
Targeting the CD47-SIRPα Axis: Present Therapies and the Future for Cutaneous T-cell Lymphoma



Abstract
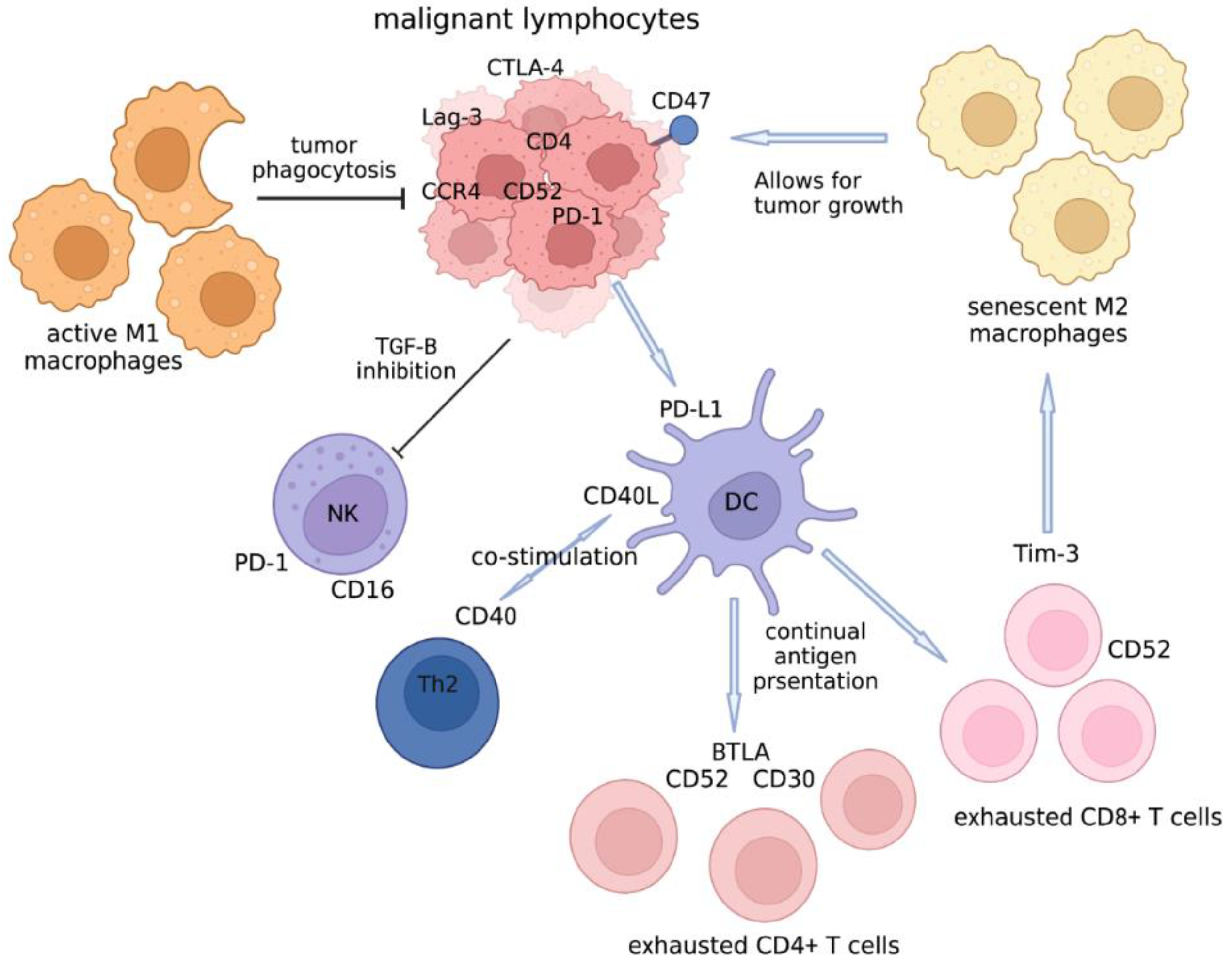
1. Introduction
2. Anti-CD47-SIRPα Agents
2.1. Anti-CD47 Antibodies
2.2. Anti-SIRPα Antibodies: cc-95251 and BI 765063
2.3. SIRPα Fusion Proteins: TTI-621, TTI-622, ALX-148
2.4. Small Molecules
2.5. Bispecific Antibody/Fusion Proteins: DSP107, HX009, IBI322, and SL-172154
3. Current and Potential Combination with Anti-CD47 Agents for CTCL
3.1. Current Biologic Agents Approved for CTCL
3.2. Current Chemotherapeutic Agents Approved for CTCL
4. Future Targets Currently Not Approved for CTCL in Clinical Trials for Other Cancer and in Preclinical Stage of Development
4.1. The Cellular Players in the Tumor Microenvironment of CTCL That Can Be Targeted
4.2. Other Checkpoint Molecules Besides Anti-PD1/PDL1
4.3. TAG-72
4.4. Immature DCs
4.5. CD40 Ligand in TME of Lymphoma
4.6. BTLA
4.7. Mannose
4.8. TSP1
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindberg, F.P.; Bullard, D.C.; Caver, T.E.; Gresham, H.D.; Beaudet, A.L.; Brown, E.J. Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science 1996, 274, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Anzengruber, F.; Ignatova, D.; Schlaepfer, T.; Chang, Y.T.; French, L.E.; Pascolo, S.; Contassot, E.; Bobrowicz, M.; Hoetzenecker, W.; Guenova, E. Divergent LAG-3 versus BTLA, TIGIT, and FCRL3 expression in Sezary syndrome. Leuk Lymphoma 2019, 60, 1899–1907. [Google Scholar] [CrossRef]
- Gao, A.G.; Lindberg, F.P.; Dimitry, J.M.; Brown, E.J.; Frazier, W.A. Thrombospondin modulates alpha v beta 3 function through integrin-associated protein. J. Cell Biol. 1996, 135, 533–544. [Google Scholar] [CrossRef]
- Liu, Y.; Merlin, D.; Burst, S.L.; Pochet, M.; Madara, J.L.; Parkos, C.A. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J. Biol. Chem. 2001, 276, 40156–40166. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Ohnishi, H.; Okazawa, H.; Tomonaga, H.; Hayashi, A.; Fujimoto, T.T.; Furuya, N.; Matozaki, T. Promotion of neurite and filopodium formation by CD47: Roles of integrins, Rac, and Cdc42. Mol. Biol. Cell. 2004, 15, 3950–3963. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, M.I.; Lindberg, F.P.; Kersh, G.J.; Allen, P.M.; Brown, E.J. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J. Exp. Med. 1997, 185, 1–11. [Google Scholar] [CrossRef]
- Okazawa, H.; Motegi, S.; Ohyama, N.; Ohnishi, H.; Tomizawa, T.; Kaneko, Y.; Oldenborg, P.A.; Ishikawa, O.; Matozaki, T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 2005, 174, 2004–2011. [Google Scholar] [CrossRef]
