Mst2, with HSalvador via C-Terminal Coiled-Coil Domains, Leads to Its Stabilization and Phosphorylation. FEBS J. 2006, 273, 4264–4276. [Google Scholar] [CrossRef] [PubMed]
- Praskova, M.; Xia, F.; Avruch, J. MOBKL1A/MOBKL1B Phosphorylation by MST1 and MST2 Inhibits Cell Proliferation. Curr. Biol. 2008, 18, 311. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Schmitz, D.; Hemmings, B.A. The Human Tumour Suppressor LATS1 Is Activated by Human MOB1 at the Membrane. Biochem. Biophys. Res. Commun. 2006, 345, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell 2013, 154, 1342. [Google Scholar] [CrossRef]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by MTOR and Autophagy Reveals a Therapeutic Target of Tuberous Sclerosis Complex. J. Exp. Med. 2014, 211, 2249. [Google Scholar] [CrossRef]
- Yamaguchi, N. Multiple Roles of Vestigial-Like Family Members in Tumor Development. Front. Oncol. 2020, 10, 1266. [Google Scholar] [CrossRef]
- Koontz, L.M.; Liu-Chittenden, Y.; Yin, F.; Zheng, Y.; Yu, J.; Huang, B.; Chen, Q.; Wu, S.; Pan, D. The Hippo Effector Yorkie Controls Normal Tissue Growth by Antagonizing Scalloped-Mediated Default Repression. Dev. Cell 2013, 25, 388. [Google Scholar] [CrossRef]
- Ma, S.; Tang, T.; Probst, G.; Konradi, A.; Jin, C.; Li, F.; Silvio Gutkind, J.; Fu, X.D.; Guan, K.L. Transcriptional Repression of Estrogen Receptor Alpha by YAP Reveals the Hippo Pathway as Therapeutic Target for ER+ Breast Cancer. Nat. Commun. 2022 131 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo Pathway Target, YAP, Promotes Metastasis through Its TEAD-Interaction Domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441. [Google Scholar] [CrossRef]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs Complex Couples Cell Density Sensing to Hippo-Dependent Control of the TGF-β-SMAD Pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, N.; Inoue, K.; Adachi, K.; Kiyonari, H.; Ota, M.; Ralston, A.; Yabuta, N.; Hirahara, S.; Stephenson, R.O.; Ogonuki, N.; et al. The Hippo Signaling Pathway Components Lats and Yap Pattern Tead4 Activity to Distinguish Mouse Trophectoderm from Inner Cell Mass. Dev. Cell 2009, 16, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.R.; Kreger, B.T.; Lien, W.H.; Klezovitch, O.; Rudakova, G.M.; Camargo, F.D.; Lantz, D.M.; Seykora, J.T.; Vasioukhin, V. α-Catenin Is a Tumor Suppressor That Controls Cell Accumulation by Regulating the Localization and Activity of the Transcriptional Coactivator Yap1. Sci. Signal. 2011, 4, ra33. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Yang, J.; Deran, M.; Wu, C.; Su, A.I.; Bonamy, G.M.C.; Liu, J.; Peters, E.C.; Wu, X. Identification of Serum-Derived Sphingosine-1-Phosphate as a Small Molecule Regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Yu, F.X.; Gong, R.; Brown, J.H.; Guan, K.L. Regulation of the Hippo–YAP Pathway by Protease-Activated Receptors (PARs). Genes Dev. 2012, 26, 2138. [Google Scholar] [CrossRef]
- Yu, F.X.; Guan, K.L. The Hippo Pathway: Regulators and Regulations. Genes Dev. 2013, 27, 355. [Google Scholar] [CrossRef]
- Gong, R.; Hong, A.W.; Plouffe, S.W.; Zhao, B.; Liu, G.; Yu, F.X.; Xu, Y.; Guan, K.L. Opposing Roles of Conventional and Novel PKC Isoforms in Hippo-YAP Pathway Regulation. Cell Res. 2015, 25, 985. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, S.; Wang, Z.; Feng, X.; Liu, P.; Lv, X.B.; Li, F.; Yu, F.X.; Sun, Y.; Yuan, H.; et al. Estrogen Regulates Hippo Signaling via GPER in Breast Cancer. J. Clin. Investig. 2015, 125, 2123. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein Coupled Receptor Signaling. Cell 2012, 150, 780. [Google Scholar] [CrossRef]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating YAP. Cancer Cell 2014, 25, 822. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of Mevalonate and Hippo Pathways Regulates RHAMM Transcription via YAP to Modulate Breast Cancer Cell Motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89. [Google Scholar] [CrossRef]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villén, J.; Becker, E.B.E.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A Conserved MST-FOXO Signaling Pathway Mediates Oxidative-Stress Responses and Extends Life Span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef]
