

Whole Transcriptome Sequencing Reveals Cancer-Related, Prognostically Significant Transcripts and Tumor-Infiltrating Immunocytes in Mantle Cell Lymphoma
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. MCL Patient Information
2.2. Total RNA Isolation from MCL Tumor Sections
2.3. FACS Sorting of Reactive Tonsil B-Cell Subtypes and RNA Isolations
2.3.1. Preparation of Reactive Tonsil Cell Suspensions
2.3.2. Immunostaining, FACS Sorting, and RNA Isolations
2.4. Whole Transcriptome Sequencing
2.5. Quantification of the Expressed Transcripts in MCL Tumor and Control Samples
2.5.1. Quality Control of the Raw WTS Data
2.5.2. Mapping Reads to a Reference Genome
2.5.3. Counting Reads
2.6. Principal Component Analysis
2.7. Identification of the Differentially Expressed mRNAs, lncRNAs, and Alternative Transcripts
2.8. Pathway Analysis of Differentially Expressed mRNAs, Alternative Transcripts, and lncRNAs
2.9. Identification of the Candidate Oncogenes and Tumor Suppressor Genes
2.10. Microenvironmental Immunocyte Ratio Estimation via CIBERSORTx Analysis
2.11. Immunohistochemistry and Hematoxylin–Eosin Staining
2.12. Survival Analysis
2.13. qRT-PCR
2.14. Comparison of WTS and qRT-PCR Results with Linear Correlation Analysis
2.15. Statistical Analyses
3. Results
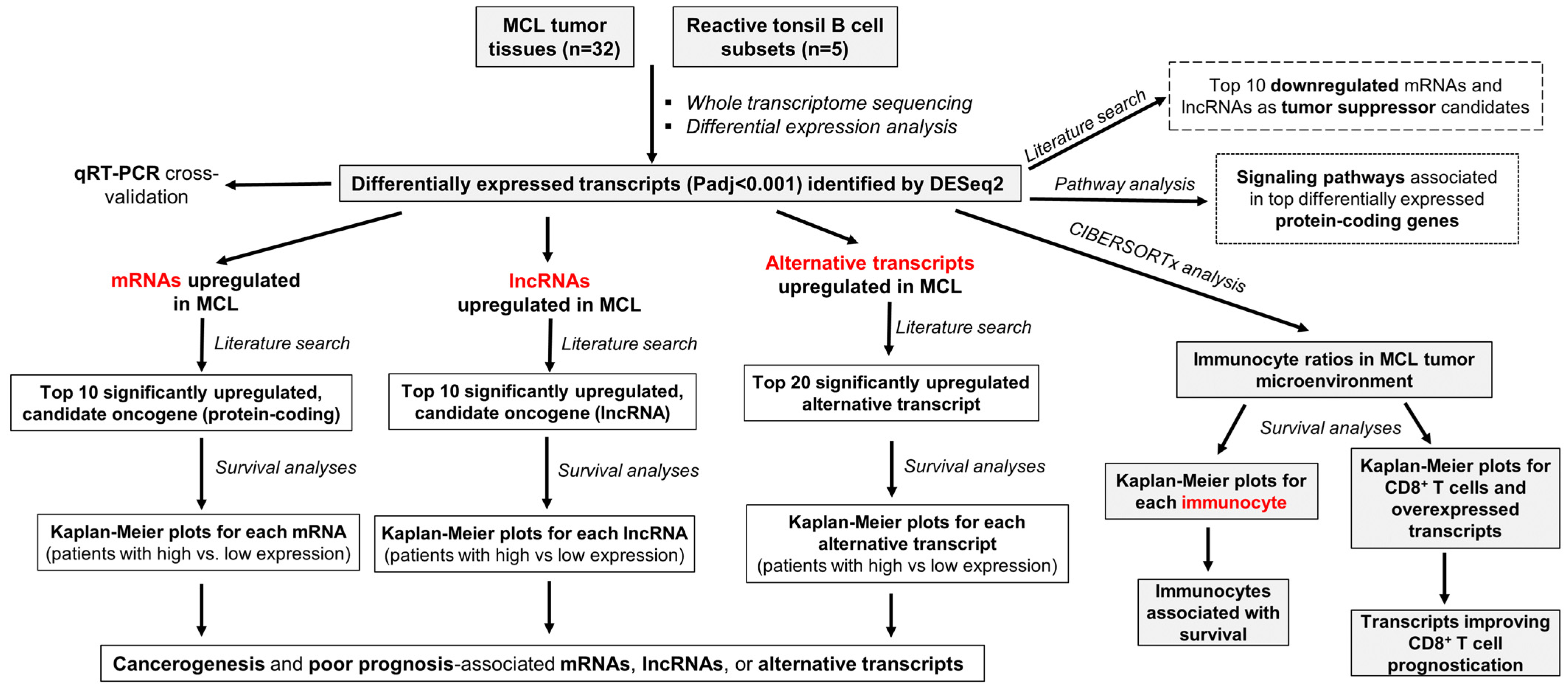
3.1. Overall Research and Analysis Plan of the Study
3.2. CCND1, SOX11, ROR1-AS1, and LINK-A Are Overexpressed in MCL Tumors
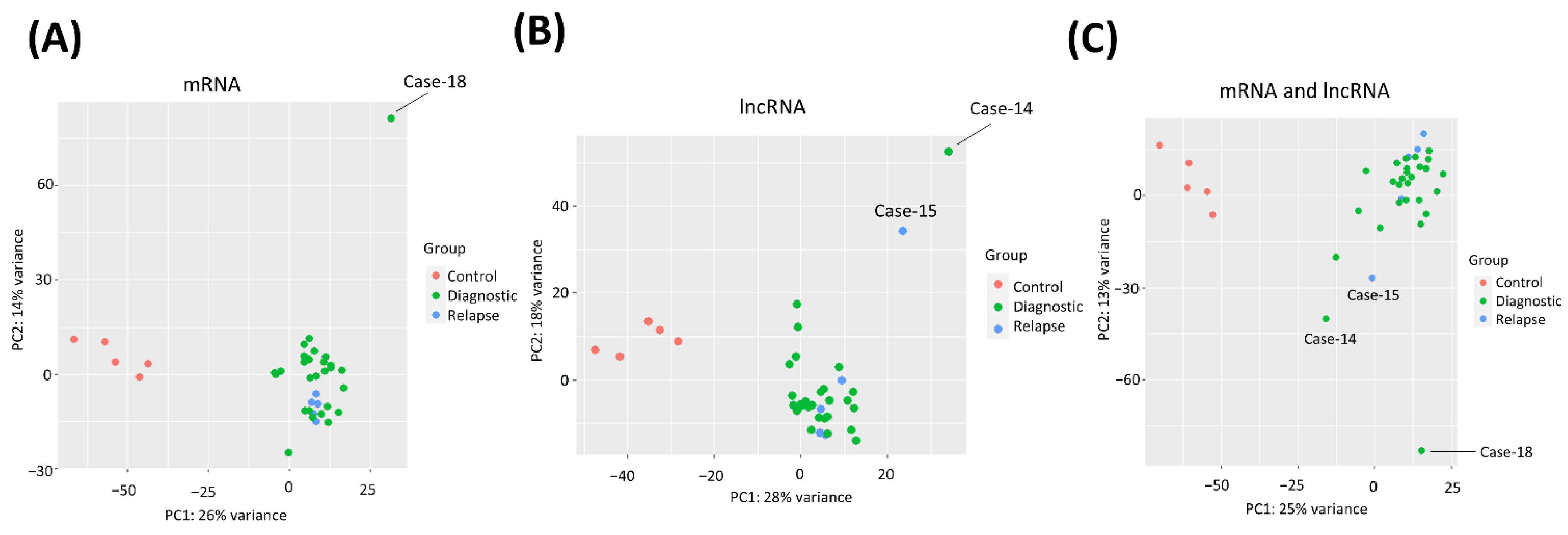
3.3. PCA Clustering of MCL and Control Samples by Transcriptome Profiles
3.4. Differentially Expressed mRNAs, lncRNAs, and Alternative Transcripts in MCL Tumor Tissues
3.5. Cancer-Related Signaling Pathways Are Enriched in Top Differentially Expressed Genes
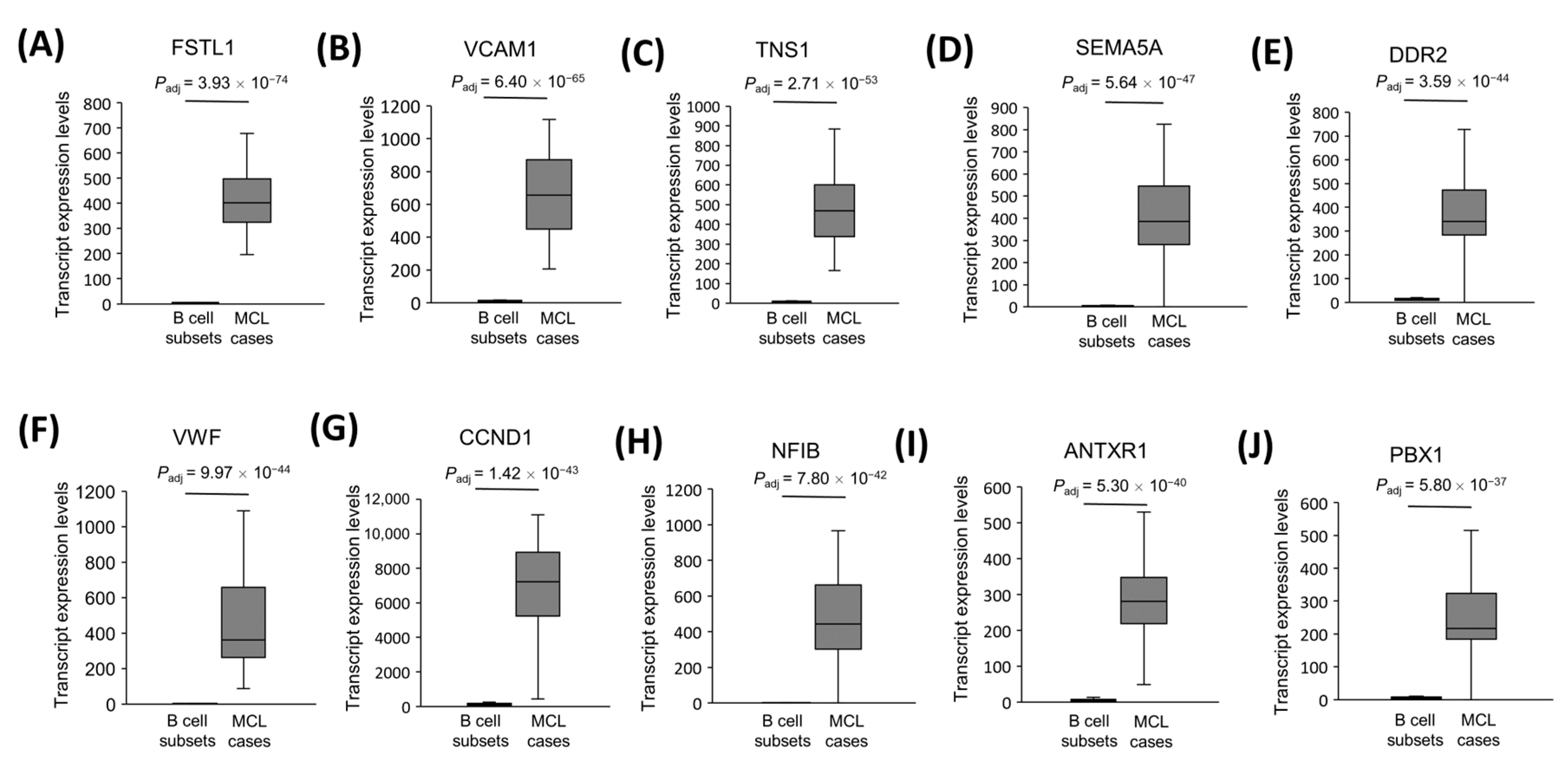
3.6. The Most Significantly Overexpressed, Cancerogenesis-Associated Gene mRNAs in MCL Tumor Tissues
3.7. The Most Significantly Underexpressed Tumor Suppressor Candidate Gene mRNAs in MCL Tumor Tissues
3.8. The Most Significantly Overexpressed Oncogenesis-Associated lncRNA Genes
3.9. The Significantly Underexpressed Tumor Suppressor-Candidate Gene lncRNAs in MCL Tumor Tissues
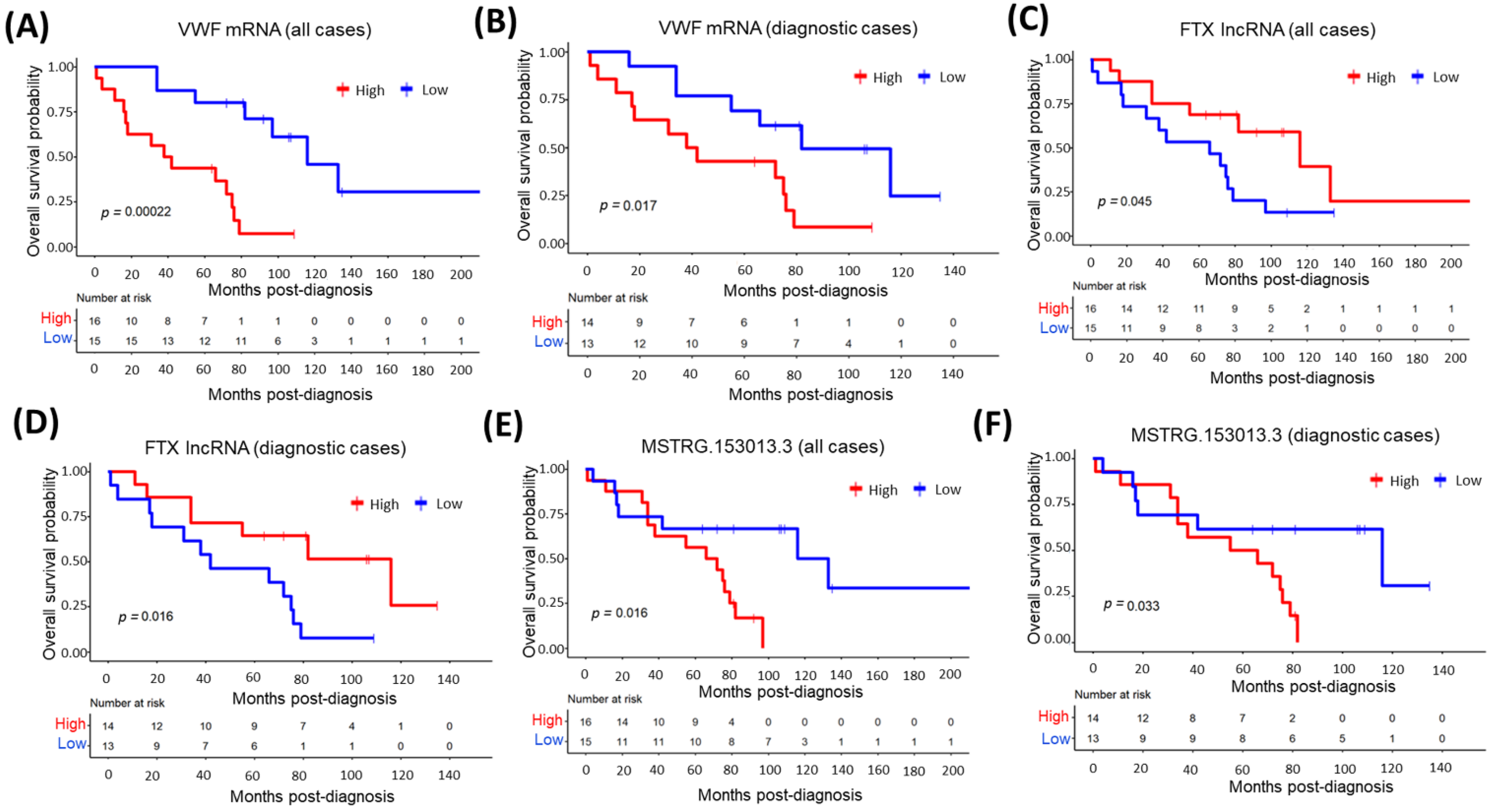
3.10. The Relationship between Top Overexpressed Transcripts and MCL Survival
3.11. The Relationship between Demographic or Clinical Variables and MCL Survival
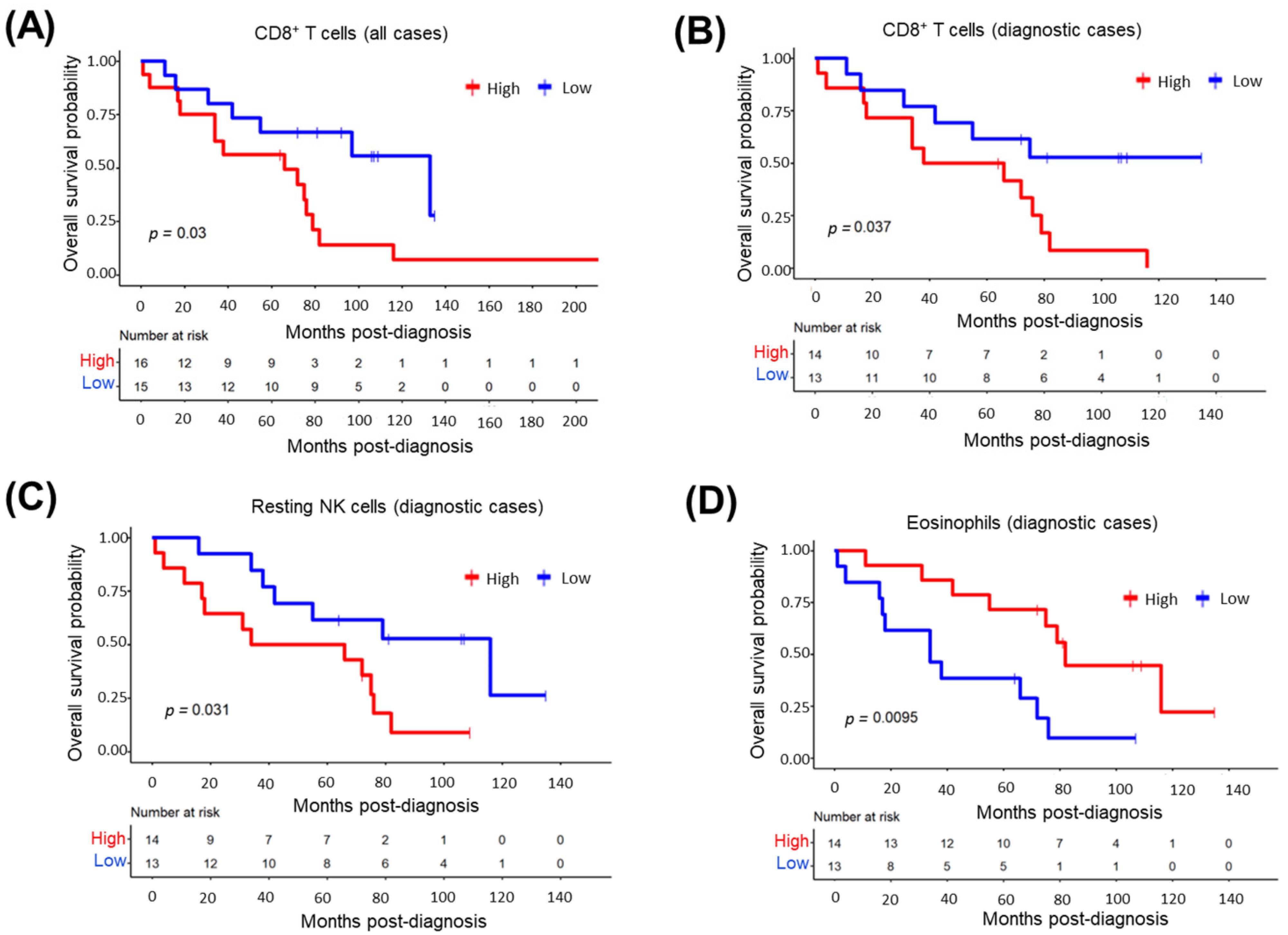
3.12. CD8+ T-Cells in Tumor Microenvironment Predict Inferior MCL Survival
3.13. Impact of Tumor-Infiltrating CD8+ T-Cell Abundance and Cancer-Associated Transcripts on MCL Survival
3.14. Multivariate Cox’s Regression Analysis Results of Overall Survival
3.15. qRT-PCR Cross-Validated WTS Data of CCND1 and SNHG5 Transcripts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armitage, J.O.; Longo, D.L. Mantle-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Jares, P.; Colomer, D.; Campo, E. Genetic and molecular pathogenesis of mantle cell lymphoma: Perspectives for new targeted therapeutics. Nat. Rev. Cancer 2007, 7, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Veloza, L.; Ribera-Cortada, I.; Campo, E. Mantle Cell Lymphoma Biology. Clin. Lymphoma Myeloma Leuk. 2018, 18, S97–S98. [Google Scholar] [CrossRef]
- Beekman, R.; Amador, V.; Campo, E. SOX11, a key oncogenic factor in mantle cell lymphoma. Curr. Opin. Hematol. 2018, 25, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Wang, M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am. J. Hematol. 2019, 94, 710–725. [Google Scholar] [CrossRef]
- Ghielmini, M.; Zucca, E. How I treat mantle cell lymphoma. Blood 2009, 114, 1469–1476. [Google Scholar] [CrossRef]
