
Palmitate Inhibits Mouse Macrophage Efferocytosis by Activating an mTORC1-Regulated Rho Kinase 1 Pathway: Therapeutic Implications for the Treatment of Obesity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Generation of BMDMs and Treatments
2.4. Generation of Apoptotic Thymocytes
2.5. Cell Viability Assay
2.6. Efferocytosis Assays
2.7. Fluorescent Microscopy
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis of mRNA Expression
2.9. Western Blot Analysis
2.10. Determination of Rac1, Cdc42 and RhoA Activity in Macrophages
2.11. Staining of Engulfing BMDMs for Confocal Lase-Scanning Microscopy
2.12. Confocal Laser Scanning Microscopy
2.13. Statistical Analysis
3. Results
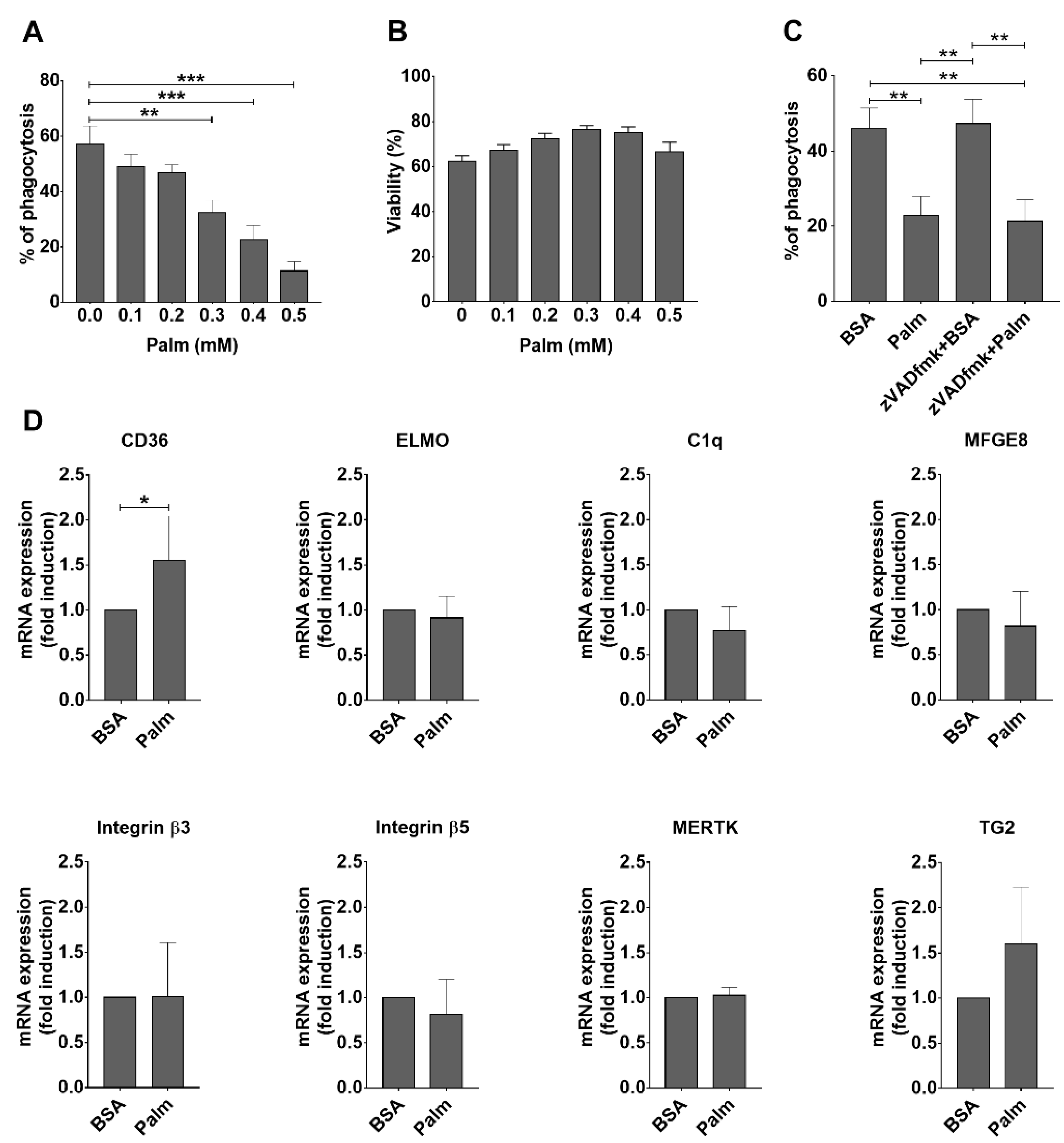
3.1. Palmitate Exposure Inhibits Phagocytosis of Apoptotic Cells by BMDMs without Significantly Affecting the mRNA Expression of Various Phagocytosis-Related Molecules
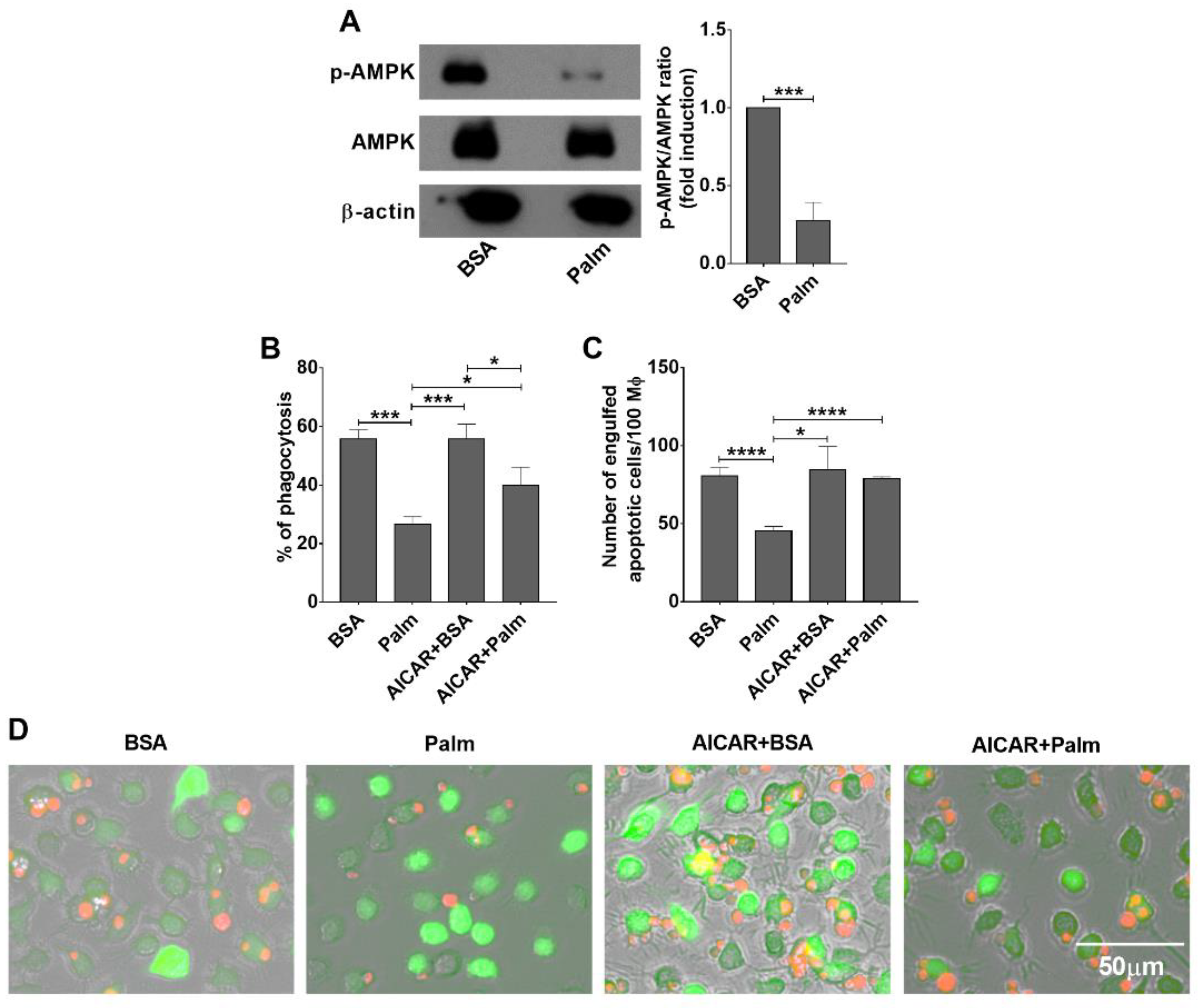
3.2. Exposure to Palmitate Affects Efferocytosis Very Likely via the Energy Sensing Mechanisms in BMDMs
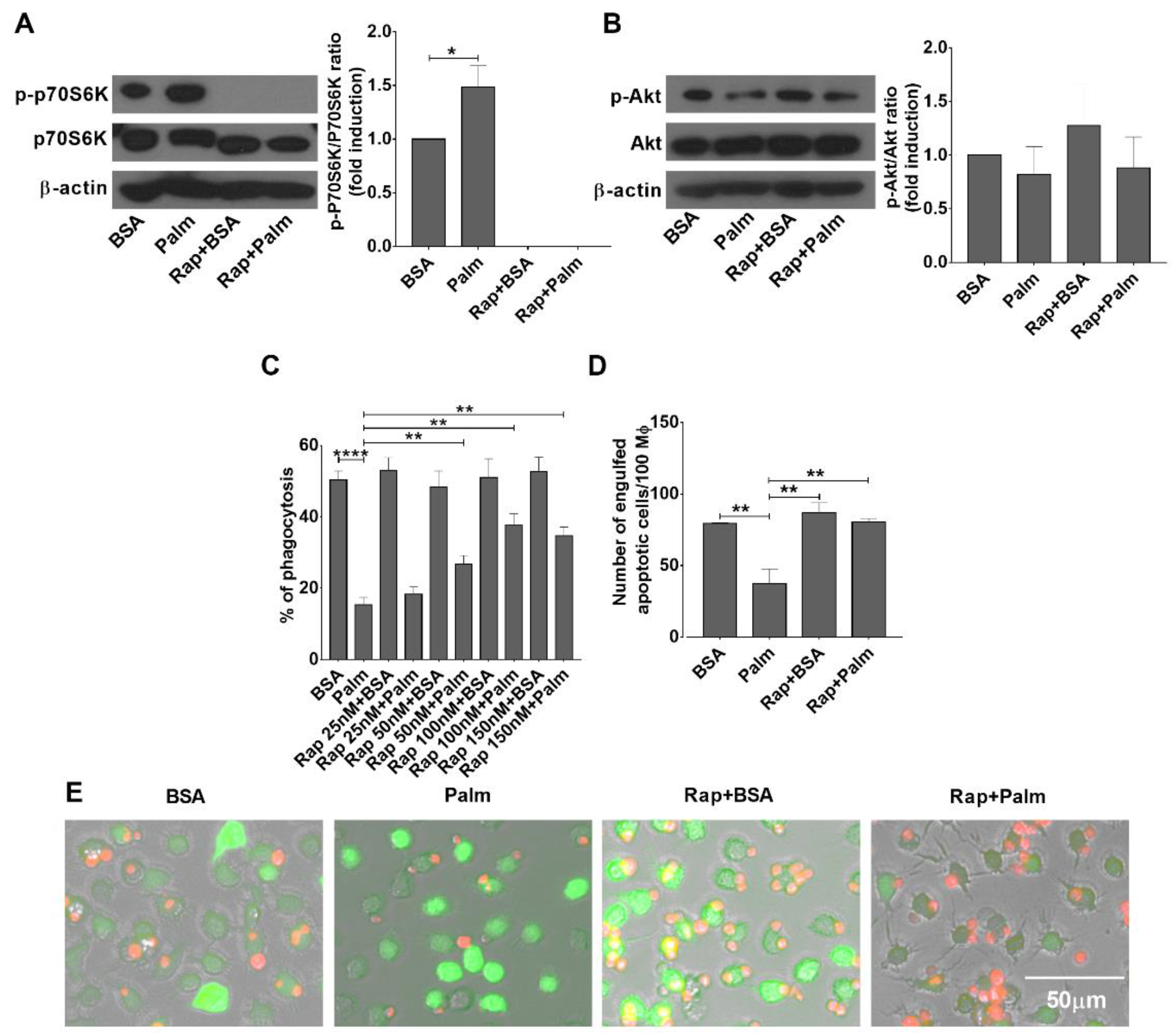
3.3. Palmitate Exposure Triggers mTORC1 Activity in BMDMs
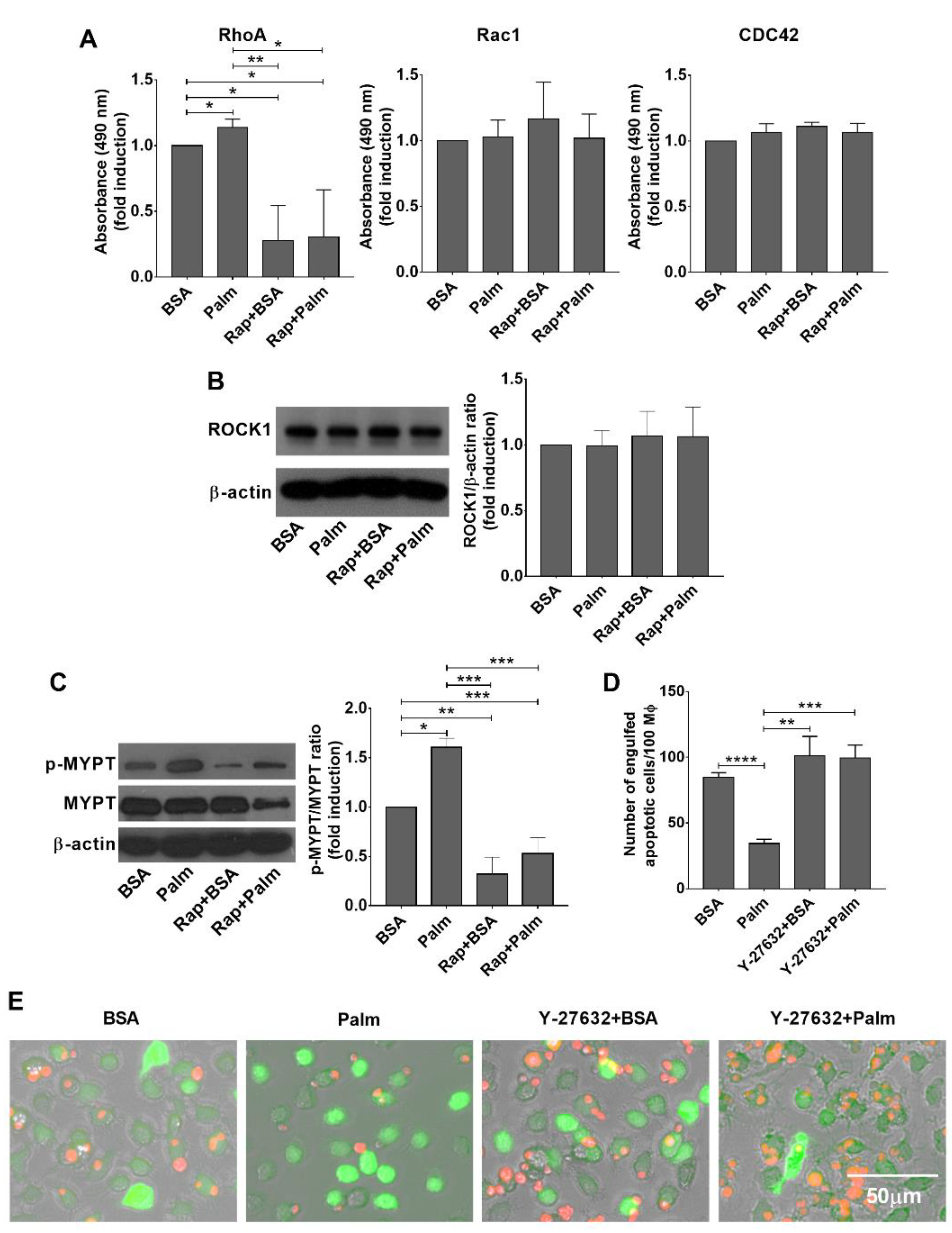
3.4. Palmitate Exposure Triggers an mTORC1 Dependent Rho Kinase Activation to Inhibit Efferocytosis
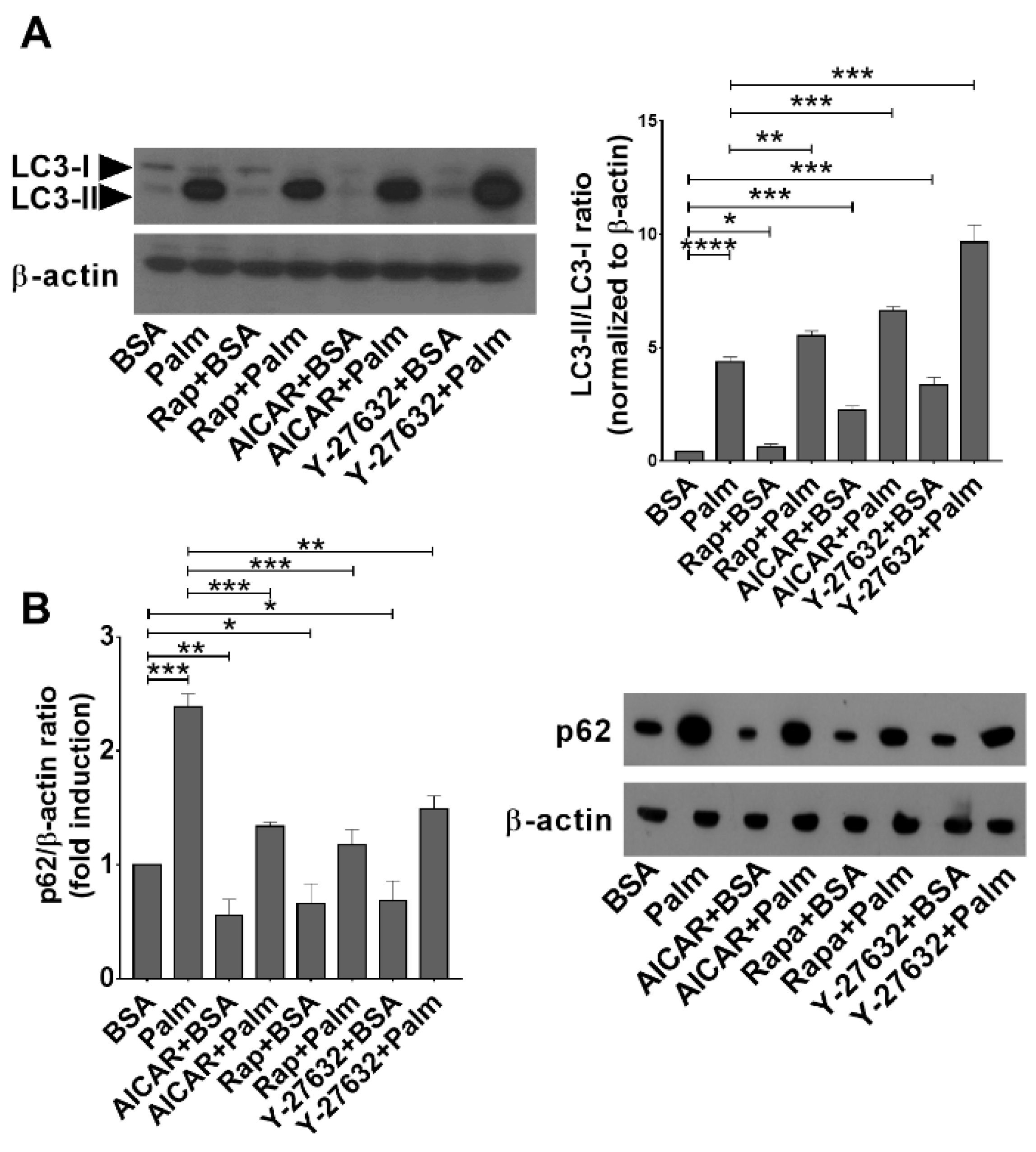
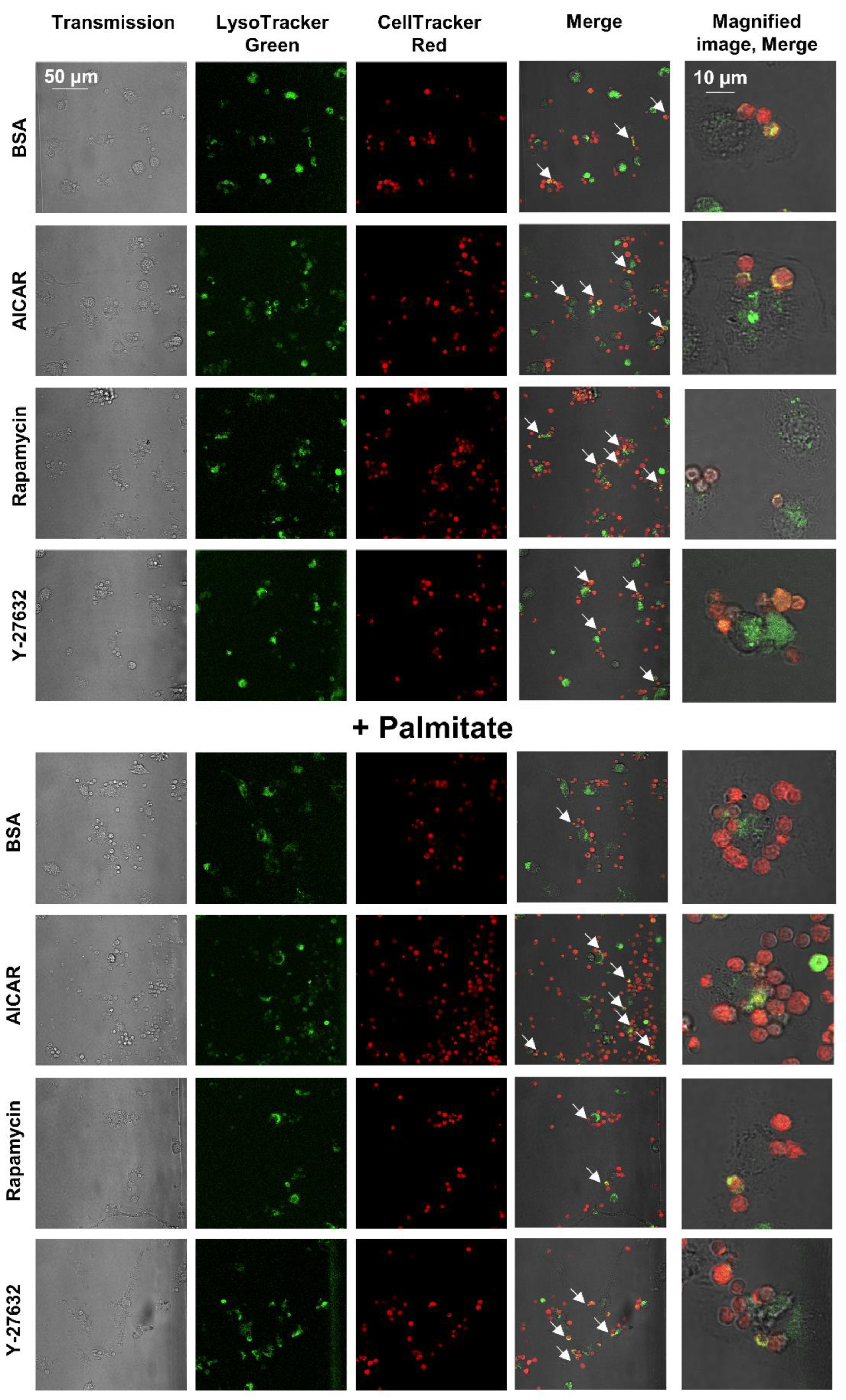
3.5. Palmitate Exposure Induces Autophagy but Inhibits the Fusion of Autophagic Vacuoles and Lysosomes
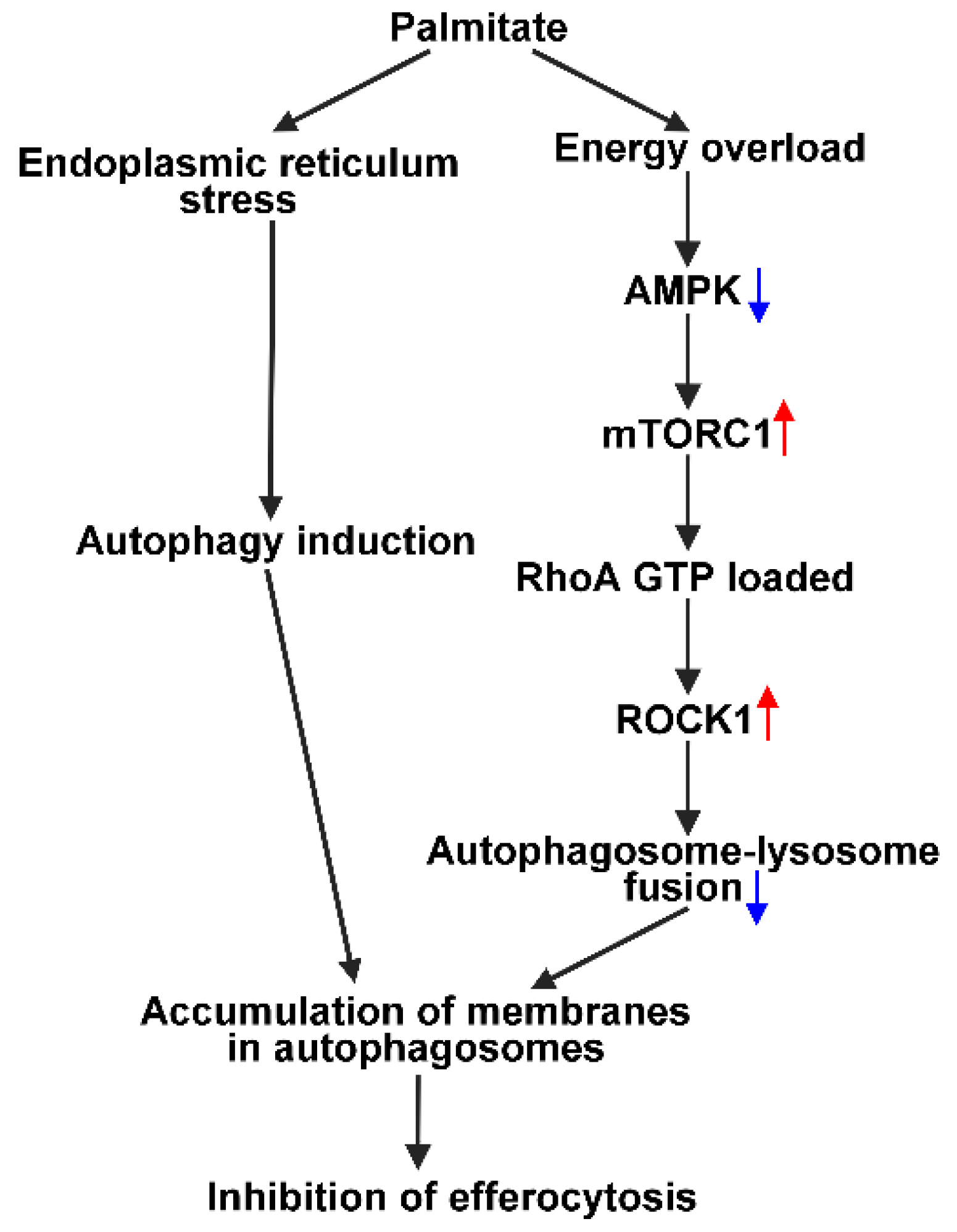