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. The Kinases Mst1 and Mst2 Positively Regulate Phagocyte ROS Induction and Bactericidal Activity. Nat. Immunol. 2015, 16, 1142. [Google Scholar] [CrossRef]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A Functional Interaction between Hippo-YAP Signaling and FoxO1 Mediates the Oxidative Stress Response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef]
- Rajesh, K.; Krishnamoorthy, J.; Gupta, J.; Kazimierczak, U.; Papadakis, A.I.; Deng, Z.; Wang, S.; Kuninaka, S.; Koromilas, A.E. The eIF2α serine 51 phosphorylation-ATF4 arm promotes HIPPO signaling and cell death under oxidative stress. Oncotarget 2016, 7, 51044–51058. [Google Scholar] [CrossRef]
- deRan, M.; Yang, J.; Shen, C.H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy Stress Regulates Hippo–YAP Signaling Involving AMPK-Mediated Regulation of Angiomotin Like-1 Protein. Cell Rep. 2014, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular Energy Stress Induces AMPK-Mediated Regulation of YAP and the Hippo Pathway. Nat. Cell Biol. 2015, 17, 500. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK Modulates Hippo Pathway Activity to Regulate Energy Homeostasis. Nat. Cell Biol. 2015, 17, 490. [Google Scholar] [CrossRef]
- Gordon, M.D.; Nusse, R. Wnt Signaling: Multiple Pathways, Multiple Receptors, and Multiple Transcription Factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef]
- Polakis, P. The Many Ways of Wnt in Cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef]
- Angers, S.; Moon, R.T. Proximal Events in Wnt Signal Transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic Wnt Signaling Promotes Protein Stabilization and Regulates Cell Size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Noori, R.; Gaspar, C.; Franken, P.; Sacchetti, A.; Rafati, H.; Mahmoudi, T.; Decraene, C.; Calin, G.A.; Merrill, B.J.; et al. Wnt Signaling Regulates the Lineage Differentiation Potential of Mouse Embryonic Stem Cells through Tcf3 Down-Regulation. PLoS Genet. 2013, 9, e1003424. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem Cell Signaling. An Integral Program for Tissue Renewal and Regeneration: Wnt Signaling and Stem Cell Control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Green, J.L.; Inoue, T.; Sternberg, P.W. Opposing Wnt Pathways Orient Cell Polarity during Organogenesis. Cell 2008, 134, 646. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Nusse, R. An Ancient Cluster of Wnt Paralogues. Trends Genet. 2001, 17, 443. [Google Scholar] [CrossRef]
- Nile, A.H.; Hannoush, R.N. Fatty Acylation of Wnt Proteins. Nat. Chem. Biol. 2016, 12, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Herr, P.; Basler, K. Porcupine-Mediated Lipidation Is Required for Wnt Recognition by Wls. Dev. Biol. 2012, 361, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural Basis of Wnt Recognition by Frizzled. Science 2012, 337, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Proffitt, K.D.; Virshup, D.M. Precise Regulation of Porcupine Activity Is Required for Physiological Wnt Signaling. J. Biol. Chem. 2012, 287, 34167–34178. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt Proteins Are Lipid-Modified and Can Act as Stem Cell Growth Factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef]
- Castro, L.F.C.; Wilson, J.M.; Gonçalves, O.; Galante-Oliveira, S.; Rocha, E.; Cunha, I. The Evolutionary History of the Stearoyl-CoA Desaturase Gene Family in Vertebrates. BMC Evol. Biol. 2011, 11, 132. [Google Scholar] [CrossRef]
- Kakugawa, S.; Langton, P.F.; Zebisch, M.; Howell, S.A.; Chang, T.H.; Liu, Y.; Feizi, T.; Bineva, G.; O’Reilly, N.; Snijders, A.P.; et al. Notum Deacylates Wnt Proteins to Suppress Signaling Activity. Nature 2015, 519, 187–192. [Google Scholar] [CrossRef]
- Kimelman, D.; Xu, W. β-Catenin Destruction Complex: Insights and Questions from a Structural Perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Behrens, J.; Jerchow, B.A.; Würtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kühl, M.; Wedlich, D.; Birchmeier, W. Functional Interaction of an Axin Homolog, Conductin, with Beta-Catenin, APC, and GSK3beta. Science 1998, 280, 596–599. [Google Scholar] [CrossRef]
- Hart, M.J.; De Los Santos, R.; Albert, I.N.; Rubinfeld, B.; Polakis, P. Downregulation of Beta-Catenin by Human Axin and Its Association with the APC Tumor Suppressor, Beta-Catenin and GSK3 Beta. Curr. Biol. 1998, 8, 573–581. [Google Scholar] [CrossRef]