- Damon, L.E.; Johnson, J.L.; Niedzwiecki, D.; Cheson, B.D.; Hurd, D.D.; Bartlett, N.L.; LaCasce, A.S.; Blum, K.A.; Byrd, J.C.; Kelly, M.; et al. Immunochemotherapy and Autologous Stem-Cell Transplantation for Untreated Patients with Mantle-Cell Lymphoma: CALGB 59909. J. Clin. Oncol. 2009, 27, 6101–6108. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sha, F.; Toure, A.; Dogan, A.; Ni, A.; Batlevi, C.L.; Palomba, M.L.M.; Portlock, C.; Straus, D.J.; Noy, A.; et al. Patterns of survival in patients with recurrent mantle cell lymphoma in the modern era: Progressive shortening in response duration and survival after each relapse. Blood Cancer J. 2019, 9, 50. [Google Scholar] [CrossRef]
- Wallace, D.; Reagan, P.M. Novel Treatments for Mantle Cell Lymphoma: From Targeted Therapies to CAR T Cells. Drugs 2021, 81, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Hoster, E.; Dreyling, M.; Klapper, W.; Gisselbrecht, C.; Van Hoof, A.; Kluin-Nelemans, J.C.; Pfreundschuh, M.; Reiser, M.; Metzner, B.; Einsele, H.; et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558–565. [Google Scholar] [CrossRef]
- Geisler, C.H.; Kolstad, A.; Laurell, A.; Jerkeman, M.; Räty, R.; Andersen, N.S.; Pedersen, L.B.; Eriksson, M.; Nordström, M.; Kimby, E.; et al. Nordic MCL2 trial update: Six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: Still very long survival but late relapses do occur. Br. J. Haematol. 2012, 158, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Husby, S.; Eskelund, C.W.; Besenbacher, S.; Fjelstrup, S.; Côme, C.; Ek, S.; Kolstad, A.; Räty, R.; Jerkeman, M.; et al. Expression patterns and prognostic potential of circular RNAs in mantle cell lymphoma: A study of younger patients from the MCL2 and MCL3 clinical trials. Leukemia 2021, 36, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Mas, R.M.V.; Navarro, A.; Salaverria, I.; Martín-Garcia, D.; Jares, P.; Giné, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [PubMed]
- Greiner, T.; Moynihan, M.; Chan, W.; Lytle, D.; Pedersen, A.; Anderson, J.; Weisenburger, D. p53 mutations in mantle cell lymphoma are associated with variant cytology and predict a poor prognosis. Blood 1996, 87, 4302–4310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jima, D.; Moffitt, A.; Liu, Q.; Czader, M.; Hsi, E.D.; Fedoriw, Y.; Dunphy, C.H.; Richards, K.L.; Gill, J.I.; et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014, 123, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Pararajalingam, P.; Coyle, K.M.; Arthur, S.E.; Thomas, N.; Alcaide, M.; Meissner, B.; Boyle, M.; Qureshi, Q.; Grande, B.M.; Rushton, C.; et al. Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood 2020, 136, 572–584. [Google Scholar] [CrossRef]
- Ferrero, S.; Rossi, D.; Rinaldi, A.; Bruscaggin, A.; Spina, V.; Eskelund, C.W.; Evangelista, A.; Moia, R.; Kwee, I.; Dahl, C.; et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: A FIL study. Haematologica 2019, 105, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunawardana, J.; Jenkins, C.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Yan, Y.; Jin, M.; Bhattacharya, S.; Wang, Y.; Wu, Y.; Yang, L.; Gine, E.; Clot, G.; Chen, L.; et al. Genomic and transcriptomic profiling reveals distinct molecular subsets associated with outcomes in mantle cell lymphoma. J. Clin. Investig. 2022, 132, e153283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, P.; Du, H.; Zhang, L. LINK-A lncRNA Promotes Proliferation and Inhibits Apoptosis of Mantle Cell Lymphoma Cell by Upregulating Survivin. Med. Sci. Monit. 2019, 25, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, P.; Zhou, Y.; Zhang, L. Inhibition of LINK-A lncRNA overcomes ibrutinib resistance in mantle cell lymphoma by regulating Akt/Bcl2 pathway. PeerJ 2021, 9, e12571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-J.; Lin, J.; Lwin, T.; Yang, H.; Guo, J.; Kong, W.; Dessureault, S.; Moscinski, L.C.; Rezania, D.; Dalton, W.S.; et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood 2010, 115, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Shen, J.; Hou, Z.; Chen, S.; Su, Y.; Fang, J. lncRNA FOXP4-AS1 predicts poor prognosis and accelerates the progression of mantle cell lymphoma through the miR-423-5p/NACC1 pathway. Oncol. Rep. 2020, 45, 469–480. [Google Scholar] [CrossRef]
