1. Introduction
Astrocytes constitute the largest portion of cells and are extremely dense in the brain [
1]. Unlike neurons, which are electrically activated and receive signals through synapses, astrocytes extend processes between the synaptic gaps of neurons and directly interact with pre- and postsynaptic neurons, thereby regulating the formation, function, and elimination of synapses [
2]. In particular, astrocytes form a gap junction that is densely and closely connected between cells. This enables the movement of ions, water, glutamate, and gamma-aminobutyric acid (GABA) between cells, and plays an important role in regulating physiological homeostasis in the brain [
3,
4,
5].
All cells have a constant membrane potential maintained by various ions in the cell. This membrane potential becomes a source of energy to operate the channel, especially in excitatory neurons or muscle cells, enabling signal transmission between cells. The membrane potential is similar to the equilibrium potential of potassium and changes sensitively in response to the potassium concentration outside the cell [
6]. This occurs because the cell membrane has very high potassium permeability due to the high density of potassium channels such as Kir4.1 and leak K
+ channels [
7]. Potassium above an appropriate level in the brain causes depolarization of the resting neuronal membrane potential and affects a variety of neuronal activity [
4]. Perturbations in potassium homeostasis can induce neurological diseases such as seizures and ataxia [
4,
6]. Therefore, maintaining potassium homeostasis is essential for the central nervous system. Although the exact mechanism of potassium homeostasis in the brain remains ambiguous, many studies have shown that astrocytes may play an important role in homeostasis maintenance.
The excess potassium accumulated outside nerve cells is taken up by the channels in astrocyte membranes, transferred to astrocytes with a low concentration of extracellular potassium through the gap junction and released [
4,
8,
9]. This process is called potassium clearance, and contributes to the maintenance of potassium homeostasis [
4]. Disruption of this process in astrocytes causes neurological diseases including epilepsy. Potassium clearance by astrocytes allows for a linear current–voltage (
I–
V) relationship that differentiates them from other cells, which is called passive conductance [
10]. The physiological regulatory mechanism of passive conductance remains unclear but is considered an important characteristic resulting from intrinsic astrocyte functions.
The tandem of pore domains in a weak inward rectifying K
+ channel (TWIK1, K2P 1.1, or KCNK1) and TWIK-related K
+ channel 1 (TREK1, K2P 2.1, or KCNK2) are members of the two pore domain potassium (K2P) channel family, consisting of 15 channels that regulate the stabilization of resting membrane potential and cellular excitability by wielding background K
+ leakage currents [
11]. Although TWIK1 is considered a non-functional channel, recent studies showed that TWIK1 can act as a heterodimeric channel [
12]. The TWIK1/TASK3 heterodimeric channel regulates the intrinsic excitability of dentate granule neurons [
13]. Recently, we reported that TWIK1 and TREK1 form TWIK1/TREK1 heterodimeric channels in a disulfide bond-dependent manner. Additionally, these channels are the major molecular identities that constitute the passive conductivity of hippocampal astrocytes and regulate glutamate release [
14]. However, the functional regulatory mechanisms of these heterodimeric channels remain poorly understood.
Among the components of TWIK1/TREK1 heterodimeric channels, the regulatory mechanisms of TREK1 have been well studied. TREK1 is a polymodal activated channel regulated by a wide variety of chemical and physiological factors [
15]. The activity is regulated by intracellular acidosis, a stretch of mechanical stimulation, polyunsaturated fatty acids (PUFA) such as omega3, general anesthesia [
16], protein kinase A (PKA), and protein kinase C (PKC). Furthermore, its expression is disturbed in pathological conditions such as depression or ischemia [
17,
18,
19,
20]. TREK1 contains various binding proteins such as microtubule-associated protein 2 (Mtap2), A-kinase anchoring protein 150 (AKAP150) [
21], and Spadin [
22]. β-coat protein (COP) is a subunit of the COP1 complex involved in protein transport between the Golgi apparatus and endoplasmic reticulum (ER) [
23]. We reported that β-COP regulates membrane trafficking by directly binding to TREK1 [
24]. Compared to TREK1, regulation mechanisms and binding proteins of TWIK1 remain poorly understood. Functional properties of K2P channels are regulated by a variety of intracellular proteins for single conductivity, open probability, and membrane transport [
25,
26]. Therefore, TWIK1 or TREK1 binding proteins may also regulate the functions of TWIK1/TREK1 heterodimeric channels, although intracellular TWIK1 binding proteins have not yet been reported.