- Munemitsu, S.; Albert, I.; Souza, B.; Rubinfeld, D.; Polakis, P. Regulation of Intracellular Beta-Catenin Levels by the Adenomatous Polyposis Coli (APC) Tumor-Suppressor Protein. Proc. Natl. Acad. Sci. USA 1995, 92, 3046. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Souza, B.; Albert, I.; Müller, O.; Chamberlain, S.H.; Masiarz, F.R.; Munemitsu, S.; Polakis, P. Association of the APC Gene Product with Beta-Catenin. Science 1993, 262, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Amit, S.; Hatzubai, A.; Birman, Y.; Andersen, J.S.; Ben-Shushan, E.; Mann, M.; Ben-Neriah, Y.; Alkalay, I. Axin-Mediated CKI Phosphorylation of Beta-Catenin at Ser 45: A Molecular Switch for the Wnt Pathway. Genes Dev. 2002, 16, 1066–1076. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of Beta-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK3beta to the APC-Beta-Catenin Complex and Regulation of Complex Assembly. Science 1996, 272, 1023–1026. [Google Scholar] [CrossRef]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-Ubiquitin Ligase Complex Associates Specifically with Phosphorylated Destruction Motifs in IkappaBalpha and Beta-Catenin and Stimulates IkappaBalpha Ubiquitination in Vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef]
- Hart, M.; Concordet, J.P.; Lassot, I.; Albert, I.; Del Los Santos, R.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-Box Protein Beta-TrCP Associates with Phosphorylated Beta-Catenin and Regulates Its Activity in the Cell. Curr. Biol. 1999, 9, 207–211. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-Catenin Is a Target for the Ubiquitin-Proteasome Pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef]
- Orford, K.; Crockett, C.; Jensen, J.P.; Weissman, A.M.; Byers, S.W. Serine Phosphorylation-Regulated Ubiquitination and Degradation of Beta-Catenin. J. Biol. Chem. 1997, 272, 24735–24738. [Google Scholar] [CrossRef]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A New Member of the Frizzled Family from Drosophila Functions as a Wingless Receptor. Nature 1996, 382, 225–231. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 Receptors for Wnt/β-Catenin Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- Yang-Snyder, J.; Miller, J.R.; Brown, J.D.; Lai, C.J.; Moon, R.T. A Frizzled Homolog Functions in a Vertebrate Wnt Signaling Pathway. Curr. Biol. 1996, 6, 1302–1306. [Google Scholar] [CrossRef]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-Receptor-Related Protein Mediates Wnt Signaling in Mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Semenov, M.; Kato, Y.; Spokony, R.; Liu, C.; Katsuyama, Y.; Hess, F.; Saint-Jeannet, J.P.; He, X. LDL-Receptor-Related Proteins in Wnt Signal Transduction. Nature 2000, 407, 530–535. [Google Scholar] [CrossRef]
- Wehrli, M.; Dougan, S.T.; Caldwell, K.; O’Keefe, L.; Schwartz, S.; Valzel-Ohayon, D.; Schejter, E.; Tomlinson, A.; DiNardo, S. Arrow Encodes an LDL-Receptor-Related Protein Essential for Wingless Signaling. Nature 2000, 407, 527–530. [Google Scholar] [CrossRef]
- Klingensmith, J.; Nusse, R.; Perrimon, N. The Drosophila Segment Polarity Gene Dishevelled Encodes a Novel Protein Required for Response to the Wingless Signal. Genes Dev. 1994, 8, 118–130. [Google Scholar] [CrossRef]
- Krasnow, R.E.; Wong, L.L.; Adler, P.N. Dishevelled Is a Component of the Frizzled Signaling Pathway in Drosophila. Development 1995, 121, 4095–4102. [Google Scholar] [CrossRef]
- Cong, F.; Schweizer, L.; Varmus, H. Wnt Signals across the Plasma Membrane to Activate the Beta-Catenin Pathway by Forming Oligomers Containing Its Receptors, Frizzled and LRP. Development 2004, 131, 5103–5115. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Jordens, I.; Kirchner, K.; Slootstra, J.W.; Kruitwagen, T.; Bouwman, B.A.M.; Noutsou, M.; Rüdiger, S.G.D.; Schwamborn, K.; Schambony, A.; et al. Wnt/β-Catenin Signaling Requires Interaction of the Dishevelled DEP Domain and C Terminus with a Discontinuous Motif in Frizzled. Proc. Natl. Acad. Sci. USA 2012, 109, E812–E820. [Google Scholar] [CrossRef]
- Tamai, K.; Zeng, X.; Liu, C.; Zhang, X.; Harada, Y.; Chang, Z.; He, X. A Mechanism for Wnt Coreceptor Activation. Mol. Cell 2004, 13, 149–156. [Google Scholar] [CrossRef]