- Olcucuoglu, E.; Sirin, M.E.; Aydog, G.; Gazel, E.; Tastemur, S.; Odabas, O. Relationship between immunohistochemical staining extent of CD47 and histopathologic features of bladder tumor. Cent. Eur. J. Urol. 2017, 70, 349–355. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Rendtlew Danielsen, J.M.; Knudsen, L.M.; Dahl, I.M.; Lodahl, M.; Rasmussen, T. Dysregulation of CD47 and the ligands thrombospondin 1 and 2 in multiple myeloma. Br. J. Haematol. 2007, 138, 756–760. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.D.S.; Banerjee, S.; Kruglov, O.; Viller, N.N.; Horwitz, S.M.; Lesokhin, A.; Zain, J.; Querfeld, C.; Chen, R.; Okada, C.; et al. Targeting CD47 in Sézary syndrome with SIRPαFc. Blood Adv. 2019, 3, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, O.; Johnson, L.D.S.; Minic, A.; Jordan, K.; Uger, R.A.; Wong, M.; Sievers, E.L.; Shou, Y.; Akilov, O.E. The pivotal role of cytotoxic NK cells in mediating the therapeutic effect of anti-CD47 therapy in mycosis fungoides. Cancer Immunol. Immunother. 2021, 71, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef]
- Puro, R.J.; Bouchlaka, M.N.; Hiebsch, R.R.; Capoccia, B.J.; Donio, M.J.; Manning, P.T.; Frazier, W.A.; Karr, R.W.; Pereira, D.S. Development of AO-176, a Next-Generation Humanized Anti-CD47 Antibody with Novel Anticancer Properties and Negligible Red Blood Cell Binding. Mol. Cancer Ther. 2020, 19, 835–846. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef]
- Burris III, H.A.; Spira, A.I.; Taylor, M.H.; Yeku, O.O.; Liu, J.F.; Munster, P.N.; Hamilton, E.P.; Thomas, J.S.; Gatlin, F.; Penson, R.T.; et al. A first-in-human study of AO-176, a highly differentiated anti-CD47 antibody, in patients with advanced solid tumors. In Proceedings of the ASCO Annnual Meeting I, Alexandria, VA, USA, 3–7 June 2021. [Google Scholar]
- Crescioli, S.; Correa, I.; Karagiannis, P.; Davies, A.M.; Sutton, B.J.; Nestle, F.O.; Karagiannis, S.N. IgG4 Characteristics and Functions in Cancer Immunity. Curr. Allergy Asthma Rep. 2016, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Graziano, R.F.; Engelhardt, J.J. Role of FcγRs in Antibody-Based Cancer Therapy. Curr. Top. Microbiol. Immunol. 2019, 423, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Asch, A.S.; Kambhampati, S.; Al Malki, M.M.; Zeidner, J.F.; Donnellan, W.; Lee, D.J.; Vyas, P.; Jeyakumar, D.; Mannis, G.N.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab Combined with Azacitidine Is Well-Tolerated and Effective in AML Patients: Phase 1b Results. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. [Google Scholar]
- Liu, B.; Guo, H.; Xu, J.; Qin, T.; Guo, Q.; Gu, N.; Zhang, D.; Qian, W.; Dai, J.; Hou, S.; et al. Elimination of tumor by CD47/PD-L1 dual-targeting fusion protein that engages innate and adaptive immune responses. MAbs 2018, 10, 315–324. [Google Scholar] [CrossRef]