- Wang, X.; Sehgal, L.; Jain, N.; Khashab, T.; Mathur, R.; Samaniego, F. LncRNA MALAT1 promotes development of mantle cell lymphoma by associating with EZH2. J. Transl. Med. 2016, 14, 346. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kang, M.; Li, J.; Qin, W.; Wang, R. Prognostic value of the mRNA expression of members of the HSP90 family in non-small cell lung cancer. Exp. Ther. Med. 2019, 17, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Fu, Z.; Li, Y.; Zhang, W.; Liu, Y. Aberrant FAM64A mRNA expression is an independent predictor of poor survival in pancreatic cancer. PLoS ONE 2019, 14, e0211291. [Google Scholar] [CrossRef]
- Chen, Y.; Bi, F.; An, Y.; Yang, Q. Identification of pathological grade and prognosis-associated lncRNA for ovarian cancer. J. Cell Biochem. 2019, 120, 14444–14454. [Google Scholar] [CrossRef]
- Li, X.; Jin, F.; Li, Y. A novel autophagy-related lncRNA prognostic risk model for breast cancer. J. Cell Mol. Med. 2020, 25, 4–14. [Google Scholar] [CrossRef]
- Lasham, A.; Knowlton, N.; Mehta, S.; Braithwaite, A.; Print, C. Breast Cancer Patient Prognosis Is Determined by the Interplay between TP53 Mutation and Alternative Transcript Expression: Insights from TP53 Long Amplicon Digital PCR Assays. Cancers 2021, 13, 1531. [Google Scholar] [CrossRef]
- Pavlidou, A.; Kroupis, C.; Dimas, K. Association of survivin splice variants with prognosis and treatment of breast cancer. World J. Clin. Oncol. 2014, 5, 883–894. [Google Scholar] [CrossRef]
- Drakes, M.L.; Stiff, P.J. Regulation of Ovarian Cancer Prognosis by Immune Cells in the Tumor Microenvironment. Cancers 2018, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Zhang, Y.; Wang, L.; Zhang, H.; Liu, H.; Liu, Y. Cellular components in tumor microenvironment of neuroblastoma and the prognostic value. PeerJ 2019, 7, e8017. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, S.; Yang, Y.; Zhao, M.; Zhu, G.; Hou, Z. The prognostic landscape of tumor-infiltrating immune cell and immunomodulators in lung cancer. Biomed. Pharmacother. 2017, 95, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-Infiltrating Immune Cells Act as a Marker for Prognosis in Colorectal Cancer. Front. Immunol. 2019, 10, 2368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ba, H.-J.; Wen, X.-Z.; Zhou, M.; Küçük, C.; Tamagnone, L.; Wei, L.; You, H. A prognostic model for melanoma patients on the basis of immune-related lncRNAs. Aging 2021, 13, 6554–6564. [Google Scholar] [CrossRef]
- Von Ahlfen, S.; Missel, A.; Bendrat, K.; Schlumpberger, M. Determinants of RNA Quality from FFPE Samples. PLoS ONE 2007, 2, e1261. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. BioSystems 2015, 12, 477–479. [Google Scholar] [CrossRef]
- Zhang, Y.; Bu, D.; Huo, P.; Wang, Z.; Rong, H.; Li, Y.; Liu, J.; Ye, M.; Wu, Y.; Jiang, Z.; et al. ncFANs v2.0: An integrative platform for functional annotation of non-coding RNAs. Nucleic Acids Res. 2021, 49, W459–W468. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T. A package for survival analysis in R. In Book a Package Survival Analysis R; R Package Version 2020, 3.1-12 ed.; Springer: New York, NY, USA, 2020. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using ‘ggplot2’, R Package Version 0.4.9; 2021. Available online: https://rpkgs.datanovia.com/survminer/index.html (accessed on 16 August 2022).
- Hatipoğlu, T.; Sönmez, E.E.; Hu, X.; Yuan, H.; Danyeli, A.E.; Şeyhanlı, A.; Önal-Süzek, T.; Zhang, W.; Akman, B.; Olgun, A.; et al. Plasma Concentrations and Cancer-Associated Mutations in Cell-Free Circulating DNA of Treatment-Naive Follicular Lymphoma for Improved Non-Invasive Diagnosis and Prognosis. Front. Oncol. 2022, 12, 870487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, C.; Shi, F.; Ma, S.; Zheng, J.; Du, X.; Zhang, L. Comparative transcriptome analysis reveals sesquiterpenoid biosynthesis among 1-, 2- and 3-year old Atractylodes chinensis. BMC Plant Biol. 2021, 21, 354. [Google Scholar] [CrossRef]
