

A Novel P53 Nanomedicine Reduces Immunosuppression and Augments Anti-PD-1 Therapy for Non-Small Cell Lung Cancer in Syngeneic Mouse Models


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Nanocomplex Preparation
2.3. Transfection
2.4. Animal Studies
2.5. ATP Assay
2.6. HMGB1 Assay
2.7. Gal-1 Immunoassay
2.8. Immunohistochemistry
2.9. Flow Cytometry
2.10. RT-qPCR
2.11. Detection of miR-22
2.12. Bioluminescence Imaging
2.13. Transcriptome Analysis
2.14. Patient Survival Data Analysis
2.15. Statistical Analysis
3. Results
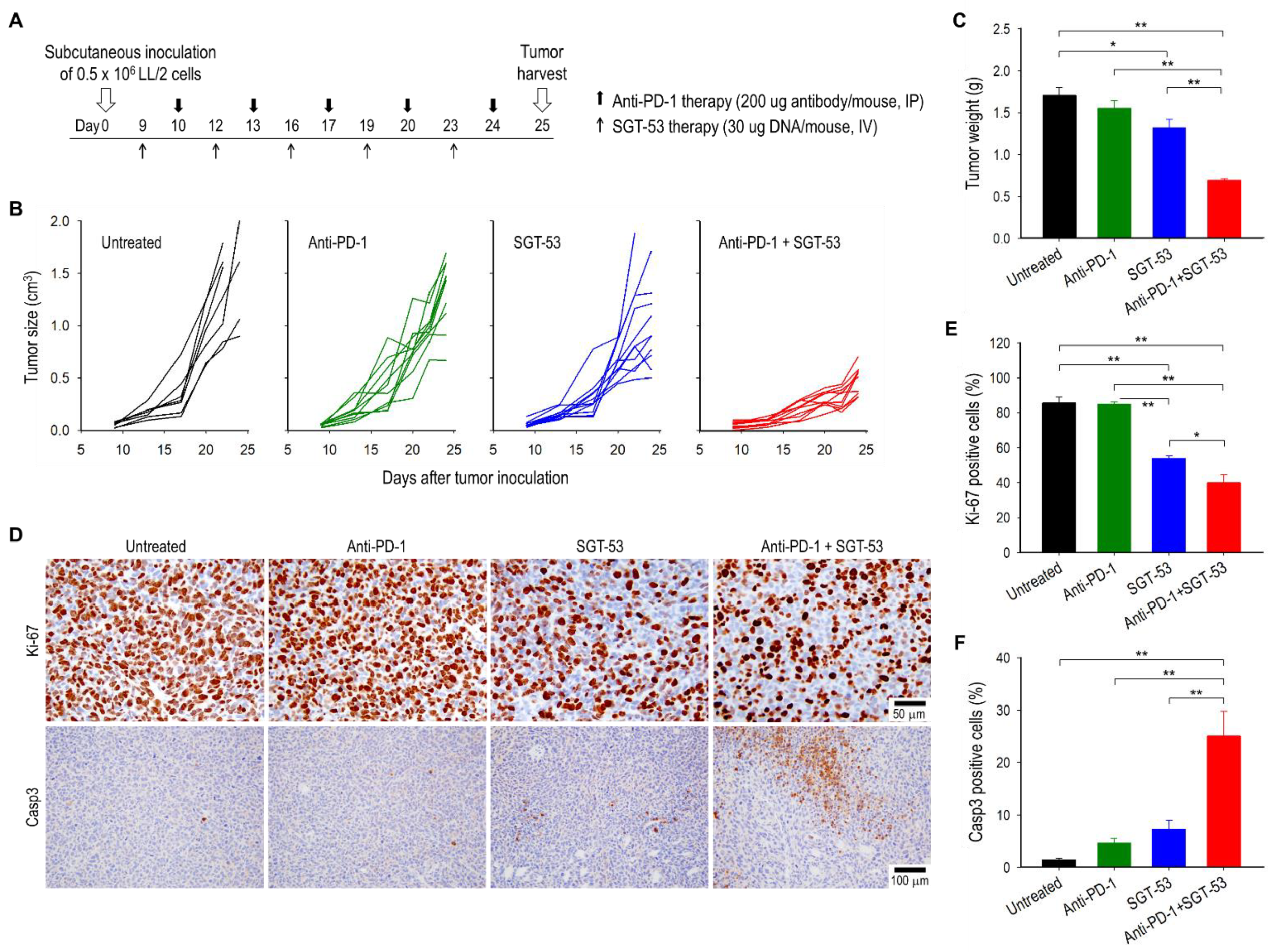
3.1. Combining Anti-PD-1 and SGT-53 Inhibits LL/2 Tumor Growth
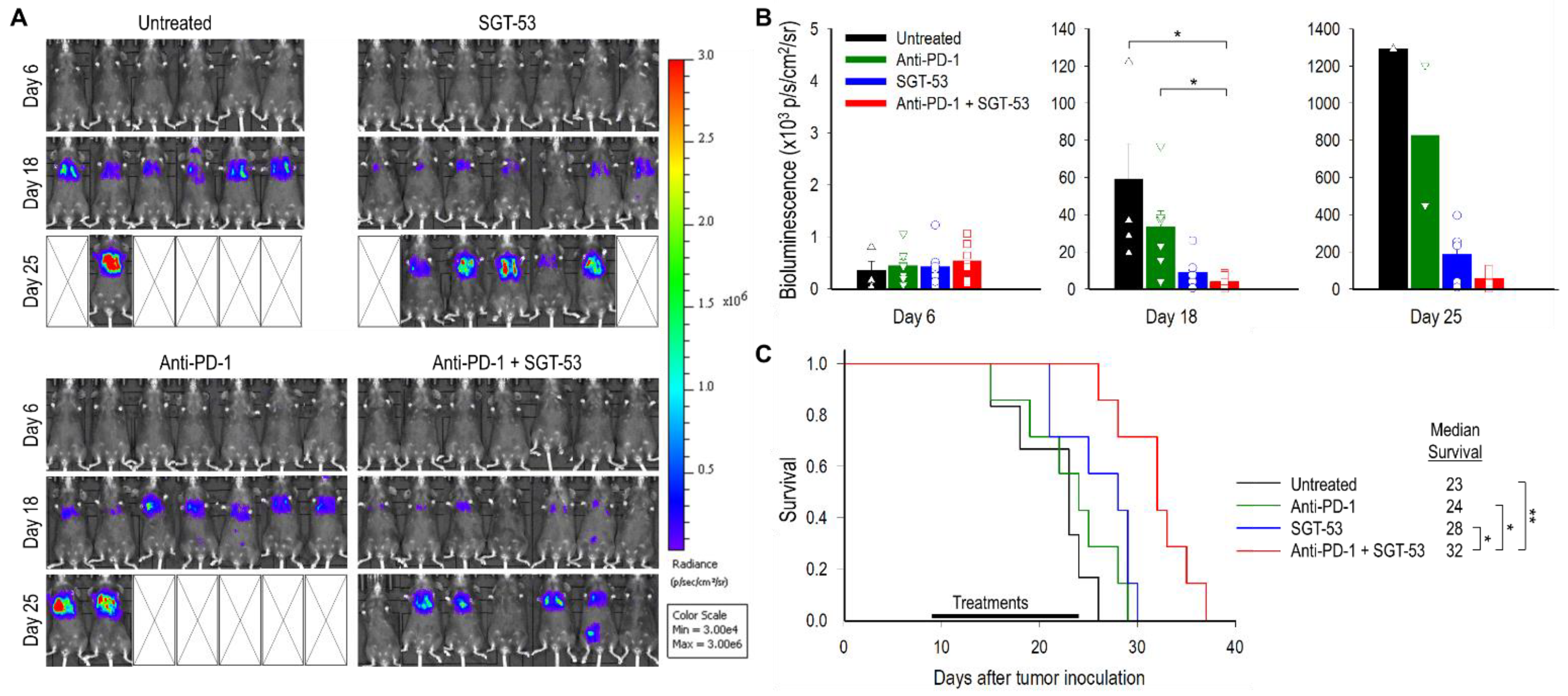
3.2. Combining Anti-PD-1 and SGT-53 Improves Survival in a Metastatic LL/2 Tumor Model
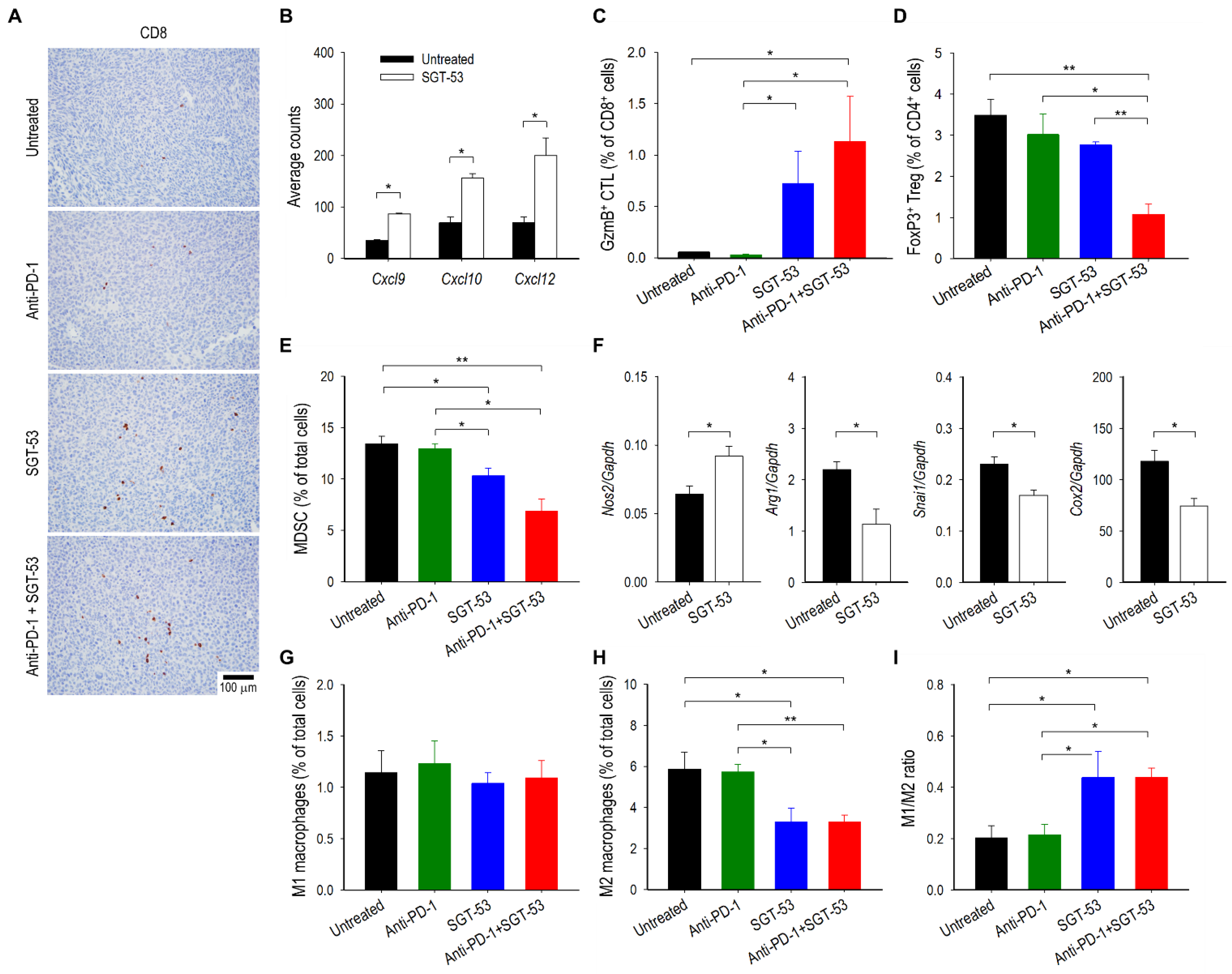
3.3. Combining Anti-PD-1 and SGT-53 Enhances Host Immune Responses
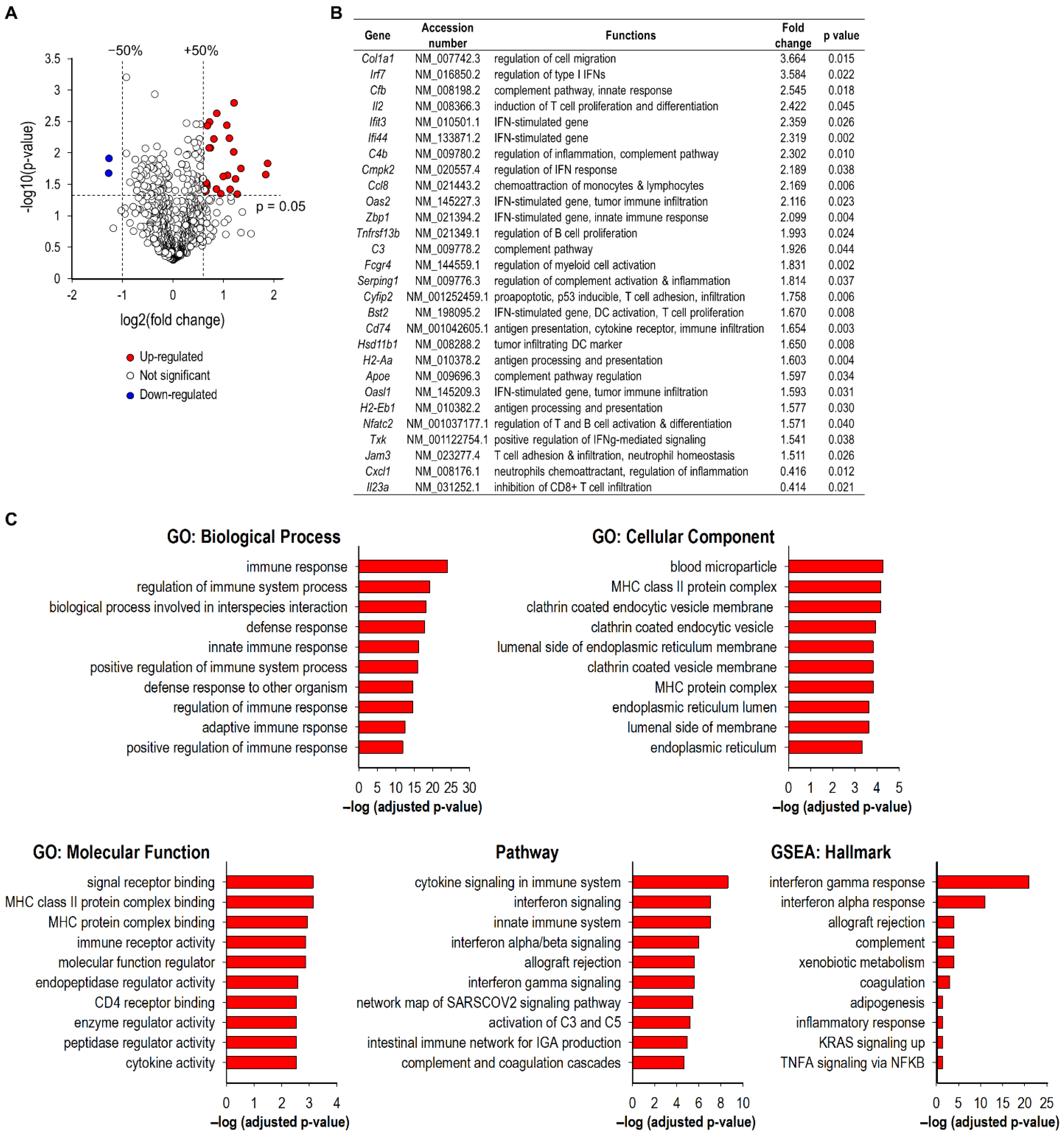
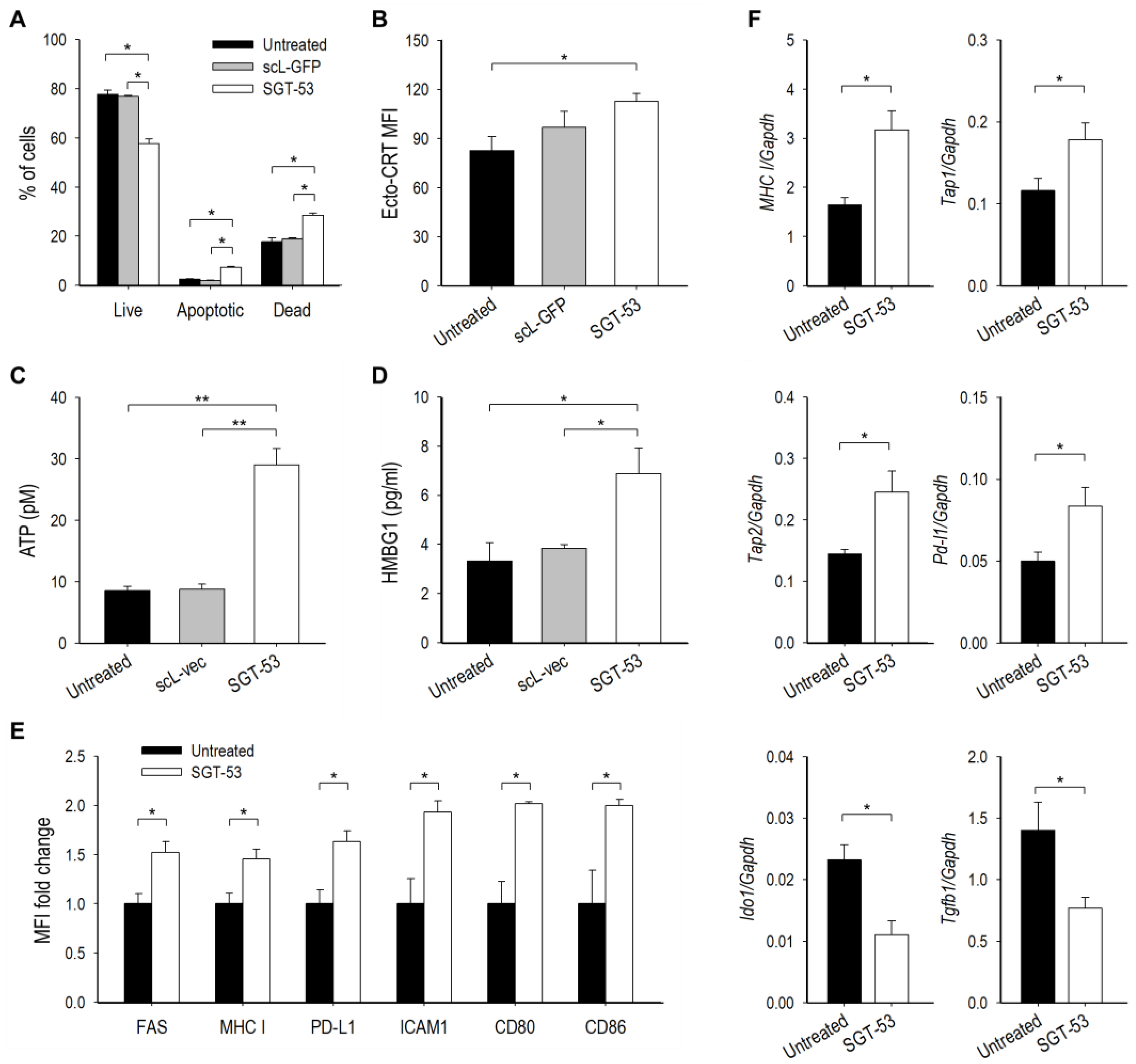
3.4. SGT-53 Increases Immunogenicity of Tumor Cells
3.5. SGT-53 Represses Immunosuppressive Gal-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Califano, R.; Gomes, F.; Ackermann, C.J.; Rafee, S.; Tsakonas, G.; Ekman, S. Immune checkpoint blockade for non-small cell lung cancer: What is the role in the special populations? Eur. J. Cancer 2019, 125, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Fenchel, K.; Dale, S.P. Programmed cell death ligand-1 (PD-L1) as a biomarker for non-small cell lung cancer (NSCLC) treatment-are we barking up the wrong tree? Transl. Lung Cancer Res. 2018, 7, S275–S279. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Smyth, M.J.; Teng, M.W. Acquired resistance to anti-PD1 therapy: Checkmate to checkpoint blockade? Genome Med. 2016, 8, 111. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Kaufman, H.L.; Wigginton, J.M.; Wolchok, J.D. Combination immunotherapy: A road map. J. Immunother. Cancer 2017, 5, 16. [Google Scholar] [CrossRef]
- Kim, S.S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef]
- Kong, N.; Tao, W.; Ling, X.; Wang, J.; Xiao, Y.; Shi, S.; Ji, X.; Shajii, A.; Gan, S.T.; Kim, N.Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, eaaw1565. [Google Scholar] [CrossRef]
- Cui, Y.; Guo, G. Immunomodulatory Function of the Tumor Suppressor p53 in Host Immune Response and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.Y.; Tang, S.J.; Sun, G.H.; Chou, T.Y.; Yeh, T.S.; Yu, S.L.; Sun, K.H. Galectin-1 promotes lung cancer progression and chemoresistance by upregulating p38 MAPK, ERK, and cyclooxygenase-2. Clin. Cancer Res. 2012, 18, 4037–4047. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Huang, C.C.; Huang, W.; Tang, W.H.; Rait, A.; Yin, Y.Z.; Cruz, I.; Xiang, L.M.; Pirollo, K.F.; Chang, E.H. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol. Cancer Ther. 2002, 1, 337–346. [Google Scholar] [PubMed]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2013. [Google Scholar]