- Davidson, G.; Shen, J.; Huang, Y.L.; Su, Y.; Karaulanov, E.; Bartscherer, K.; Hassler, C.; Stannek, P.; Boutros, M.; Niehrs, C. Cell Cycle Control of Wnt Receptor Activation. Dev. Cell 2009, 17, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Wu, W.; Shen, J.; Bilic, J.; Fenger, U.; Stannek, P.; Glinka, A.; Niehrs, C. Casein Kinase 1 Gamma Couples Wnt Receptor Activation to Cytoplasmic Signal Transduction. Nature 2005, 438, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Tamai, K.; Doble, B.; Li, S.; Huang, H.; Habas, R.; Okamura, H.; Woodgett, J.; He, X. A Dual-Kinase Mechanism for Wnt Co-Receptor Phosphorylation and Activation. Nature 2005, 438, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Wang, J.; Liu, B.; Pan, W.; Farr, G.H.; Flynn, C.; Yuan, H.; Takada, S.; Kimelman, D.; Li, L.; et al. Low-Density Lipoprotein Receptor-Related Protein-5 Binds to Axin and Regulates the Canonical Wnt Signaling Pathway. Mol. Cell 2001, 7, 801–809. [Google Scholar] [CrossRef]
- Cselenyi, C.S.; Jernigan, K.K.; Tahinci, E.; Thorne, C.A.; Lee, L.A.; Lee, E. LRP6 Transduces a Canonical Wnt Signal Independently of Axin Degradation by Inhibiting GSK3’s Phosphorylation of Beta-Catenin. Proc. Natl. Acad. Sci. USA 2008, 105, 8032–8037. [Google Scholar] [CrossRef]
- Piao, S.; Lee, S.H.; Kim, H.; Yum, S.; Stamos, J.L.; Xu, Y.; Lee, S.J.; Lee, J.; Oh, S.; Han, J.K.; et al. Direct Inhibition of GSK3β by the Phosphorylated Cytoplasmic Domain of LRP6 in Wnt/β-Catenin Signaling. PLoS ONE 2008, 3, e4046. [Google Scholar] [CrossRef]
- Stamos, J.L.; Chu, M.L.H.; Enos, M.D.; Shah, N.; Weis, W.I. Structural Basis of GSK-3 Inhibition by N-Terminal Phosphorylation and by the Wnt Receptor LRP6. elife 2014, 3, e01998. [Google Scholar] [CrossRef]
- Cavallo, R.A.; Cox, R.T.; Moline, M.M.; Roose, J.; Polevoy, G.A.; Clevers, H.; Peifer, M.; Bejsovec, A. Drosophila Tcf and Groucho Interact to Repress Wingless Signaling Activity. Nature 1998, 395, 604–608. [Google Scholar] [CrossRef]
- Roose, J.; Molenaar, M.; Peterson, J.; Hurenkamp, J.; Brantjes, H.; Moerer, P.; Van De Wetering, M.; Destrée, O.; Clevers, H. The Xenopus Wnt Effector XTcf-3 Interacts with Groucho-Related Transcriptional Repressors. Nature 1998, 395, 608–612. [Google Scholar] [CrossRef]
- Behrens, J.; Von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional Interaction of Beta-Catenin with the Transcription Factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.; Van De Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 Transcription Factor Mediates Beta-Catenin-Induced Axis Formation in Xenopus Embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Cavallo, R.; Dooijes, D.; Van Beest, M.; Van Es, J.; Loureiro, J.; Ypma, A.; Hursh, D.; Jones, T.; Bejsovec, A.; et al. Armadillo Coactivates Transcription Driven by the Product of the Drosophila Segment Polarity Gene DTCF. Cell 1997, 88, 789–799. [Google Scholar] [CrossRef]
- Nakamura, Y.; De Paiva Alves, E.; Veenstra, G.J.C.; Hoppler, S. Tissue- and Stage-Specific Wnt Target Gene Expression Is Controlled Subsequent to β-Catenin Recruitment to Cis-Regulatory Modules. Development 2016, 143, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Imajo, M.; Miyatake, K.; Iimura, A.; Miyamoto, A.; Nishida, E. A Molecular Mechanism That Links Hippo Signaling to the Inhibition of Wnt/β-Catenin Signaling. EMBO J. 2012, 31, 1109–1122. [Google Scholar] [CrossRef]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The Hippo Pathway Regulates Wnt/Beta-Catenin Signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as Mediator of Wnt Signaling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-Desaturase 1 Regulates Lung Cancer Stemness via Stabilization and Nuclear Localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Mueller, K.A.; Glajch, K.E.; Huizenga, M.N.; Wilson, R.A.; Granucci, E.J.; Dios, A.M.; Tousley, A.R.; Iuliano, M.; Weisman, E.; LaQuaglia, M.J.; et al. Hippo Signaling Pathway Dysregulation in Human Huntington’s Disease Brain and Neuronal Stem Cells. Sci. Rep. 2018, 8, 11355. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A Novel Form of Necrosis, TRIAD, Occurs in Human Huntington’s Disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Chen, X.; Xu, M.; Fujita, K.; Motoki, K.; Sasabe, T.; Homma, H.; Murata, M.; Tagawa, K.; Tamura, T.; et al. Targeting TEAD/YAP-Transcription-Dependent Necrosis, TRIAD, Ameliorates Huntington’s Disease Pathology. Hum. Mol. Genet. 2016, 25, 4749–4770. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; Sugars, K.L.; Bao, Y.P.; Rubinsztein, D.C. Glycogen Synthase Kinase-3beta Inhibitors Prevent Cellular Polyglutamine Toxicity Caused by the Huntington’s Disease Mutation. J. Biol. Chem. 2002, 277, 33791–33798. [Google Scholar] [CrossRef]