- Gu, C.; Wang, X.; Long, T.; Wang, X.; Zhong, Y.; Ma, Y.; Hu, Z.; Li, Z. FSTL1 interacts with VIM and promotes colorectal cancer metastasis via activating the focal adhesion signalling pathway. Cell Death Dis. 2018, 9, 654. [Google Scholar] [CrossRef]
- Wang, P.-C.; Weng, C.-C.; Hou, Y.-S.; Jian, S.-F.; Fang, K.-T.; Hou, M.-F.; Cheng, K.-H. Activation of VCAM-1 and Its Associated Molecule CD44 Leads to Increased Malignant Potential of Breast Cancer Cells. Int. J. Mol. Sci. 2014, 15, 3560–3579. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Wu, L.; Xie, W.; Li, L.; Yuan, Y.; Chen, Y.; Lin, Y.; He, X. Elevated transgelin/TNS1 expression is a potential biomarker in human colorectal cancer. Oncotarget 2017, 9, 1107–1113. [Google Scholar] [CrossRef]
- Sadanandam, A.; Rosenbaugh, E.G.; Singh, S.; Varney, M.; Singh, R.K. Semaphorin 5A promotes angiogenesis by increasing endothelial cell proliferation, migration, and decreasing apoptosis. Microvasc. Res. 2010, 79, 1–9. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, W.; Liu, X.; Zhao, H.; Zhang, J.; Li, X.; Zhang, Y.; Bu, X.; Shi, M.; Yao, L.; et al. Discoidin domain receptor 2 (DDR2) promotes breast cancer cell metastasis and the mechanism implicates epithelial-mesenchymal transition programme under hypoxia. J. Pathol. 2014, 234, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.-J.; Wang, M.; Wang, Y.; Cai, W.; Li, Q.; Zhao, T.-T.; Zhang, L.-H.; Houck, K.; Chen, X.; Jin, Y.-L.; et al. Cancer cell-derived von Willebrand factor enhanced metastasis of gastric adenocarcinoma. Oncogenesis 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lv, X.-B.; Wang, X.-P.; Sang, Y.; Xu, S.; Hu, K.; Wu, M.; Liang, Y.; Liu, P.; Tang, J.; et al. MiR-138 suppressed nasopharyngeal carcinoma growth and tumorigenesis by targeting the CCND1 oncogene. Cell Cycle 2012, 11, 2495–2506. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, R.-Z.; Vo, T.M.; Jain, S.; Choi, W.-S.; Garcia, E.; Monckton, E.A.; Mackey, J.R.; Godbout, R. NFIB promotes cell survival by directly suppressing p21 transcription in TP53-mutated triple-negative breast cancer. J. Pathol. 2018, 247, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Lu, G.-F.; Ji, M.-H.; Kong, D.-Y.; Wang, S.-Y.; Tian, H.; Xie, Z.-M.; Pan, M.; Gong, N.-L. MicroRNA-26b-3p/ANTXR1 signaling modulates proliferation, migration, and apoptosis of glioma. Am. J. Transl. Res. 2019, 11, 7568–7578. [Google Scholar] [PubMed]
- Park, J.T.; Shih, I.-M.; Wang, T.-L. Identification of Pbx1, a Potential Oncogene, as a Notch3 Target Gene in Ovarian Cancer. Cancer Res. 2008, 68, 8852–8860. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; Tan, B.; Mu, R.; Chang, Y.; Wu, M.; Tu, H.-Q.; Zhang, Y.-C.; Guo, S.-S.; Qin, X.-H.; Li, T.; et al. Ubiquitin-associated Domain-containing Ubiquitin Regulatory X (UBX) Protein UBXN1 is a Negative Regulator of Nuclear Factor κB (NF-κB) Signaling. J. Biol. Chem. 2015, 290, 10395–10405. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, J.; Harvey, S.E.; Hu, X.; Cheng, C. RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev. 2017, 31, 2296–2309. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Sheikh, M.S.; Yu, K.; Zhan, Q.; Iglesias, M.; Woodworth, C.; Fornace, A.J., Jr. Activation of Gadd34 by diverse apoptotic signals and suppression of its growth inhibitory effects by apoptotic inhibitors. Int. J. Cancer 2001, 96, 22–31. [Google Scholar] [CrossRef]