- Cai, L.; Lin, S.; Girard, L.; Zhou, Y.; Yang, L.; Ci, B.; Zhou, Q.; Luo, D.; Yao, B.; Tang, H.; et al. LCE: An open web portal to explore gene expression and clinical associations in lung cancer. Oncogene 2019, 38, 2551–2564. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Ozga, A.J.; Servis, R.L.; Frederick, D.T.; Lo, J.A.; Fisher, D.E.; Freeman, G.J.; Boland, G.M.; Luster, A.D. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019, 50, 1498–1512.e5. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Liang, C.H.; Yu, J.H.; Huang, K.C.; Tung, C.H.; Wu, J.E.; Wu, Y.Y.; Chang, C.H.; Hong, T.M.; Chen, Y.L. A DNA Aptamer Targeting Galectin-1 as a Novel Immunotherapeutic Strategy for Lung Cancer. Mol. Nucleic Acids 2019, 18, 991–998. [Google Scholar] [CrossRef]
- Rubinstein, N.; Alvarez, M.; Zwirner, N.W.; Toscano, M.A.; Ilarregui, J.M.; Bravo, A.; Mordoh, J.; Fainboim, L.; Podhajcer, O.L.; Rabinovich, G.A. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell 2004, 5, 241–251. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; von Eyben, R.; et al. Galectin-1-driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Investig. 2019, 129, 5553–5567. [Google Scholar] [CrossRef]