- Godin, J.D.; Poizat, G.; Hickey, M.A.; Maschat, F.; Humbert, S. Mutant Huntingtin-Impaired Degradation of β-Catenin Causes Neurotoxicity in Huntington’s Disease. EMBO J. 2010, 29, 2433. [Google Scholar] [CrossRef]
- Gines, S.; Ivanova, E.; Seong, I.S.; Saura, C.A.; MacDonald, M.E. Enhanced Akt Signaling Is an Early Pro-Survival Response That Reflects N-Methyl-D-Aspartate Receptor Activation in Huntington’s Disease Knock-in Striatal Cells. J. Biol. Chem. 2003, 278, 50514–50522. [Google Scholar] [CrossRef]
- Ghatak, S.; Raha, S. Micro RNA-214 Contributes to Proteasome Independent Downregulation of Beta Catenin in Huntington’s Disease Knock-in Striatal Cell Model STHdhQ111/Q111. Biochem. Biophys. Res. Commun. 2015, 459, 509–514. [Google Scholar] [CrossRef]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s Disease IPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef]
- Smith-Geater, C.; Hernandez, S.J.; Lim, R.G.; Adam, M.; Wu, J.; Stocksdale, J.T.; Wassie, B.T.; Gold, M.P.; Wang, K.Q.; Miramontes, R.; et al. Aberrant Development Corrected in Adult-Onset Huntington’s Disease IPSC-Derived Neuronal Cultures via WNT Signaling Modulation. Stem Cell Rep. 2020, 14, 406–419. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Li, F.; Lin, Y.; Zhang, X.; Lv, Z.; Jiang, J. MiR-214 Inhibits Cell Growth in Hepatocellular Carcinoma through Suppression of β-Catenin. Biochem. Biophys. Res. Commun. 2012, 428, 525–531. [Google Scholar] [CrossRef]
- Xia, H.; Ooi, L.L.P.J.; Hui, K.M. MiR-214 Targets β-Catenin Pathway to Suppress Invasion, Stem-Like Traits and Recurrence of Human Hepatocellular Carcinoma. PLoS ONE 2012, 7, e44206. [Google Scholar] [CrossRef]
- Sahu, M.R.; Mondal, A.C. The Emerging Role of Hippo Signaling in Neurodegeneration. J. Neurosci. Res. 2020, 98, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhao, X.; Fu, H.; Gao, Y. The Effects of YAP and Its Related Mechanisms in Central Nervous System Diseases. Front. Neurosci. 2020, 14, 595. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Homma, H.; Fujita, K.; Kondo, K.; Yamada, S.; Jin, X.; Waragai, M.; Ohtomo, G.; Iwata, A.; Tagawa, K.; et al. YAP-Dependent Necrosis Occurs in Early Stages of Alzheimer’s Disease and Regulates Mouse Model Pathology. Nat. Commun. 2020, 11, 507. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, N.; Nagai, M.; Miyazaki, K.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; Okazawa, H.; Abe, K. Progressive Decrease in the Level of YAPdeltaCs, Prosurvival Isoforms of YAP, in the Spinal Cord of Transgenic Mouse Carrying a Mutant SOD1 Gene. J. Neurosci. Res. 2009, 87, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Vakili, G.; Mueller, K.; Granucci, E.; Dios, A.; Berry, J.; Vakili, K. Alterations in Hippo Pathway Signaling as a Mechanism of Neuronal Death in Amyotrophic Lateral Sclerosis (P4.436). Neurology 2018, 90 (15 supplement). Available online: https://n.neurology.org/content/90/15_Supplement/P4.436.
- Azuma, Y.; Tokuda, T.; Kushimura, Y.; Yamamoto, I.; Mizuta, I.; Mizuno, T.; Nakagawa, M.; Ueyama, M.; Nagai, Y.; Iwasaki, Y.; et al. Hippo, Drosophila MST, Is a Novel Modifier of Motor Neuron Degeneration Induced by Knockdown of Caz, Drosophila FUS. Exp. Cell Res. 2018, 371, 311–321. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, J.H.; Hwang, S.G.; Gwag, B.J.; McKee, A.C.; Lee, J.; Kowall, N.W.; Ryu, H.; Lim, D.S.; Choi, E.J. MST1 Functions as a Key Modulator of Neurodegeneration in a Mouse Model of ALS. Proc. Natl. Acad. Sci. USA 2013, 110, 12066–12071. [Google Scholar] [CrossRef]
- Matsumoto, H.; Murakami, Y.; Kataoka, K.; Lin, H.; Connor, K.M.; Miller, J.W.; Zhou, D.; Avruch, J.; Vavvas, D.G. Mammalian STE20-like Kinase 2, Not Kinase 1, Mediates Photoreceptor Cell Death during Retinal Detachment. Cell Death Dis. 2014, 5, e1269. [Google Scholar] [CrossRef]