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hart, S.P.; Dransfield, I.; Rossi, A.G. Phagocytosis of apoptotic cells. Methods 2008, 44, 280–285. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Barth, N.D.; Marwick, J.A.; Vendrell, M.; Rossi, A.G.; Dransfield, I. Phagocytic synapse” and clearance of apoptotic cells. Front. Immunol. 2017, 8, 1708. [Google Scholar] [CrossRef]
- Tóth, B.; Garabuczi, E.; Sarang, Z.; Vereb, G.; Vámosi, G.; Aeschlimann, D.; Blaskó, B.; Bécsi, B.; Erdődi, F.; Lacy-Hulbert, A.; et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J. Immunol. 2009, 82, 2084–2092. [Google Scholar] [CrossRef]
- Nakaya, M.; Kitano, M.; Matsuda, M.; Nagata, S. Spatiotemporal activation of Rac1 for engulfment of apoptotic cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9198–9203. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Nakada-Tsukui, K.; Ravichandran, K.S. Engulfment of apoptotic cells is negatively regulated by Rho-mediated signaling. J. Biol. Chem. 2003, 278, 49911–49919. [Google Scholar] [CrossRef]
- Kobayashi, S.; Shirai, T.; Kiyokawa, E.; Mochizuki, N.; Matsuda, M.; Fukui, Y. Membrane recruitment of DOCK180 by binding to PtdIns(3,4,5)P3. Biochem. J. 2001, 354, 73–78. [Google Scholar] [CrossRef]
- Cote, J.F.; Motoyama, A.B.; Bush, J.A.; Vuori, K. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat. Cell Biol. 2005, 7, 797–807. [Google Scholar] [CrossRef]
- Croisé, P.; Estay-Ahumada, C.; Gasman, S.; Ory, S. Rho GTPases, phosphoinositides, and actin: A tripartite framework for efficient vesicular trafficking. Small GTPases 2014, 5, e29469. [Google Scholar] [CrossRef]
- Thi, E.P.; Reiner, N.E. Phosphatidylinositol 3-kinases and their roles in phagosome maturation. J. Leukoc. Biol. 2012, 92, 553–566. [Google Scholar] [CrossRef]