- Rodriguez-Aguayo, C.; Monroig, P.D.C.; Redis, R.S.; Bayraktar, E.; Almeida, M.I.; Ivan, C.; Fuentes-Mattei, E.; Rashed, M.R.; Chavez-Reyes, A.; Ozpolat, B.; et al. Regulation of hnRNPA1 by microRNAs controls the miR-18a–K-RAS axis in chemotherapy-resistant ovarian cancer. Cell Discov. 2017, 3, 17029. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic Investigations Reveal a Role for RNA Processing Factor THRAP3 in the DNA Damage Response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Hayami, Y.; Iida, S.; Nakazawa, N.; Hanamura, I.; Kato, M.; Komatsu, H.; Miura, I.; Dave, B.J.; Sanger, W.G.; Lim, B.; et al. Inactivation of the E3/LAPTm5 gene by chromosomal rearrangement and DNA methylation in human multiple myeloma. Leukemia 2003, 17, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 Mediate Oncogene-Induced p16INK4a Activation. Cancer Res. 2009, 69, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-E.; Yao, Y.; Dong, Y.; Lin, F.; Zhao, H.; Shen, Z.; Sun, Y.-J.; Tang, L.-N. Down-regulation of ribosomal protein L7A in human osteosarcoma. J. Cancer Res. Clin. Oncol. 2009, 135, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhu, Z.; Johnson, C.; Stoops, J.; Eaker, A.E.; Bowen, W.; DeFrances, M.C. PIK3IP1, a Negative Regulator of PI3K, Suppresses the Development of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 5591–5598. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2–mTOR signaling and tumor suppression through REDD1-mediated 14–3–3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, C.; Zhou, Y. LncRNA MIR100HG promotes cancer cell proliferation, migration and invasion in laryngeal squamous cell carcinoma through the downregulation of miR-204-5p. OncoTargets Ther. 2019, 12, 2967–2973. [Google Scholar] [CrossRef]
- Chen, B.; Li, Y.; Nie, Y.; Tang, A.; Zhou, Q. Long non-coding RNA LINC01268 promotes cell growth and inhibits cell apoptosis by modulating miR-217/SOS1 axis in acute myeloid leukemia. Braz. J. Med. Biol. Res. 2020, 53, e9299. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, G.; Zhai, J.; Hu, D.; Fan, Y. LncRNA FTX Promotes Proliferation and Invasion of Gastric Cancer via miR-144/ZFX Axis. OncoTargets Ther. 2019, 12, 11701–11713. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gupta, S.K.; Troska, T.P.; Nair, A.; Gupta, M. Long non-coding RNA profile in mantle cell lymphoma identifies a functional lncRNA ROR1-AS1 associated with EZH2/PRC2 complex. Oncotarget 2017, 8, 80223–80234. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ni, B.; Zhang, Z.; Wang, C.; Wo, L.; Zhou, C.; Zhao, Q.; Zhao, E. Long non-coding RNA DNM3OS promotes tumor progression and EMT in gastric cancer by associating with Snail. Biochem. Biophys. Res. Commun. 2019, 511, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Jia, Y.; Wang, Q.; Zhao, Q.; Song, M.; Ni, R.; Wang, J. Long Noncoding RNA KCNQ1OT1 Promotes the Progression of Non-Small Cell Lung Cancer via Regulating miR-204-5p/ATG3 Axis. OncoTargets Ther. 2019, 12, 10787–10797. [Google Scholar] [CrossRef]
- Gao, H.; Li, X.; Zhan, G.; Zhu, Y.; Yu, J.; Wang, J.; Li, L.; Wu, W.; Liu, N.; Guo, X. Long noncoding RNA MAGI1-IT1 promoted invasion and metastasis of epithelial ovarian cancer via the miR-200a/ZEB axis. Cell Cycle 2019, 18, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Qin, C. Downregulation of long non-coding RNA NR2F2-AS1 inhibits proliferation and induces apoptosis of nasopharyngeal carcinoma cells by upregulating the expression of PTEN. Oncol. Lett. 2019, 19, 1145–1150. [Google Scholar] [CrossRef]
- Li, Y.; Wan, Q.; Wang, W.; Mai, L.; Sha, L.; Mashrah, M.; Lin, Z.; Pan, C. LncRNA ADAMTS9-AS2 promotes tongue squamous cell carcinoma proliferation, migration and EMT via the miR-600/EZH2 axis. Biomed. Pharmacother. 2019, 112, 108719. [Google Scholar] [CrossRef]