- Osborne, P.B.; Halliday, G.M.; Cooper, H.M.; Keast, J.R. Localization of Immunoreactivity for Deleted in Colorectal Cancer (DCC), the Receptor for the Guidance Factor Netrin-1, in Ventral Tier Dopamine Projection Pathways in Adult Rodents. Neuroscience 2005, 131, 671–681. [Google Scholar] [CrossRef]
- Lin, L.; Isacson, O. Axonal Growth Regulation of Fetal and Embryonic Stem Cell-Derived Dopaminergic Neurons by Netrin-1 and Slits. Stem Cells 2006, 24, 2504. [Google Scholar] [CrossRef]
- Li, J.; Duarte, T.; Kocabas, A.; Works, M.; McConnell, S.K.; Hynes, M.A. Evidence for Topographic Guidance of Dopaminergic Axons by Differential Netrin-1 Expression in the Striatum. Mol. Cell. Neurosci. 2014, 61, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Kang, S.S.; Qi, Q.; Liu, X.; Ye, K. Netrin1 Deficiency Activates MST1 via UNC5B Receptor, Promoting Dopaminergic Apoptosis in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 24503–24513. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt Signaling in the Nervous System and in Alzheimer’s Disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef]
- De Ferrari and, G.V.; Inestrosa, N.C. Wnt Signaling Function in Alzheimer’s Disease. Brain Res. Brain Res. Rev. 2000, 33, 1–12. [Google Scholar] [CrossRef]
- Cerpa, W.; Toledo, E.M.; Varela-Nallar, L.; Inestrosa, N.C. The Role of Wnt Signaling in Neuroprotection. Drug News Perspect. 2009, 22, 579–591. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Moon, R.T. The Ups and Downs of Wnt Signaling in Prevalent Neurological Disorders. Oncogene 2006, 25, 7545–7553. [Google Scholar] [CrossRef]
- Al-Harthi, L. Wnt/β-Catenin and Its Diverse Physiological Cell Signaling Pathways in Neurodegenerative and Neuropsychiatric Disorders. J. Neuroimmune Pharmacol. 2012, 7, 725. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Montecinos-Oliva, C.; Fuenzalida, M. Wnt Signaling: Role in Alzheimer Disease and Schizophrenia. J. Neuroimmune Pharmacol. 2012, 7, 788–807. [Google Scholar] [CrossRef]
- Boonen, R.A.C.M.; van Tijn, P.; Zivkovic, D. Wnt Signaling in Alzheimer’s Disease: Up or down, That Is the Question. Ageing Res. Rev. 2009, 8, 71–82. [Google Scholar] [CrossRef]
- Toledo, E.M.; Colombres, M.; Inestrosa, N.C. Wnt Signaling in Neuroprotection and Stem Cell Differentiation. Prog. Neurobiol. 2008, 86, 281–296. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Chacón, M.A.; Barría, M.I.; Garrido, J.L.; Godoy, J.A.; Olivares, G.; Reyes, A.E.; Alvarez, A.; Bronfman, M.; Inestrosa, N.C. Activation of Wnt Signaling Rescues Neurodegeneration and Behavioral Impairments Induced by Beta-Amyloid Fibrils. Mol. Psychiatry 2003, 8, 195–208. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Avila, M.E.; Medina, M.A.; Pérez-Palma, E.; Bustos, B.I.; Alarcón, M.A. Wnt/β-Catenin Signaling in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2014, 13, 745–754. [Google Scholar] [CrossRef]
- Alvarez, A.R.; Godoy, J.A.; Mullendorff, K.; Olivares, G.H.; Bronfman, M.; Inestrosa, N.C. Wnt-3a Overcomes Beta-Amyloid Toxicity in Rat Hippocampal Neurons. Exp. Cell Res. 2004, 297, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimers. Dis. 2013, 33, S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a Negative Modulator of the Wnt Pathway, Is Associated with Neuronal Degeneration in Alzheimer’s Brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef]
- Purro, S.A.; Dickins, E.M.; Salinas, P.C. The Secreted Wnt Antagonist Dickkopf-1 Is Required for Amyloid β-Mediated Synaptic Loss. J. Neurosci. 2012, 32, 3492–3498. [Google Scholar] [CrossRef]
- Scali, C.; Caraci, F.; Gianfriddo, M.; Diodato, E.; Roncarati, R.; Pollio, G.; Gaviraghi, G.; Copani, A.; Nicoletti, F.; Terstappen, G.C.; et al. Inhibition of Wnt Signaling, Modulation of Tau Phosphorylation and Induction of Neuronal Cell Death by DKK1. Neurobiol. Dis. 2006, 24, 254–265. [Google Scholar] [CrossRef]
- Dickins, E.M.; Salinas, P.C. Wnts in Action: From Synapse Formation to Synaptic Maintenance. Front. Cell. Neurosci. 2013, 7, 162. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, W.K.; Shi, X.D.; Li, Y. Curcumin Activates Wnt/β-Catenin Signaling Pathway through Inhibiting the Activity of GSK-3β in APPswe Transfected SY5Y Cells. Eur. J. Pharm. Sci. 2011, 42, 540–546. [Google Scholar] [CrossRef]
- Lin, C.H.; Tsai, P.I.; Wu, R.M.; Chien, C.T. LRRK2 G2019S Mutation Induces Dendrite Degeneration through Mislocalization and Phosphorylation of Tau by Recruiting Autoactivated GSK3β. J. Neurosci. 2010, 30, 13138. [Google Scholar] [CrossRef]