- de Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Brierley, C.K.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.T.; Murphy, M.F. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, Z.; Guo, B.; Cao, W.; Shen, H. TJC4, a Differentiated Anti-CD47 Antibody with Novel Epitope and RBC Sparing Properties. Blood 2019, 134, 4063. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Qiao, J.; Al-Tamimi, M.; Baker, R.I.; Andrews, R.K.; Gardiner, E.E. The platelet Fc receptor, FcγRIIa. Immunol. Rev. 2015, 268, 241–252. [Google Scholar] [CrossRef]
- Kang, T.H.; Lee, C.H.; Delidakis, G.; Jung, J.; Richard-Le Goff, O.; Lee, J.; Kim, J.E.; Charab, W.; Bruhns, P.; Georgiou, G. An Engineered Human Fc variant With Exquisite Selectivity for FcγRIIIa(V158) Reveals That Ligation of FcγRIIIa Mediates Potent Antibody Dependent Cellular Phagocytosis with GM-CSF-Differentiated Macrophages. Front. Immunol. 2019, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Stefanidakis, M.; Newton, G.; Lee, W.Y.; Parkos, C.A.; Luscinskas, F.W. Endothelial CD47 interaction with SIRPgamma is required for human T-cell transendothelial migration under shear flow conditions in vitro. Blood 2008, 112, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Vermi, W.; Boles, K.S.; Fuchs, A.; Strader, C.A.; Facchetti, F.; Cella, M.; Colonna, M. Adhesion of human T cells to antigen-presenting cells through SIRPbeta2-CD47 interaction costimulates T-cell proliferation. Blood 2005, 105, 2421–2427. [Google Scholar] [CrossRef]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPαFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W.; Flinn, I.W.; Sawas, A.; Minden, M.D.; Villa, D.; Percival, M.M.; Advani, A.S.; et al. Phase I Study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2021, 27, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Thompson, J.A.; Taylor, M.H.; DeSimone, J.A.; Zain, J.M.; Shustov, A.R.; Johns, C.; McCann, S.; Lin, G.H.Y.; Petrova, P.S.; et al. Intralesional TTI-621, a novel biologic targeting the innate immune checkpoint CD47, in patients with relapsed or refractory mycosis fungoides or Sezary syndrome: A multicentre, phase 1 study. Lancet Haematol. 2021, 8, e808–e817. [Google Scholar] [CrossRef]
- Rubio Gonzalez, B.; Zain, J.; Rosen, S.T.; Querfeld, C. Tumor microenvironment in mycosis fungoides and Sezary syndrome. Curr. Opin. Oncol. 2016, 28, 88–96. [Google Scholar] [CrossRef]
- Jiang, T.T.; Kruglov, O.; Lin, G.H.Y.; Minic, A.; Jordan, K.; Uger, R.A.; Wong, M.; Shou, Y.; Akilov, O.E. Clinical Response to Anti-CD47 Immunotherapy Is Associated with Rapid Reduction of Exhausted Bystander CD4(+) BTLA(+) T Cells in Tumor Microenvironment of Mycosis Fungoides. Cancers 2021, 13, 5982. [Google Scholar] [CrossRef]
- Kauder, S.E.; Kuo, T.C.; Harrabi, O.; Chen, A.; Sangalang, E.; Doyle, L.; Rocha, S.S.; Bollini, S.; Han, B.; Sim, J.; et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS ONE 2018, 13, e0201832. [Google Scholar] [CrossRef]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://ir.shattucklabs.com/news-and-events/press-releases (accessed on 10 October 2022).