- Teplova, I.; Lozy, F.; Price, S.; Singh, S.; Barnard, N.; Cardiff, R.D.; Birge, R.; Karantza, V. ATG proteins mediate efferocytosis and suppress inflammation in mammary involution. Autophagy 2013, 4, 459–475. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-associated phagocytosis and inflammation. J. Mol. Biol. 2017, 429, 3561–3576. [Google Scholar] [CrossRef]
- Dossou, A.S.; Basu, A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef]
- Mellman, I.S.; Plutner, H.; Steinman, R.M.; Unkeless, J.C.; Cohn, Z.A. Internalization and degradation of macrophage Fc receptors during receptor-mediated phagocytosis. J. Cell Biol. 1983, 96, 887–895. [Google Scholar] [CrossRef]
- Hackam, D.J.; Rotstein, O.D.; Sjolin, C.; Schreiber, A.D.; Trimble, W.S.; Grinstein, S. v-SNARE-dependent secretion is required for phagocytosis. Proc. Natl. Acad. Sci. USA 1998, 95, 11691–11696. [Google Scholar] [CrossRef]
- Czibener, C.; Sherer, N.M.; Becker, S.M.; Pypaert, M.; Hui, E.; Chapman, E.R.; Mothes, W.; Andrews, N.W. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol. 2006, 174, 997–1007. [Google Scholar] [CrossRef]
- Yin, C.; Argintaru, D.; Heit, B. Rab17 mediates intermixing of phagocytosed apoptotic cells with recycling endosomes. Small GTPases 2019, 10, 218–226. [Google Scholar] [CrossRef]
- Haobam, B.; Nozawa, T.; Minowa-Nozawa, A.; Tanaka, M.; Oda, S.; Watanabe, T.; Aikawa, C.; Maruyama, F.; Nakagawa, I. Rab17-mediated recycling endosomes contribute to autophagosome formation in response to Group A Streptococcus invasion. Cell Microbiol. 2014, 16, 1806–1821. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Li, S.; Sun, Y.; Liang, C.P.; Thorp, E.B.; Han, S.; Jehle, A.W.; Saraswathi, V.; Pridgen, B.; Kanter, J.E.; Li, R.; et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ. Res. 2009, 105, 1072–1082. [Google Scholar] [CrossRef]