To test this hypothesis, we examined the role of β-COP on the function of TWIK1/TREK1 heterodimeric channels. In this study, we found that β-COP interacts with the TWIK1/TREK1 heterodimeric channel in a TREK1-dependent manner and enhances passive conductance in astrocytes. We also identified the exact binding motifs of TREK1 that are recognized by β-COP. These results clearly demonstrate that β-COP is a pivotal binding protein of TWIK1/TREK1 heterodimeric channels, which regulates astrocytic passive conductance in the brain.
2. Materials and Methods
2.1. Chemical
Spadin was obtained from Peptron (Peptide Custom Service, Daejeon, Korea). Stock solutions of all materials were stored at −20 °C and diluted in water to the required concentration immediately before the experiment.
2.2. Genetic Material and Vectors Used in Experiments
cDNAs encoding the full-length mouse TREK1 isotype 2 (GenBank accession no. NM_010607), TREK1 isotype 1 (NM_001159850), mouse TWIK1 (NM_008430), and mouse β-COP (NM_033370) cDNAs were generated by reverse transcription polymerase chain reaction (RT-PCR). Several mutants of TREK1 including TREK1ΔN1, TREK1ΔN1,2, TREK1ΔN1,3, TREK1ΔN1,4, and TREK1ΔN4 were generated using mouse TREK1 isotype 2 cDNA as a template using an EZchangeTM site-directed mutagenesis kit (Enzynomics). All expression vectors used in the experiment were cloned into various destination vectors, including pDEST-FLAG-C, pDEST-EGFP-N, and pDEST-IRES2-GFP using the gateway cloning method. TWIK1 shRNA and TREK1 shRNA were constructed for gene silencing in cultured astrocytes and have been previously validated for their effectiveness. The target regions of shRNA were as follows: mouse TWIK1: 5’-GCATCATCTACTCTGTCATCG-3’; mouse TREK1: 5’-GCGTGGAGATCTACGACAAGT-3’; mouse β-COP: 5’-GCAGTTAGCACTGGATCTTGT-3’; mouse α-COP: 5’- atagttaccctaatcgcaatt-3’; mouse γ-COP: 5’- gctcgggtctttaacgaaact-3’; and mouse ADP-ribosylation factor 1 (Arf1): 5’- gatgctgttctcttggtgttt-3’. pSicoR β-COP, α-COP, γ-COP, and Arf1 shRNAs were constructed using the EZchangeTM site-directed mutagenesis kit (Enzynomics) and confirmed by fluorescence imaging and immunoblotting. Co-transfection with the two types of shRNAs was accomplished using different pSicoR vectors expressing mCherry and GFP fluorescent proteins.
2.3. Primary Astrocyte Culture and Electroporation
Primary cortical astrocytes were obtained from 1-day-old male and female f C57BL/6 mouse pups. The isolated cerebral cortex was dissociated into a single cell suspension by gentle pipetting after the meninges were removed. The resulting single cell suspension was plated on glass coverslips coated with 0.1 mg/mL poly D-lysine (PDL) and used four days later. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum, 10% heat-inactivated horse serum, and 100 units per mL of penicillin-streptomycin. Cell plates were maintained at 37 °C in a humidified 5% CO2 incubator. On day four of incubation, the cells were gently washed to remove other suspended cell types and debris. Occasionally, the purity of cultured astrocytes was monitored by immunocytochemical staining with a specific antibody against glial fibrillary acidic protein (GFAP). Above 95% of primary cultured cells normally showed GFAP-positive signals. On the following day (day five of incubation), cells were placed on glass coverslips coated with PDL, and an optimized voltage protocol was used (Thermo Fisher, Waltham, MA, USA) using a Neon Electroporation instrument (Thermo Fisher, Waltham, MA, USA). A total of 1200 V, 20 ms pulse width, and 2 pulse counts were used to transfect cultured astrocytes with various shRNAs.