- Shen, K.; Liu, Y.; Cao, X.; Zhou, D.; Li, J. Successful treatment of refractory Sezary syndrome by anti-PD-1 antibody (nivolumab). Ann. Hematol. 2017, 96, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in Relapsed and Refractory Mycosis Fungoides and Sezary Syndrome: A Multicenter Phase II Study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Saulite, I.; Ignatova, D.; Chang, Y.T.; Fassnacht, C.; Dimitriou, F.; Varypataki, E.; Anzengruber, F.; Nägeli, M.; Cozzio, A.; Dummer, R.; et al. Blockade of programmed cell death protein 1 (PD-1) in Sézary syndrome reduces Th2 phenotype of non-tumoral T lymphocytes but may enhance tumor proliferation. Oncoimmunology 2020, 9, 1738797. [Google Scholar] [CrossRef]
- Knol, A.C.; Quereux, G.; Brocard, A.; Ballanger, F.; Khammari, A.; Nguyen, J.M.; Dreno, B. About the cutaneous targets of bexarotene in CTCL patients. Exp. Dermatol. 2010, 19, e299–e301. [Google Scholar] [CrossRef]
- Gaunt, C.M.; Rainbow, D.B.; Mackenzie, R.J.; Jarvis, L.B.; Mousa, H.S.; Cunniffe, N.; Georgieva, Z.; Brown, J.W.; Coles, A.J.; Jones, J.L. The MS Remyelinating Drug Bexarotene (an RXR Agonist) Promotes Induction of Human Tregs and Suppresses Th17 Differentiation In Vitro. Front. Immunol. 2021, 10, 712241. [Google Scholar] [CrossRef]
- Kelly-Sell, M.J.; Kim, Y.H.; Strauss, S.; Benoit, B.; Harrison, C.; Sutherland, K.; Armstrong, R.; Weng, W.K.; Showe, L.C.; Wysocka, M.; et al. The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T cell lymphoma patients. Am. J. Hematol. 2012, 87, 354–360. [Google Scholar] [CrossRef]
- Wollina, U.; Dummer, R.; Brockmeyer, N.H.; Konrad, H.; Busch, J.O.; Kaatz, M.; Knopf, B.; Koch, H.J.; Hauschild, A. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer 2003, 98, 993–1001. [Google Scholar] [CrossRef]
- Straus, D.J.; Duvic, M.; Kuzel, T.; Horwitz, S.; Demierre, M.F.; Myskowski, P.; Steckel, S. Results of a phase II trial of oral bexarotene (Targretin) combined with interferon alfa-2b (Intron-A) for patients with cutaneous T-cell lymphoma. Cancer 2007, 109, 1799–1803. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47 +/CD73 +/PDL1 + immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef]
- Feliz-Mosquea, Y.R.; Christensen, A.A.; Wilson, A.S.; Westwood, B.; Varagic, J.; Meléndez, G.C.; Schwartz, A.L.; Chen, Q.R.; Mathews Griner, L.; Guha, R.; et al. Combination of anthracyclines and anti-CD47 therapy inhibit invasive breast cancer growth while preventing cardiac toxicity by regulation of autophagy. Breast Cancer Res. Treat 2018, 172, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Jidar, K.; Ingen-Housz-Oro, S.; Beylot-Barry, M.; Paul, C.; Chaoui, D.; Sigal-Grinberg, M.; Morel, P.; Dubertret, L.; Bachelez, H. Gemcitabine treatment in cutaneous T-cell lymphoma: A multicentre study of 23 cases. Br. J. Dermatol. 2009, 161, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Welborn, M.; Duvic, M. Antibody-Based Therapies for Cutaneous T-Cell Lymphoma. Am. J. Clin. Dermatol. 2019, 20, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ollila, T.A.; Sahin, I.; Olszewski, A.J. Mogamulizumab: A new tool for management of cutaneous T-cell lymphoma. Onco Targets Ther. 2019, 12, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Langridge, T.; Duvic, M. Depletion of regulatory T cells by targeting CC chemokine receptor type 4 with mogamulizumab. Oncoimmunology 2015, 4, e1011524. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hirotsu, K.E.; Neal, T.M.; Raghavan, S.S.; Kwong, B.Y.; Khodadoust, M.S.; Brown, R.A.; Novoa, R.A.; Kim, Y.H.; Rieger, K.E. Histopathologic Characterization of Mogamulizumab-associated Rash. Am. J. Surg. Pathol. 2020, 44, 1666–1676. [Google Scholar] [CrossRef]