- Rocha, V.; Libby, P. Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, J.K.; Heo, E.Y.; Kim, D.K.; Chung, H.S. The effect of obesity on patients with mild chronic obstructive pulmonary disease: Results from KNHANES 2010 to 2012. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 2, 757–763. [Google Scholar] [CrossRef][Green Version]
- Gremese, E.; Tolusso, B.; Gigante, M.R.; Ferraccioli, G. Obesity as a risk and severity factor in rheumatic diseases (autoimmune chronic inflammatory diseases). Front. Immunol. 2014, 5, 576. [Google Scholar] [CrossRef]
- Fige, E.; Sarang, Z.; Sós, L.; Szondy, Z. Retinoids promote mouse bone marrow-derived macrophage differentiation and efferocytosis via upregulating bone morphogenetic protein-2 and Smad3. Cells 2022, 11, 2928. [Google Scholar] [CrossRef]
- Available online: http://www.wklab.org/wp-content/uploads/2016/02/Palmitate-BSA_Prep_SOP_v080624.pdf (accessed on 30 March 2016).
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Liao, J.K.; Seto, M.; Noma, K. Rho kinase (ROCK) inhibitors. J. Cardiovasc. Pharmacol. 2007, 50, 17–24. [Google Scholar] [CrossRef]
- Slee, E.A.; Zhu, H.; Chow, S.C.; MacFarlane, M.; Nicholson, D.W.; Cohen, G.M. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD-FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J. 1996, 315, 21–24. [Google Scholar] [CrossRef]