- You, Y.; Tan, J.X.; Dai, H.S.; Chen, H.W.; Xu, X.J.; Yang, A.G.; Zhang, Y.J.; Bai, L.H.; Bie, P. MiRNA-22 inhibits oncogene galectin-1 in hepatocellular carcinoma. Oncotarget 2016, 7, 57099–57116. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.; Masui, O.; Newsted, D.; Scorilas, A.; Romaschin, A.D.; Bjarnason, G.A.; Siu, K.W.; Yousef, G.M. Galectin-1 has potential prognostic significance and is implicated in clear cell renal cell carcinoma progression through the HIF/mTOR signaling axis. Br. J. Cancer 2014, 110, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Chen, T.T.; Xia, L.; Guo, M.; Xu, Y.; Yue, F.; Jiang, Y.; Chen, G.Q.; Zhao, K.W. Hypoxia inducible factor-1 mediates expression of galectin-1: The potential role in migration/invasion of colorectal cancer cells. Carcinogenesis 2010, 31, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Ajina, R.; Zahavi, D.J.; Zhang, Y.W.; Weiner, L.M. Overcoming malignant cell-based mechanisms of resistance to immune checkpoint blockade antibodies. Semin. Cancer Biol. 2020, 65, 28–37. [Google Scholar] [CrossRef]
- Malhotra, J.; Jabbour, S.K.; Aisner, J. Current state of immunotherapy for non-small cell lung cancer. Transl. Lung Cancer Res. 2017, 6, 196–211. [Google Scholar] [CrossRef]
- Moya-Horno, I.; Viteri, S.; Karachaliou, N.; Rosell, R. Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (NSCLC). Ther. Adv. Med. Oncol. 2018, 10, 1758834017745012. [Google Scholar] [CrossRef]