2.4. Cell Lines and Transfection
Human embryonic kidney (HEK) 293T and COS7 cells were purchased from the Korea Cell Line Bank (Seoul National University, Seoul, Korea). Cells were grown at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) added with 10% fetal bovine serum (Invitrogen) and 100 units per mL of penicillin-streptomycin. The humidified atmosphere consisted of 95% air and 5% CO2. Cells were transfected with polyetherimide (PEI; Sigma-Aldrich).
2.5. Immunocytochemistry (ICC)
Cultured astrocytes on coverslips were fixed in 4% paraformaldehyde (PFA) at room temperature for 20 min and permeabilized with phosphate buffered saline (PBS) containing 0.5% NP-40 for 10 min. Non-specific binding caused by the antibody was prevented by incubation for 2 h in 3% donkey serum (GeneTex). Thereafter, cells were incubated at 4 °C overnight in donkey serum-containing anti-TREK1 (Alomone Labs, # APC-047, 1: 200) and anti-β-COP (Santa Cruz Biotechnology; sc-393615, 1: 200). After washing the next day, Dye Light 488, 549 conjugated secondary antibodies (Jackson Labs, 1:500) were treated and incubated at room temperature for 1 h. After that, the cells were mounted, and observed under a Nikon A1 confocal microscope to determine the surface expression of TREK1 according to the decrease in β-COP expression. Anti-TREK1 was used in cultured astrocytes transfected with β-COP shRNA in the same manner as above. Before permeation, plasma membrane staining was performed with WGA-647 conjugate (1: 200; W32466, Thermo) through additional culture at 4 °C for 15 min.
2.6. Duolink Proximity Ligation Analysis
A Duolink Proximity ligation assay (PLA) was performed using anti-TREK1 (Alomone Labs, # APC-047, 1:100) or anti-TWIK1 antibody (Santa Cruz Biotechnology; sc-11483, 1:100) with β-COP (1:200; D- 10, Santa Cruz Biotechnology) using Bioscience’s in situ PLA kit according to the manufacturer’s instructions. The PLA probe anti-rabbit minus bound to anti-TREK1 and TWIK1 antibodies, and the PLA probe anti-mouse plus bound to anti-β-COP antibodies. Cultured astrocytes were transfected with scrambled (Sc) or β-COP shRNA and incubated for 24 h before the experiments. The cells were observed under a Nikon A1 confocal microscope.
2.7. Co-Immunoprecipitation (Co-IP) and Immunoblotting
For Co-IP, cultured astrocytes and HEK293T cells were washed with PBS and then lysed with RIPA buffer composed of 50 mM Tris-Cl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and protease inhibitor cocktail (Tech & Innovation, Kangwon, Korea), and incubated for 1 h at 4 °C. Then, the lysate was centrifuged at 13,000× g for 20 min at 4 °C, and only the supernatant was transferred to a new tube. For co-IP, the reaction was carried out overnight at 4 °C on a rocking mixer with the corresponding antibody (anti-TREK1, Alomone Labs; anti-β-COP, Santa Cruz Biotechnology; anti-Flag, Sigma-Aldrich). Subsequently, the reaction was mixed with Protein G Agarose (Santa Cruz Biotechnology) for 1 h and gently washed three times with LIPA buffer. For immunoblotting, protein samples were separated by SDS–polyacrylamide gel electrophoresis (PAGE) using 10% gels. The separated proteins on the gel were transferred onto polyvinylidene fluoride membranes. The blots were incubated overnight at 4 °C with an anti-TREK1 antibody (Alomone Labs; 1:1000), anti-FLAG antibody (Sigma-Aldrich, 1:1000), or anti-β-COP antibody (Santa Cruz Biotechnology, 1:500) anti-GFP antibody (Santa Cruz Biotechnology, 1:1000). Blots were then washed and incubated with horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, or anti-rabbit IgG, followed by washing and detection of immunoreactivity using enhanced chemiluminescence (Amersham Biosciences).