- Köröskényi, K.; Duró, E.; Pallai, A.; Sarang, Z.; Kloor, D.; Ucker, D.S.; Beceiro, S.; Castrillo, A.; Chawla, A.; Ledent, C.A.; et al. Involvement of adenosine A2A receptors in engulfment-dependent apoptotic cell suppression of inflammation. J. Immunol. 2011, 186, 7144–7155. [Google Scholar] [CrossRef]
- Schilling, J.D.; Machkovech, H.M.; He, L.; Diwan, A.; Schaffer, J.E. TLR4 Activation under lipotoxic conditions leads to synergistic macrophage cell death through a TRIF-dependent pathway. J. Immunol. 2013, 190, 1285–1296. [Google Scholar] [CrossRef]
- Brookheart, R.T.; Michel, C.I.; Schaffer, J.E. As a matter of fat. Cell Metab. 2009, 10, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Hornberger, T.A.; Sukhija, K.B.; Wang, X.R.; Chien, S. mTOR is the Rapamycin-Sensitive Kinase that Confers Mechanically-Induced Phosphorylation of the Hydrophobic Motif Site Thr(389) in p70S6k. FEBS Lett. 2007, 581, 4562–4566. [Google Scholar] [CrossRef]
- Bozulic, L.; Hemmings, B.A. PIKKing on PKB: Regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol. 2009, 21, 256–261. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Fox, R.; Nhan, T.Q.; Law, G.L.; Morris, D.R.; Liles, W.C.; Schwartz, S.M. PSGL-1 and mTOR regulate translation of ROCK-1 and physiological functions of macrophages. EMBO J. 2007, 26, 505–515. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [PubMed]
- Woodworth-Hobbs, M.E.; Perry, B.D.; Rahnert, J.A.; Hudson, M.B.; Zheng, B.; Russ Price, S. Docosahexaenoic acid counteracts palmitate-induced endoplasmic reticulum stress in C2C12 myotubes: Impact on muscle atrophy. Physiol. Rep. 2017, 5, e13530. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yan, D.-Y.; Wang, C.; Ma, Z.; Deng, Y.; Liu, W.; Xu, B. IRE1 signaling pathway mediates protective autophagic response against manganese-induced neuronal apoptosis in vivo and in vitro. Sci. Total Environ. 