- Kim, S.S.; Rait, A.; Rubab, F.; Rao, A.K.; Kiritsy, M.C.; Pirollo, K.F.; Wang, S.; Weiner, L.M.; Chang, E.H. The clinical potential of targeted nanomedicine: Delivering to cancer stem-like cells. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 278–291. [Google Scholar] [CrossRef]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase I Study of a Systemically Delivered p53 Nanoparticle in Advanced Solid Tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef]
- Pirollo, K.F.; Nemunaitis, J.; Leung, P.K.; Nunan, R.; Adams, J.; Chang, E.H. Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1697–1706. [Google Scholar] [CrossRef]
- Leung, C.P.; Barve, M.A.; Wu, M.-S.; Pirollo, K.F.; Strauss, J.F.; Liao, W.-C.; Yang, S.-H.; Nunan, R.A.; Adams, J.; Harford, J.B.; et al. A phase II trial combining tumor-targeting TP53 gene therapy with gemcitabine/nab-paclitaxel as a second-line treatment for metastatic pancreatic cancer. J. Clin. Oncol. 2021, 39, 4139. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Komarova, E.A. Pathologies associated with the p53 response. Cold Spring Harb. Perspect. Biol. 2010, 2, a001180. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; van Miltenburg, M.H.; Slagter, M.; de Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.C.; Sun, L.; Clavijo, P.E.; Friedman, J.; Harford, J.B.; Saleh, A.D.; Van Waes, C.; Chang, E.H.; Allen, C.T. Nanocomplex-based TP53 gene therapy promotes anti-tumor immunity through TP53- and STING-dependent mechanisms. Oncoimmunology 2018, 7, e1404216. [Google Scholar] [CrossRef]