2.8. Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
RNA was isolated from cultured mouse astrocytes according to the manufacturer’s instructions using an RNA purification kit (Geneol, Korea). As soon as RNA was extracted, cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Invitrogen) according to the manufacturer’s instructions. The SYBR® Select Master Mix (Invitrogen, New York) was used, and qRT-PCR was performed using a Step One Plus instrument (Life Technologies, New York). Each assay was performed on astrocytes cultured from different mice. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal reference and run in parallel with the target gene. Data were obtained as cycle threshold (Ct) values (strike cycles). The expression level of the target gene was expressed as 2-ΔCT, where ΔCT represents the difference in Ct between the gene of interest and GAPDH.
2.9. Bimolecular Fluorescence Complementation (BiFC) Experiment
For BiFC analysis, TREK1 isotype 1, TREK1 isotype 2, and all deficient mutants of TREK1 were cloned into all possible combinations of bimolecular fluorescence complement (pBiFC)- VN173 and pBiFC-VC155 vectors. Vectors containing each of the two genes for the binding test were transfected into cells and fixed with 4% paraformaldehyde for 20 min at room temperature, 24 h after transfection. The nuclei were then stained with DAPI for 3 min and mounted with Dako fluorescence mounting medium. Fluorescence was observed using a confocal microscope.
2.10. Electrophysiological Recording
For electrophysiological data, cultured astrocytes were plated on coverslips 24 h after transfection with the target gene, and COS7 cells were plated on coverslips 12 h after transfection. To measure currents, cells were immersed in a standard bath solution containing: 150 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM N-2-Hydroxyethylpiperazine-N’-2-Ethanesulfonic Acid (HEPES), 5.5 mM D-glucose, and 20 mM sucrose (pH 7.4, adjusted with NaOH). A patch pipette was made using a borosilicate glass capillary (Warner Instruments, Washington, DC, USA), and filled with a standard solution containing:150 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM EGTA, and 10 mM HEPES (pH 7.2, adjusted with KOH). A Digidata 1550 A interface (Axon Instruments, Union City, CA, USA) was used to convert the digital-to-analog signals between the computer and amplifier, and Clampfit software (Axon Instruments, Union City, CA, USA) was used to analyze the currents. The current−voltage (I−V) curves were measured by applying 1-s ramp pulse (from −150 mV to +50 mV) from a holding potential of −60 mV while continuously perfusing the bathing solution into the chamber (RC-25 chamber, Warner Instruments, Washington, DC, USA) at a rate of 1 mL/min. Data was sampled at 5 kHz and filtered at 1 kHz. All experiments were performed at room temperature of 20−22 °C.
2.11. Immunohistochemistry (IHC)
Brain slices were washed with PBS at room temperature for 20 min, followed by antigen retrieval at 85 °C for 30 min using 10 mM sodium citrate buffer. Slices were permeabilized with 0.4% Triton X-100 in PBS at RT for 20 min. The slices were then blocked with 10% donkey serum and 0.1% Triton X-100 in PBS at RT for 3 h, followed by incubation with primary antibodies, 5% donkey serum, and 0.1% Triton X-100 in PBS at 4 ◦C overnight. After washing three times with 0.1% Triton X-100 in PBS at RT for 15 min, secondary antibodies, 5% donkey serum, and 0.1% Triton X-100 in PBS were added for 2 h at 4 °C. The slices were counterstained with DAPI and mounted with a mounting medium (Vectashield, Vector Laboratories Inc., Burlingame, CA, USA). The following antibodies were used: rabbit anti-TREK1 (Alomone Labs, Jerusalem, Israel, 1:200; RRID: APC-047, 1:200), mouse anti-β-COP (Santa Cruz Biotechnology, D-10, 1:200), rat anti-GFAP (Thermo Fisher, Waltham, MA, USA; 1:500, RRID:13-0300, 1:500), and Alexa Fluor 488-, 594-, and 647-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA; 1:300). All images were acquired using a Nikon Ti2 confocal microscope (Nikon Instruments, Inc., Melville, NY, USA).