- Moore, D.C.; Elmes, J.B.; Shibu, P.A.; Larck, C.; Park, S.I. Mogamulizumab: An Anti-CC Chemokine Receptor 4 Antibody for T-Cell Lymphomas. Ann. Pharmacother. 2020, 54, 371–379. [Google Scholar] [CrossRef]
- Oka, T.; Miyagaki, T. Novel and Future Therapeutic Drugs for Advanced Mycosis Fungoides and Sézary Syndrome. Front. Med. (Lausanne) 2019, 6, 116. [Google Scholar] [CrossRef]
- Bagot, M.; Porcu, P.; Marie-Cardine, A.; Battistella, M.; William, B.M.; Vermeer, M.; Whittaker, S.; Rotolo, F.; Ram-Wolff, C.; Khodadoust, M.S.; et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: An international, first-in-human, open-label, phase 1 trial. Lancet Oncol. 2019, 20, 1160–1170. [Google Scholar] [CrossRef]
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schön, G.; Grenzi, P.C.; Bachurski, D.; Aquino, B.; et al. CD30 on extracellular vesicles from malignant Hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with Brentuximab-Vedotin, in vitro. Oncotarget 2016, 7, 30523–30535. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Gautam, A.; Kim, Y.H. Brentuximab vedotin: Targeting CD30 as standard in CTCL. Oncotarget 2018, 9, 11887–11888. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Hordinsky, M.; Lazaryan, A. Impressive response of CD30-negative, treatment-refractory mycosis fungoides to brentuximab vedotin. Dermatol. Ther. 2019, 32, e12835. [Google Scholar] [CrossRef]
- Warren, S.; Kheterpal, M.; Myskowski, P.L.; Moskowitz, A.; Horwitz, S.M.; Pulitzer, M.P. Unrelated immunodeficiency states may impact outcomes and immune checkpoint molecule expression in patients with mycosis fungoides: A clinicopathologic case-control study. J. Am. Acad. Dermatol. 2018, 78, 530–539. [Google Scholar] [CrossRef]
- Cetinözman, F.; Jansen, P.M.; Vermeer, M.H.; Willemze, R. Differential expression of programmed death-1 (PD-1) in Sézary syndrome and mycosis fungoides. Arch. Dermatol. 2012, 148, 1379–1385. [Google Scholar] [CrossRef]
- Katoh, M. Combination immuno-oncology therapy with pembrolizumab, an anti-PD-1 monoclonal antibody targeting immune evasion, and standard chemotherapy for patients with the squamous and non-squamous subtypes of non-small cell lung cancer. J. Thorac. Dis. 2018, 10, 5178–5183. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Marchi, E.; Alinari, L.; Tani, M.; Stefoni, V.; Pimpinelli, N.; Berti, E.; Pagano, L.; Bernengo, M.G.; Zaja, F.; Rupoli, S.; et al. Gemcitabine as frontline treatment for cutaneous T-cell lymphoma: Phase II study of 32 patients. Cancer 2005, 104, 2437–2441. [Google Scholar] [CrossRef]
- Bagot, M.; Nikolova, M.; Schirm-Chabanette, F.; Wechsler, J.; Boumsell, L.; Bensussan, A. Crosstalk between tumor T lymphocytes and reactive T lymphocytes in cutaneous T cell lymphomas. Ann. N. Y. Acad. Sci. 2001, 941, 31–38. [Google Scholar] [CrossRef]