- Kim, S.S.; Harford, J.B.; Moghe, M.; Rait, A.; Chang, E.H. Combination with SGT-53 overcomes tumor resistance to a checkpoint inhibitor. Oncoimmunology 2018, 7, e1484982. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Harford, J.B.; Moghe, M.; Slaughter, T.; Doherty, C.; Chang, E.H. A tumor-targeting nanomedicine carrying the p53 gene crosses the blood-brain barrier and enhances anti-PD-1 immunotherapy in mouse models of glioblastoma. Int. J. Cancer 2019, 145, 2535–2546. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef]
- Li, L.; Ng, D.S.; Mah, W.C.; Almeida, F.F.; Rahmat, S.A.; Rao, V.K.; Leow, S.C.; Laudisi, F.; Peh, M.T.; Goh, A.M.; et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. 2015, 22, 1081–1093. [Google Scholar] [CrossRef]
- Kawashima, H.; Takatori, H.; Suzuki, K.; Iwata, A.; Yokota, M.; Suto, A.; Minamino, T.; Hirose, K.; Nakajima, H. Tumor suppressor p53 inhibits systemic autoimmune diseases by inducing regulatory T cells. J. Immunol. 2013, 191, 3614–3623. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef]
- Fu, S.; Zhang, N.; Yopp, A.C.; Chen, D.; Mao, M.; Zhang, H.; Ding, Y.; Bromberg, J.S. TGF-beta induces Foxp3 + T-regulatory cells from CD4 + CD25—Precursors. Am. J. Transplant. 2004, 4, 1614–1627. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Suano, A.; Hamilton, A.B.; Betz, A.G. Gimme shelter: The immune system during pregnancy. Immunol. Rev. 2011, 241, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Wu, C.Y.; Hung, J.Y.; Lin, Y.S.; Huang, M.S.; Kuo, P.L. Galectin-1 promotes lung cancer tumor metastasis by potentiating integrin alpha6beta4 and Notch1/Jagged2 signaling pathway. Carcinogenesis 2013, 34, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Puchades, M.; Nilsson, C.L.; Emmett, M.R.; Aldape, K.D.; Ji, Y.; Lang, F.F.; Liu, T.J.; Conrad, C.A. Proteomic investigation of glioblastoma cell lines treated with wild-type p53 and cytotoxic chemotherapy demonstrates an association between galectin-1 and p53 expression. J. Proteome Res. 2007, 6, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Huo, R.; Xiao, L.; Zhu, X.; Xie, J.; Sun, S.; He, Y.; Zhang, J.; Sun, Y.; Zhou, Z.; et al. A novel p53/microRNA-22/Cyr61 axis in synovial cells regulates inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 49–59. [Google Scholar] [CrossRef]
- Leung, Z.; Ko, F.C.F.; Tey, S.K.; Kwong, E.M.L.; Mao, X.; Liu, B.H.M.; Ma, A.P.Y.; Fung, Y.M.E.; Che, C.M.; Wong, D.K.H.; et al. Galectin-1 promotes hepatocellular carcinoma and the combined therapeutic effect of OTX008 galectin-1 inhibitor and sorafenib in tumor cells. J. Exp. Clin. Cancer Res. CR 2019, 38, 423. [Google Scholar] [CrossRef]