2020, 712, 136480. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Alkhouri, N.; Gornicka, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Kashyap, S.; Schauer, P.R.; Feldstein, A.E. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 2010, 285, 3428–3438. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Pischon, N.; Heng, N.; Bernimoulin, J.P.; Kleber, B.M.; Willich, S.N.; Pischon, T. Obesity, inflammation, and periodontal disease. J. Dent. Res. 2007, 86, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Chazaud, B. Inflammation and skeletal muscle regeneration: Leave it to the macrophages. Trends Immunol. 2020, 6, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaeed, N.; Budai, Z.; Szondy, Z.; Sarang, Z. TAM kinase signaling is indispensable for the proper skeletal muscle regeneration process. Cell Death Dis. 2021, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, D.; Berdeaux, R. The effects of obesity on the skeletal muscle regeneration. Front. Physiol. 2013, 4, 371. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, Y.N.; Dinh, T.P.; Salas, R.E.; Johnson, E.L.; Wright, T.G.; Robson, M.C.; Payne, W.G. Obesity and surgical wound healing: A current review. ISRN Obes. 2014, 2014, 638936. [Google Scholar] [CrossRef]
- Wu, Y.; Song, P.; Xu, J.; Zhang, M.; Zou, M.H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 9777–9788. [Google Scholar] [CrossRef]
- Kwon, B.; Querfurth, H.W. Palmitate activates mTOR/p70S6K through AMPK inhibition and hypophosphorylation of raptor in skeletal muscle cells: Reversal by oleate is similar to metformin. Biochimie 2015, 118, 141–150. [Google Scholar] [CrossRef]