- Querfeld, C.; Leung, S.; Myskowski, P.L.; Curran, S.A.; Goldman, D.A.; Heller, G.; Wu, X.; Kil, S.H.; Sharma, S.; Finn, K.J.; et al. Primary T Cells from Cutaneous T-cell Lymphoma Skin Explants Display an Exhausted Immune Checkpoint Profile. Cancer Immunol. Res. 2018, 6, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Maredia, H.; Cozzio, A.; Dummer, R.; Ramelyte, E.; Kim, E.J.; Rozati, S. Acute progression of the leukemic phase in mycosis fungoides and Sézary syndrome. JAAD Case Rep. 2021, 15, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; Digifico, E.; Anfray, C.; Ummarino, A.; Torres Andon, F. Targeting Tumor-Associated Macrophages in Anti-Cancer Therapies: Convincing the Traitors to Do the Right Thing. J. Clin. Med. 2020, 9, 3226. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef]
- Kulkarni, A.; Chandrasekar, V.; Natarajan, S.K.; Ramesh, A.; Pandey, P.; Nirgud, J.; Bhatnagar, H.; Ashok, D.; Ajay, A.K.; Sengupta, S. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat. Biomed. Eng. 2018, 2, 589–599. [Google Scholar] [CrossRef]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.A.; Brown, N.A.; Wilcox, R.A. Expression of the checkpoint receptors LAG-3, TIM-3 and VISTA in peripheral T cell lymphomas. J. Clin. Pathol. 2020, 73, 197–203. [Google Scholar] [CrossRef]
- Avery, L.; Filderman, J.; Szymczak-Workman, A.L.; Kane, L.P. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. USA 2018, 115, 2455–2460. [Google Scholar] [CrossRef]
- Evtimov, V.; Boyd, R.; Nisbet, I.; Prince, M.; Tounson, A. T Cell Disease Treatment Targeting Tag-72. WIPO (PCT) WO2019161439A1, 29 August 2019. [Google Scholar]
- Shu, R.; Evtimov, V.J.; Hammett, M.V.; Nguyen, N.N.; Zhuang, J.; Hudson, P.J.; Howard, M.C.; Pupovac, A.; Trounson, A.O.; Boyd, R.L. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol. Ther. Oncolytics 2021, 20, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.L.; Hanlon, D.; Kanada, D.; Dhodapkar, M.; Lombillo, V.; Wang, N.; Christensen, I.; Howe, G.; Crouch, J.; El-Fishawy, P.; et al. The growth of cutaneous T-cell lymphoma is stimulated by immature dendritic cells. Blood 2002, 99, 2929–2939. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Mangsbo, S.M.; Broos, S.; Fletcher, E.; Veitonmäki, N.; Furebring, C.; Dahlén, E.; Norlén, P.; Lindstedt, M.; Tötterman, T.H.; Ellmark, P. The human agonistic CD40 antibody ADC-1013 eradicates bladder tumors and generates T-cell-dependent tumor immunity. Clin. Cancer Res. 2015, 21, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- French, R.R.; Chan, H.T.; Tutt, A.L.; Glennie, M.J. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat. Med. 1999, 5, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Cai, H.; Peng, X.; Zhang, P.; Wu, X.; Tian, R. Targeted Imaging of Tumor-Associated Macrophages by Cyanine 7-Labeled Mannose in Xenograft Tumors. Mol. Imaging 2017, 16, 1536012116689499. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wang, Y.; Kang, X.; Wu, A.; Yin, W.; Tang, Y.; Wang, J.; Zhang, M.; Duan, Y.; Huang, Y. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem. Sci. 2018, 9, 2674–2689. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef]