- Zhang, G.; He, X.; Ren, C.; Lin, J.; Wang, Q. Retracted: Long noncoding RNA PCA3 regulates prostate cancer through sponging miR-218-5p and modulating high mobility group box 1. J. Cell Physiol. 2018, 234, 13097–13109. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Zhu, B.; Huynh, T.-T.; Meuter, L.; Jha, A.; Talvacchio, S.; Knue, M.; Patel, M.; Prodanov, T.; Das, S.; et al. A long noncoding RNA–microRNA expression signature predicts metastatic signature in pheochromocytomas and paragangliomas. Endocrine 2021, 75, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, X.; Wang, F.; Yang, Q.; Luo, X.; Kong, J.; Ju, S. Down-regulated lncRNA SLC25A5-AS1 facilitates cell growth and inhibits apoptosis via miR-19a-3p/PTEN/PI3K/AKT signalling pathway in gastric cancer. J. Cell Mol. Med. 2019, 23, 2920–2932. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wang, J.; Cui, N.; Liu, X.; Wang, H. Autophagy-related long non-coding RNA signature for potential prognostic biomarkers of patients with cervical cancer: A study based on public databases. Ann. Transl. Med. 2021, 9, 1668. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lan, J.; Ye, Z.; Duan, S.; Hu, Y.; Zou, Y.; Zhou, J. Long noncoding RNA LRRC75A-AS1 inhibits cell proliferation and migration in colorectal carcinoma. Exp. Biol. Med. 2019, 244, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, Y.; Zhang, H.; Lin, F.; Tan, Q.; Qin, Q.; Bao, W.; Liu, Y.; Xie, J.; Zeng, Q. Long intergenic non-protein coding RNA 324 prevents breast cancer progression by modulating miR-10b-5p. Aging 2020, 12, 6680–6699. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, J.; Chen, L.; Liu, X.; Yao, M.; Zhang, H. LncRNA CD27-AS1 promotes acute myeloid leukemia progression through the miR-224-5p/PBX3 signaling circuit. Cell Death Dis. 2021, 12, 510. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Fan, C.; Liu, N.; Huang, K.; Fang, X.; Wang, K. Downregulation of the long non-coding RNA ZFAS1 is associated with cell proliferation, migration and invasion in breast cancer. Mol. Med. Rep. 2018, 17, 6405–6412. [Google Scholar] [CrossRef]
- Zhao, L.; Guo, H.; Zhou, B.; Feng, J.; Li, Y.; Han, T.; Liu, L.; Li, L.; Zhang, S.; Liu, Y.; et al. Long non-coding RNA SNHG5 suppresses gastric cancer progression by trapping MTA2 in the cytosol. Oncogene 2016, 35, 5770–5780. [Google Scholar] [CrossRef]
- Wang, J.; Tian, Y.; Zheng, H.; Ding, Y.; Wang, X. An integrated analysis reveals the oncogenic function of lncRNA LINC00511 in human ovarian cancer. Cancer Med. 2019, 8, 3026–3035. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Zhang, X. TP53 Mutation Related and Directly Regulated lncRNA Prognosis Markers in Hepatocellular Carcinoma. OncoTargets Ther. 2021, 14, 4427–4437. [Google Scholar] [CrossRef]
- Nadeu, F.; Martin-Garcia, D.; Clot, G.; Díaz-Navarro, A.; Duran-Ferrer, M.; Navarro, A.; Vilarrasa-Blasi, R.; Kulis, M.; Royo, R.; Gutiérrez-Abril, J.; et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood 2020, 136, 1419–1432. [Google Scholar] [CrossRef]
- Hu, G.; Zhang, Y.; Gupta, M. RIP sequencing in mantle cell lymphoma identifies functional long non-coding RNAs associated with translation machinery. Blood Cancer J. 2019, 9, 55. [Google Scholar] [CrossRef]
- Farhana, A.; Lappin, S.L. Biochemistry, lactate dehydrogenase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Tao, Q.; Qi, Y.; Gu, J.; Yu, D.; Lu, Y.; Liu, J.; Liang, X. Breast cancer cells-derived Von Willebrand Factor promotes VEGF-A-related angiogenesis through PI3K/Akt-miR-205-5p signaling pathway. Toxicol. Appl. Pharmacol. 2022, 440, 115927. [Google Scholar] [CrossRef]