- Mordier, S.; Iynedjian, P.B. Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochem. Biophys. Res. Commun. 2007, 362, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Tanaka, Y.; Kume, S.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Koya, D.; Haneda, M.; et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim. Biophys. Acta 2014, 1842, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; van Veen, E.; van Vugt, M.; Thoreen, C.C.; Sabatini, D.M. mTORC1 regulates cytokinesis through activation of Rho-ROCK signaling. arXiv 2015, arXiv:1506.04437. [Google Scholar]
- Huang, H.; Lee, S.H.; Sousa-Lima, I.; Kim, S.S.; Hwang, W.M.; Dagon, Y.; Yang, W.M.; Cho, S.; Kang, M.C.; Seo, J.A.; et al. Rho-kinase/AMPK axis regulates hepatic lipogenesis during overnutrition. J. Clin. Investig. 2018, 128, 5335–5350. [Google Scholar] [CrossRef]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef]
- Huang, H.; Ouyang, Q.; Zhu, M.; Yu, H.; Mei, K.; Liu, R. mTOR-mediated phosphorylation of VAMP8 and SCFD1 regulates autophagosome maturation. Nat. Commun. 2021, 12, 6622. [Google Scholar] [CrossRef]
- Miyazaki, M.; Hiramoto, M.; Takano, N.; Kokuba, H.; Takemura, J.; Tokuhisa, M.; Hino, H.; Kazama, H.; Miyazawa, K. Targeted disruption of GAK stagnates autophagic flux by disturbing lysosomal dynamics. Int. J. Mol. Med. 2021, 48, 195. [Google Scholar] [CrossRef]
- Rontogianni, S.; Iskit, S.; van Doorn, S.; Peeper, D.S.; Altelaar, M. Combined EGFR and ROCK inhibition in triple-negative breast cancer leads to cell death via impaired autophagic flux. Mol. Cell Proteom. 2020, 19, 261–277. [Google Scholar] [CrossRef]