2.12. Electrophysiological Recording in Hippocampal Slices
The CA1 region of the hippocampus was sliced (350 mm) using a vibrating blade microtome (Leica VT1000 S) 55 into the brains of virus-infected mice. One prepared slice was recovered in oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (130 mM NaCl, 24 mM NaHCO3, 3.5 mM KCl, 1.25 mM NaH2PO4, 1.5 mM CaCl2, 1.5 mM MgCl2, and 10 mM glucose saturated with 95% O2–5% CO2 at pH 7.4) for 1 h. Hippocampal slices were incubated in a chamber containing artificial cerebrospinal fluid (ACSF) consisting of 130 mM NaCl, 24 mM NaHCO3, 3.5 mM KCl, 1.25 mM NaH2PO4, 1.5 mM CaCl2, 1.5 mM MgCl2, and 10 mM glucose 95% O2–5% CO2 was continuously supplied during the experiment. Additionally, to determine the localization of astrocytes in the hippocampus, brain slices were treated with SR-101 (Sigma; 1 μM) dye for 30 min prior to current measurement. The patch pipette was filled with a solution containing 140 mM KCl, 10 mM HEPES, 5 mM EGTA, 2 mM Mg-ATP and 0.2 mM Na-GTP adjusted to pH 7.4 with KOH. Axopatch 200 A (Axon Instruments, Union City, CA, USA) was used to perform whole-cell patch recordings of astrocytes in the hippocampus. Whole-cell currents were recorded after applying 1-s voltage steps from −160 mV to +40 mV in 10 mV increments from a holing potential of −60 mV.
2.13. Statistics
Analysis of the experiment was performed using the Clampfit and SigmaPlot software. Images analyzed by confocal microscopy were analyzed in Image J, and quantification of all data was expressed as mean ± standard error of the mean (SEM). Significance was evaluated using Student’s t-test (paired t-test) or one-way ANOVA followed by Tukey’s post hoc test, and the significance levels were specified as follows. N.S: not significant, * p < 0.05, ** p < 0.01, and *** p < 0.001. Prism9.0 software (GraphPad Software, San Diego, CA, USA) was used for carrying out the statistical analysis.
4. Discussion
This study was conducted based on previously reported experimental results that β-COP directly binds to the N-terminus of TREK1 [
24]. β-COP is one of the seven subunits (α, β’, β, ε, γ, δ, ζ) of the COP1 complex that forms transport vesicles [
23]. In the presence of Arf1, a small GTPase, the COP1 complex recognizes a specific sequence of target proteins, forms a complex with coatomers, and is involved in material transport [
30,
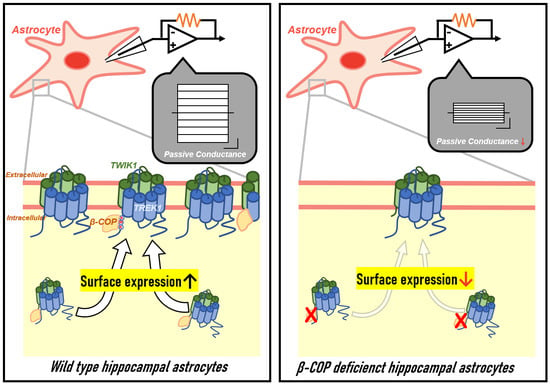
32]. However, our current results show that β-COP binds to TREK1 and enhances the surface expression and channel activity of TREK1 in astrocytes without the involvement of other coatomers of the COP1 complex to control surface expression of the channel (
Figure 1,
Figure 4 and
Figure S2). These data strongly suggest that β-COP acts as a monomer to regulate TREK1 function in astrocytes. β-COP is involved in retrograde transport or anterograde transport of various channel proteins [
24,
34,
35,
36,
37,
38]. Therefore, β-COP-mediated channel transport may also be regulated by β-COP monomers, but not the COP1 complex. Additionally, other members of the coatomer may be able to perform transport substances as monomers similar toβ-COP. These hypotheses should be tested in future studies.