- Sancho, R.M.; Law, B.M.H.; Harvey, K. Mutations in the LRRK2 Roc-COR Tandem Domain Link Parkinson’s Disease to Wnt Signaling Pathways. Hum. Mol. Genet. 2009, 18, 3955–3968. [Google Scholar] [CrossRef] [PubMed]
- Berwick, D.C.; Harvey, K. LRRK2 Functions as a Wnt Signaling Scaffold, Bridging Cytosolic Proteins and Membrane-Localized LRP6. Hum. Mol. Genet. 2012, 21, 4966–4979. [Google Scholar] [CrossRef] [PubMed]
- Dun, Y.; Yang, Y.; Xiong, Z.; Feng, M.; Zhang, Y.; Wang, M.; Xiang, J.; Li, G.; Ma, R. Induction of Dickkopf-1 Contributes to the Neurotoxicity of MPP+ in PC12 Cells via Inhibition of the Canonical Wnt Signaling Pathway. Neuropharmacology 2013, 67, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zu, G.; Zhang, X.; Wang, X.; Li, S.; Gong, X.; Liang, Z.; Zhao, J. Neuroprotective Effects of Ginsenoside Rg1 through the Wnt/β-Catenin Signaling Pathway in Both in Vivo and in Vitro Models of Parkinson’s Disease. Neuropharmacology 2016, 101, 480–489. [Google Scholar] [CrossRef]
- Dun, Y.; Li, G.; Yang, Y.; Xiong, Z.; Feng, M.; Wang, M.; Zhang, Y.; Xiang, J.; Ma, R. Inhibition of the Canonical Wnt Pathway by Dickkopf-1 Contributes to the Neurodegeneration in 6-OHDA-Lesioned Rats. Neurosci. Lett. 2012, 525, 83–88. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 Regulated Frizzled-1/β-Catenin Signaling Pathway as a Candidate Regulatory Circuit Controlling Mesencephalic Dopaminergic Neuron-Astrocyte Crosstalk: Therapeutical Relevance for Neuron Survival and Neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.C.; et al. Reactive Astrocytes and Wnt/β-Catenin Signaling Link Nigrostriatal Injury to Repair in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Parkinson’s Disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef]
- Yu, L.; Guan, Y.; Wu, X.; Chen, Y.; Liu, Z.; Du, H.; Wang, X. Wnt Signaling Is Altered by Spinal Cord Neuronal Dysfunction in Amyotrophic Lateral Sclerosis Transgenic Mice. Neurochem. Res. 2013, 38, 1904. [Google Scholar] [CrossRef]
- Wang, S.; Guan, Y.; Chen, Y.; Li, X.; Zhang, C.; Yu, L.; Zhou, F.; Wang, X. Role of Wnt1 and Fzd1 in the Spinal Cord Pathogenesis of Amyotrophic Lateral Sclerosis-Transgenic Mice. Biotechnol. Lett. 2013, 35, 1199–1207. [Google Scholar] [CrossRef]
- Chen, Y.; Guan, Y.; Zhang, Z.; Liu, H.; Wang, S.; Yu, L.; Wu, X.; Wang, X. Wnt Signaling Pathway Is Involved in the Pathogenesis of Amyotrophic Lateral Sclerosis in Adult Transgenic Mice. Neurol. Res. 2012, 34, 390–399. [Google Scholar] [CrossRef]
- Chen, Y.; Guan, Y.; Liu, H.; Wu, X.; Yu, L.; Wang, S.; Zhao, C.; Du, H.; Wang, X. Activation of the Wnt/β-Catenin Signaling Pathway Is Associated with Glial Proliferation in the Adult Spinal Cord of ALS Transgenic Mice. Biochem. Biophys. Res. Commun. 2012, 420, 397–403. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, C.; Gonzalez, P.; Andres-Benito, P.; Ferrer, I.; Rodríguez, F.J. Wnt Signaling Alterations in the Human Spinal Cord of Amyotrophic Lateral Sclerosis Cases: Spotlight on Fz2 and Wnt5a. Mol. Neurobiol. 2019, 56, 6777–6791. [Google Scholar] [CrossRef] [PubMed]
- Libro, R.; Bramanti, P.; Mazzon, E. The Role of the Wnt Canonical Signaling in Neurodegenerative Diseases. Life Sci. 2016, 158, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Takeda, N.; Kondo, M.; Kubota, M.; Saito, T.; Maruyama, J.; Fujiwara, T.; Maemura, S.; Ito, M.; Naito, A.T.; et al. Discovery of a Small Molecule to Increase Cardiomyocytes and Protect the Heart After Ischemic Injury. JACC Basic Transl. Sci. 2018, 3, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; He, Z.; Kong, L.L.; Chen, Q.; Yuan, Q.; Zhang, S.; Ye, J.; Liu, H.; Sun, X.; Geng, J.; et al. Pharmacological Targeting of Kinases MST1 and MST2 Augments Tissue Repair and Regeneration. Sci. Transl. Med. 2016, 8, 352ra108. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shen, X.; Wang, J.; Feng, W.; Wang, M.; Miao, X.; Wu, Q.; Wu, L.; Wang, X.; Ma, Y.; et al. YAP Prevents Premature Senescence of Astrocytes and Cognitive Decline of Alzheimer’s Disease through Regulating CDK6 Signaling. Aging Cell 2021, 20, e13465. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.; Liu, H.; Jin, L.; Shen, X.; Xie, C.; Xiang, W.; Yang, D.; Feng, W.; Wang, J.; et al. Astrocytic YAP Prevents the Demyelination through Promoting Expression of Cholesterol Synthesis Genes in Experimental Autoimmune Encephalomyelitis. Cell Death Dis. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Xie, C.; Shen, X.; Xu, X.; Liu, H.; Li, F.; Lu, S.; Gao, Z.; Zhang, J.; Wu, Q.; Yang, D.; et al. Astrocytic YAP Promotes the Formation of Glia Scars and Neural Regeneration after Spinal Cord Injury. J. Neurosci. 2020, 40, 2644–2662. [Google Scholar] [CrossRef] [PubMed]