- Sano, M.; Takahashi, R.; Ijichi, H.; Ishigaki, K.; Yamada, T.; Miyabayashi, K.; Kimura, G.; Mizuno, S.; Kato, H.; Fujiwara, H.; et al. Blocking VCAM-1 inhibits pancreatic tumour progression and cancer-associated thrombosis/thromboembolism. Gut 2020, 70, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Z.; Gao, A.; Wen, Q.; Sun, Y. The prognostic landscape of tumor-infiltrating immune cells in cervical cancer. Biomed. Pharmacother. 2019, 120, 109444. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, W.; Xu, P. NK cell and macrophages confer prognosis and reflect immune status in osteosarcoma. J. Cell Biochem. 2018, 120, 8792–8797. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Wu, W.; Amei, A.; Yan, X.; Lu, L.; Wang, Z. Regulation and characterization of tumor-infiltrating immune cells in breast cancer. Int. Immunopharmacol. 2020, 90, 107167. [Google Scholar] [CrossRef]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 331. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Q.; Wang, Z.; Zhang, H.; Zeng, H.; Huang, Q.; Chen, Y.; Jiang, W.; Lin, Z.; Qu, Y.; et al. Intratumoral TIGIT+ CD8+ T-cell infiltration determines poor prognosis and immune evasion in patients with muscle-invasive bladder cancer. J. Immunother. Cancer 2020, 8, e000978. [Google Scholar] [CrossRef]
- Simon, S.C.S.; Utikal, J.; Umansky, V. Opposing roles of eosinophils in cancer. Cancer Immunol. Immunother. 2018, 68, 823–833. [Google Scholar] [CrossRef]
- Hu, G.; Wang, S.; Zhong, K.; Xu, F.; Huang, L.; Chen, W.; Cheng, P. Tumor-associated tissue eosinophilia predicts favorable clinical outcome in solid tumors: A meta-analysis. BMC Cancer 2020, 20, 454. [Google Scholar] [CrossRef]
- Hansen, S.V.; Hansen, M.H.; Cédile, O.; Møller, M.B.; Haaber, J.; Abildgaard, N.; Nyvold, C.G. Detailed characterization of the transcriptome of single B cells in mantle cell lymphoma suggesting a potential use for SOX4. Sci. Rep. 2021, 11, 19092. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Kern, W.; Haferlach, C.; Schnittger, S. SOX11 overexpression is a specific marker for mantle cell lymphoma and correlates with t(11;14) translocation, CCND1 expression and an adverse prognosis. Leukemia 2013, 27, 2388–2391. [Google Scholar] [CrossRef]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom. 2014, 15, 419. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Transcript Type | Associated Cancer Type | Cancer-Related Functions or Processes Promoted | Reference |
|---|---|---|---|---|
| FSTL1 | mRNA | Colorectal cancer | Metastasis | Gu et al., 2018 [50] |
| VCAM1 | mRNA | Breast cancer | Epithelial–mesenchymal transition (EMT) | Wang et al., 2014 [51] |
| TNS1 | mRNA | Colorectal cancer | Proliferation and invasion | Zhou et al., 2018 [52] |
| SEMA5A | mRNA | Pancreatic cancer | Angiogenesis, proliferation, migration, anti-apoptosis | Sadanandam et al., 2010 [53] |
| DDR2 | mRNA | Breast cancer | Metastasis, migration, invasion, EMT | Ren et al., 2014 [54] |
| VWF | mRNA | Gastric adenocarcinoma | Metastasis | Yang et al., 2018 [55] |
| CCND1 | mRNA | Nasopharyngeal carcinoma | Cell cycle | Liu et al., 2012 [56] |
| NFIB | mRNA | Breast cancer | Cell survival | Liu et al., 2019 [57] |
| ANTXR1 | mRNA | Glioma | Proliferation, migration, anti-apoptosis | Geng et al., 2019 [58] |
| PBX1 | mRNA | Ovarian cancer | Proliferation | Park et al., 2008 [59] |
| Gene | Transcript Type | Associated Cancer Type | Cancer-Related Functions or Processes Regulated | Reference |
|---|---|---|---|---|
| UBXN1 | mRNA | Osteosarcoma | Apoptosis | Wang et al., 2015 [60] |
| HNRNPF | mRNA | Breast cancer | EMT * suppression | Huang et al., 2017 [61] |
| PPP1R15A | mRNA | Burkitt’s lymphoma | Apoptosis | Hollander et al., 2001 [62] |
| HNRNPA1 | mRNA | Ovarian cancer | Proliferation, motility, angiogenesis, and apoptosis | Rodriguez-Aguayo et al., 2017 [63] |
| THRAP3 | mRNA | Cervical cancer | DNA damage response | Beli et al., 2012 [64] |
| LAPTM5 | mRNA | Multiple myeloma | Cellular differentiation, apoptosis | Hayami et al., 2003 [65] |
| DDB1 | mRNA | HPV-associated cancers | Cellular senescence | Kotake et al., 2009 [66] |
| RPL7A | mRNA | Osteosarcoma | Cell growth, differentiation | Zheng et al., 2009 [67] |
| PIK3IP1 | mRNA | Hepatocellular carcinoma | Proliferation, motility | He et al., 2008 [68] |
| DDIT4 | mRNA | Breast cancer | Proliferation, cell growth | DeYoung et al., 2008 [69] |