- Qiu, B.Q.; Zhang, P.F.; Xiong, D.; Xu, J.J.; Long, X.; Zhu, S.Q.; Ye, X.D.; Wu, Y.; Pei, X.; Zhang, X.M.; et al. CircRNA fibroblast growth factor receptor 3 promotes tumor progression in non-small cell lung cancer by regulating Galectin-1-AKT/ERK1/2 signaling. J. Cell Physiol. 2019, 234, 11256–11264. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Yagi, S.; Ito, T.; Lowenstein, C.J. MicroRNA-22 regulates hypoxia signaling in colon cancer cells. PLoS ONE 2011, 6, e20291. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-S.; Harford, J.B.; Moghe, M.; Doherty, C.; Chang, E.H. A Novel P53 Nanomedicine Reduces Immunosuppression and Augments Anti-PD-1 Therapy for Non-Small Cell Lung Cancer in Syngeneic Mouse Models. Cells 2022, 11, 3434. https://doi.org/10.3390/cells11213434
Kim S-S, Harford JB, Moghe M, Doherty C, Chang EH. A Novel P53 Nanomedicine Reduces Immunosuppression and Augments Anti-PD-1 Therapy for Non-Small Cell Lung Cancer in Syngeneic Mouse Models. Cells. 2022; 11(21):3434. https://doi.org/10.3390/cells11213434
Chicago/Turabian StyleKim, Sang-Soo, Joe B. Harford, Manish Moghe, Caroline Doherty, and Esther H. Chang. 2022. "A Novel P53 Nanomedicine Reduces Immunosuppression and Augments Anti-PD-1 Therapy for Non-Small Cell Lung Cancer in Syngeneic Mouse Models" Cells 11, no. 21: 3434. https://doi.org/10.3390/cells11213434
APA StyleKim, S.-S., Harford, J. B., Moghe, M., Doherty, C., & Chang, E. H. (2022). A Novel P53 Nanomedicine Reduces Immunosuppression and Augments Anti-PD-1 Therapy for Non-Small Cell Lung Cancer in Syngeneic Mouse Models. Cells, 11(21), 3434. https://doi.org/10.3390/cells11213434