- Kastan, N.; Gnedeva, K.; Alisch, T.; Petelski, A.A.; Huggins, D.J.; Chiaravalli, J.; Aharanov, A.; Shakked, A.; Tzahor, E.; Nagiel, A.; et al. Small-Molecule Inhibition of Lats Kinases May Promote Yap-Dependent Proliferation in Postmitotic Mammalian Tissues. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Aihara, A.; Iwawaki, T.; Abe-Fukasawa, N.; Otsuka, K.; Saruhashi, K.; Mikashima, T.; Nishino, T. Small Molecule LATS Kinase Inhibitors Block the Hippo Signaling Pathway and Promote Cell Growth under 3D Culture Conditions. J. Biol. Chem. 2022, 298, 101779. [Google Scholar] [CrossRef]
- Shalhout, S.Z.; Yang, P.Y.; Grzelak, E.M.; Nutsch, K.; Shao, S.; Zambaldo, C.; Iaconelli, J.; Ibrahim, L.; Stanton, C.; Chadwick, S.R.; et al. YAP-Dependent Proliferation by a Small Molecule Targeting Annexin A2. Nat. Chem. Biol. 2021, 17, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Mejuch, T.; Chakraborty, S.; Karatas, H.; Bharath, S.R.; Guéret, S.M.; Goy, P.A.; Hahne, G.; Pahl, A.; Sievers, S.; et al. Identification of Quinolinols as Activators of TEAD-Dependent Transcription. ACS Chem. Biol. 2019, 14, 2909–2921. [Google Scholar] [CrossRef] [PubMed]
- Adihou, H.; Gopalakrishnan, R.; Förster, T.; Guéret, S.M.; Gasper, R.; Geschwindner, S.; Carrillo García, C.; Karatas, H.; Pobbati, A.V.; Vazquez-Chantada, M.; et al. A Protein Tertiary Structure Mimetic Modulator of the Hippo Signaling Pathway. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, F.M.; Kastan, N.R.; Haremaki, T.; Tian, Q.; Laundos, T.L.; De Santis, R.; Beaudoin, A.J.; Carroll, T.S.; Luo, J.D.; Gnedeva, K.; et al. Role of YAP in Early Ectodermal Specification and a Huntington’s Disease Model of Human Neurulation. elife 2022, 11, 73075. [Google Scholar] [CrossRef] [PubMed]
- Norflus, F.; Nanje, A.; Gutekunst, C.A.; Shi, G.; Cohen, J.; Bejarano, M.; Fox, J.; Ferrante, R.J.; Hersch, S.M. Anti-Inflammatory Treatment with Acetylsalicylate or Rofecoxib Is Not Neuroprotective in Huntington’s Disease Transgenic Mice. Neurobiol. Dis. 2004, 17, 319–325. [Google Scholar] [CrossRef]
- Schilling, G.; Savonenko, A.V.; Coonfield, M.L.; Morton, J.L.; Vorovich, E.; Gale, A.; Neslon, C.; Chan, N.; Eaton, M.; Fromholt, D.; et al. Environmental, Pharmacological, and Genetic Modulation of the HD Phenotype in Transgenic Mice. Exp. Neurol. 2004, 187, 137–149. [Google Scholar] [CrossRef]
- Bayle, E.D.; Svensson, F.; Atkinson, B.N.; Steadman, D.; Willis, N.J.; Woodward, H.L.; Whiting, P.; Vincent, J.P.; Fish, P.V. Carboxylesterase Notum Is a Druggable Target to Modulate Wnt Signaling. J. Med. Chem. 2021, 64, 4289–4311. [Google Scholar] [CrossRef]
- Zhao, Y.; Jolly, S.; Benvegnu, S.; Yvonne Jones, E.; Fish, P.V. Small-Molecule Inhibitors of Carboxylesterase Notum. Future Med. Chem. 2021, 13, 1001–1015. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sileo, P.; Simonin, C.; Melnyk, P.; Chartier-Harlin, M.-C.; Cotelle, P. Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington’s Disease and Other Neurodegenerative Disorders. Cells 2022, 11, 3631. https://doi.org/10.3390/cells11223631
Sileo P, Simonin C, Melnyk P, Chartier-Harlin M-C, Cotelle P. Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington’s Disease and Other Neurodegenerative Disorders. Cells. 2022; 11(22):3631. https://doi.org/10.3390/cells11223631
Chicago/Turabian StyleSileo, Pasquale, Clémence Simonin, Patricia Melnyk, Marie-Christine Chartier-Harlin, and Philippe Cotelle. 2022. "Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington’s Disease and Other Neurodegenerative Disorders" Cells 11, no. 22: 3631. https://doi.org/10.3390/cells11223631
APA StyleSileo, P., Simonin, C., Melnyk, P., Chartier-Harlin, M.-C., & Cotelle, P. (2022). Crosstalk between the Hippo Pathway and the Wnt Pathway in Huntington’s Disease and Other Neurodegenerative Disorders. Cells, 11(22), 3631. https://doi.org/10.3390/cells11223631