- Huynh, K.K.; Kay, J.G.; Stow, J.L.; Grinstein, S. Fusion, fission, and secretion during phagocytosis. Physiology 2007, 22, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Arraiz, N.; Aguirre, M.; Velasco, M.; Bermúdez, V. AMPK as target for intervention in childhood and adolescent obesity. J. Obes. 2011, 2011, 252817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An old drug with new applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sós, L.; Garabuczi, É.; Sághy, T.; Mocsár, G.; Szondy, Z. Palmitate Inhibits Mouse Macrophage Efferocytosis by Activating an mTORC1-Regulated Rho Kinase 1 Pathway: Therapeutic Implications for the Treatment of Obesity. Cells 2022, 11, 3502. https://doi.org/10.3390/cells11213502
Sós L, Garabuczi É, Sághy T, Mocsár G, Szondy Z. Palmitate Inhibits Mouse Macrophage Efferocytosis by Activating an mTORC1-Regulated Rho Kinase 1 Pathway: Therapeutic Implications for the Treatment of Obesity. Cells. 2022; 11(21):3502. https://doi.org/10.3390/cells11213502
Chicago/Turabian StyleSós, László, Éva Garabuczi, Tibor Sághy, Gábor Mocsár, and Zsuzsa Szondy. 2022. "Palmitate Inhibits Mouse Macrophage Efferocytosis by Activating an mTORC1-Regulated Rho Kinase 1 Pathway: Therapeutic Implications for the Treatment of Obesity" Cells 11, no. 21: 3502. https://doi.org/10.3390/cells11213502
APA StyleSós, L., Garabuczi, É., Sághy, T., Mocsár, G., & Szondy, Z. (2022). Palmitate Inhibits Mouse Macrophage Efferocytosis by Activating an mTORC1-Regulated Rho Kinase 1 Pathway: Therapeutic Implications for the Treatment of Obesity. Cells, 11(21), 3502. https://doi.org/10.3390/cells11213502