| Gene | Transcript Type | Associated Cancer Type | Cancer-Related Functions or Processes Promoted | Reference |
|---|---|---|---|---|
| MIR100HG | lncRNA | Laryngeal squamous cell carcinoma | Proliferation, migration, invasion | Huang et al., 2019 [70] |
| LINC01268 | lncRNA | Acute myeloid leukemia | Cell growth, anti-apoptosis | Chen et al.,2020 [71] |
| FTX | lncRNA | Gastric cancer | Proliferation, migration, invasion | Li et al., 2019 [72] |
| ROR1-AS1 | lncRNA | Mantle cell lymphoma | Cell growth | Hu et al., 2017 [73] |
| DNM3OS | lncRNA | Gastric cancer | Proliferation, migration, invasion, EMT * | Wang et al., 2019 [74] |
| KCNQ1OT1 | lncRNA | Non-small-cell lung cancer | Proliferation, anti-apoptosis | Kang et al., 2019 [75] |
| MAGI1-IT1 | lncRNA | Epithelial ovarian cancer | Invasion, metastasis | Gao et al., 2019 [76] |
| NR2F2-AS1 | lncRNA | Nasopharyngeal carcinoma | Proliferation, anti-apoptosis | Qin and Qin, 2020 [77] |
| ADAMTS9-AS2 | lncRNA | Tongue squamous cell carcinoma | Proliferation, migration, EMT * | Li et al., 2019 [78] |
| PCA3 | lncRNA | Prostate cancer | Proliferation, migration, invasion, anti-apoptosis | Zhang et al., 2019 [79] |
| Gene | Transcript Type | Associated Cancer Type | Cancer-Related Functions or Processes Regulated | Reference |
|---|---|---|---|---|
| LINC00877 | lncRNA | Pheochromocytomas and paragangliomas | Metastasis | Ghosal et al., 2022 [80] |
| SLC25A5-AS1 | lncRNA | Gastric cancer | Cell growth, apoptosis | Li et al., 2019 [81] |
| ILF3-DT | lncRNA | Cervical cancer | Autophagy | Feng et al., 2021 [82] |
| LRRC75A-AS1 | lncRNA | Colorectal cancer | Proliferation, migration | Chen et al., 2019 [83] |
| LINC00324 | lncRNA | Breast cancer | Proliferation, invasion, migration, apoptosis | Wang et al., 2020 [84] |
| CD27-AS1 | lncRNA | Acute myeloid leukemia | Proliferation, cellular senescence, apoptosis | Tao et al., 2021 [85] |
| ZFAS1 | lncRNA | Breast cancer | Proliferation, migration, invasion | Fan et al., 2018 [86] |
| SNHG5 | lncRNA | Gastric cancer | Proliferation, metastasis | Zhao et al., 2016 [87] |
| MIR762HG | lncRNA | Ovarian cancer | Downregulated in ovarian cancer | Wang et al., 2019 [88] |
| SNRK-AS1 | lncRNA | Hepatocellular carcinoma | Downregulated in hepatocellular carcinoma | Zhang et al., 2021 [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esmeray Sönmez, E.; Hatipoğlu, T.; Kurşun, D.; Hu, X.; Akman, B.; Yuan, H.; Erşen Danyeli, A.; Alacacıoğlu, İ.; Özkal, S.; Olgun, A.; et al. Whole Transcriptome Sequencing Reveals Cancer-Related, Prognostically Significant Transcripts and Tumor-Infiltrating Immunocytes in Mantle Cell Lymphoma. Cells 2022, 11, 3394. https://doi.org/10.3390/cells11213394
Esmeray Sönmez E, Hatipoğlu T, Kurşun D, Hu X, Akman B, Yuan H, Erşen Danyeli A, Alacacıoğlu İ, Özkal S, Olgun A, et al. Whole Transcriptome Sequencing Reveals Cancer-Related, Prognostically Significant Transcripts and Tumor-Infiltrating Immunocytes in Mantle Cell Lymphoma. Cells. 2022; 11(21):3394. https://doi.org/10.3390/cells11213394
Chicago/Turabian StyleEsmeray Sönmez, Esra, Tevfik Hatipoğlu, Deniz Kurşun, Xiaozhou Hu, Burcu Akman, Hongling Yuan, Ayça Erşen Danyeli, İnci Alacacıoğlu, Sermin Özkal, Aybüke Olgun, and et al. 2022. "Whole Transcriptome Sequencing Reveals Cancer-Related, Prognostically Significant Transcripts and Tumor-Infiltrating Immunocytes in Mantle Cell Lymphoma" Cells 11, no. 21: 3394. https://doi.org/10.3390/cells11213394
APA StyleEsmeray Sönmez, E., Hatipoğlu, T., Kurşun, D., Hu, X., Akman, B., Yuan, H., Erşen Danyeli, A., Alacacıoğlu, İ., Özkal, S., Olgun, A., Erdağ, T. K., You, H., & Küçük, C. (2022). Whole Transcriptome Sequencing Reveals Cancer-Related, Prognostically Significant Transcripts and Tumor-Infiltrating Immunocytes in Mantle Cell Lymphoma. Cells, 11(21), 3394. https://doi.org/10.3390/cells11213394