- Martin-Manso, G.; Galli, S.; Ridnour, L.A.; Tsokos, M.; Wink, D.A.; Roberts, D.D. Thrombospondin 1 promotes tumor macrophage recruitment and enhances tumor cell cytotoxicity of differentiated U937 cells. Cancer Res. 2008, 68, 7090–7099. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, J.H.; He, L.; Boyd, D.A.; Hardwick, J.M.; Hung, C.F.; Wu, T.C. Modification of professional antigen-presenting cells with small interfering RNA in vivo to enhance cancer vaccine potency. Cancer Res. 2005, 65, 309–316. [Google Scholar] [CrossRef]
- Folkes, A.S.; Feng, M.; Zain, J.M.; Abdulla, F.; Rosen, S.T.; Querfeld, C. Targeting CD47 as a cancer therapeutic strategy: The cutaneous T-cell lymphoma experience. Curr. Opin. Oncol. 2018, 30, 332–337. [Google Scholar] [CrossRef]
- Sims, S.; Willberg, C.; Klenerman, P. MHC-peptide tetramers for the analysis of antigen-specific T cells. Expert Rev. Vaccines 2010, 9, 765–774. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Company | FortySeven/Gilead | Arch Oncology | I-MAB Biopharma | Cellgene | Surface Oncology | Jiangsu HengRui Medicine | |
|---|---|---|---|---|---|---|---|
| Candidate | Margolimab (5F9) | AO-176 | Ti-061 | TJ011133 (TJC4) | CC-90002 | SRF231 | SHR 1603 |
| Fc isotype | IgG4 | IgG2 | IgG4 | IgG4 | IgG4-PE | IgG4 | IgG4 |
| Lead indication | MDS/AML; DLBL; Solid tumors; Colorectal CA; Hematologic malignancies | Solid tumors; MM; Preclinical: lymphoma and TLL | Solid tumors | R/R solid tumors and lymphoma | Not been used as monotherapy R/R NHL in combination | B cell lymphoma, R/R solid tumors | Advanced CA; hematologic malignancies |
| SIRPα Antibody | SIRPα Proteins | Bispecific Antibody | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Company | Celgene | OSE Immunotherapeutic | Weissman’s group | ALX Oncology | Trillium Therapeutics | Kahr Medical | Waterstone Hanxbio Pty Ltd. (Wuhan, China) | Invent Biologics | Shattuck Labs | |
| Candidate | CC-95251 | BI 765063 (OSE-172) | CV1 | ALX-148 | TTI-621 | TTI-622 | DSP107 | HX009 | IBI322 | SL-172154 |
| Molecule | mAb | mAb IgG4 | Truncated SIRPα protein | WT SIRPα-IgG1 fusion with inactive Fc | WT SIRPα-IgG1 Fc fusion | WT SIRPα-IgG4 Fc fusion | SIRPα/41BB | CD47/PD1 | CD47/PDL1 | SIRPα/40L |
| Lead indication | Solids tumor, leukemia/lymphoma | Advanced solid tumors | Lymphoma; breast CA | HNSCC, gastric CA, breast CA, NHL, MDS, AML | Hematologic malignancies | NSCLC, SCC, advanced solid tumors | Advanced solid tumors | NSCLC, cervical, esophageal, and liver CA, HNSCC | Ovarian CA | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, A.; Akilov, O.E. Targeting the CD47-SIRPα Axis: Present Therapies and the Future for Cutaneous T-cell Lymphoma. Cells 2022, 11, 3591. https://doi.org/10.3390/cells11223591
Xiao A, Akilov OE. Targeting the CD47-SIRPα Axis: Present Therapies and the Future for Cutaneous T-cell Lymphoma. Cells. 2022; 11(22):3591. https://doi.org/10.3390/cells11223591
Chicago/Turabian StyleXiao, Amy, and Oleg E. Akilov. 2022. "Targeting the CD47-SIRPα Axis: Present Therapies and the Future for Cutaneous T-cell Lymphoma" Cells 11, no. 22: 3591. https://doi.org/10.3390/cells11223591
APA StyleXiao, A., & Akilov, O. E. (2022). Targeting the CD47-SIRPα Axis: Present Therapies and the Future for Cutaneous T-cell Lymphoma. Cells, 11(22), 3591. https://doi.org/10.3390/cells11223591