Although β-COP is involved in retrograde or anterograde transport of various channels, it remains unclear whether the amino acid sequence of target proteins causes the difference in transport direction by β-COP. In this study, we attempted to determine the exact binding site of β-COP within TREK1 to address this question. First, TREK1 in mice has two isomers with different N-terminal amino acid sequences. Therefore, we compared the interactions between the two TREK1 isoforms and β-COP, although no difference was detected (
Figure 2). However, based on the qRT-PCR results, we found that the expression of TREK1 isotype 2 in astrocytes was higher than that of TREK1 isotype 1 (
Figure S2) and confirmed the binding site of β-COP within the N-terminal region of TREK1 (
Figure 3). The COP1 complex or β-COP is known to recognize and interact with the target protein through the following sites: 1) di-basic motif [
39], 2) K(X)KXX motif [
27,
28] (where X represents any amino acid), 3) RXR motif [
36,
40], and 4) KDEL motif [
41]. Using the deleted TREK1 mutants, we showed that β-COP binds to the di-lysine (KXKXX) motif at the end of the TREK1 N-terminus. Therefore, we could not find any differences in the binding motif of βCOP showing forward transport of TREK1 compared that showing retrograde transport of other channels. The relationship between the transport direction and the motif recognized by β-COP complex must be examined, comparing exact binding sites of other channels controlled by β-COP.
TWIK1 functions as a heterodimer through TREK1 and disulfide bonds in astrocytes [
14]. In HEK293T cells, co-IP was performed to determine whether there was an interaction between the heterodimer members, TWIK1, TREK1, and β-COP. A strong signal was observed for the binding of TREK1 to β-COP, as in a previous study [
24], but no signal was observed for TWIK1(
Figure S1B). However, based on the Duolink results in astrocytes, unlike HEK293T cells, strong PLA signals were found with anti-TWIK1 and anti-β-COP, which were abolished when treated with TREK1 shRNA (
Figure S1A). Thus, we revealed that β-COP does not interact with a single TWIK1. In astrocytes, where TWIK1 forms a heterodimer with TREK1, it interacts with β-COP in a TREK1 dependent manner and consequently interacts with the TWIK1/TREK1 heterodimeric channel (
Figure 4). Since K2P channels mainly function as dimers [
6,
12], further studies are required to determine whether other single-channel binding partners interact with heterodimers containing these channels. Investigating the interaction of single-channel binding partners with multichannel complexes is essential to reveal the underlying mechanisms of protein transport. Additionally, this interaction suggests a more diverse pathway in the regulation of surface expression of membrane proteins. It also becomes an important mediator in regulating the pathological conditions associated with that channel protein without changing the number of channel proteins.
Here, we suggest that the passive conductance of astrocytes, which is mediated by a TWIK1/TREK1 heterodimeric channel, is consequently regulated by β-COP. When excess K
+ accumulates extracellularly, the resting membrane potential changes, which greatly affects the synaptic transmission and activation of neurons [
4,
8,
9]. Astrocytes maintain brain homeostasis by removing the excess extracellular K
+ through K clearance, a process manifested by a linear current–voltage (
I–
V) relationship, that is, passive conductance [
4,
10]. Understanding the exact mechanism underlying this process will enable research on the various functions of astrocytes in the context of disease. β-COP regulates passive conductance through protein–protein interactions, and when β-COP is silenced in astrocytes, the membrane expression of TREK1 channels decreases (
Figure 4), currents mediated by TREK1 or TWIK1 are significantly reduced, and astrocytic passive conductance is significantly reduced. Since TWIK1 mRNA expression is upregulated in the hippocampal neurons and astrocytes in the kainic acid-induced seizure mouse model [
42], it seems that the increased TWIK1 expression might be a consequence of the defense mechanism against kainic acid-induced epileptic seizures. Therefore, our current study showing the β-COP-mediated increment of the TWIK1/TREK1 surface expression could help to develop therapeutic approaches for epileptic seizures.
We previously reported that TWIK1 and TREK1 among K2P channels are dominantly expressed in hippocampal astrocytes and the hippocampal astrocytic passive conductance is reduced by the deficiency of TWIK1 or TREK1 but not by the deficiency of TREK2 [
14]. However, it has been reported that the expression patterns of K2P channel are regionally dependent in the brain and other K2P members such as TREK2 or TASK1 are also expressed in astrocytes [
6]. In addition, TWIK1 can form heterodimeric channels with TREK2 or TASK1 in a heterologous expression system [
14]. Therefore, it is also plausible that astrocytic K
+ conductance in diverse regions of the brain might be controlled by different K2P channels. In addition to K2P channels, inwardly rectifying Kir4.1 channels in astrocytes also can be involved in passive conductance of hippocampal astrocytes. Since we previously found that deficiency of Kir4.1 also slightly reduced passive conductance of hippocampal astrocytes and significantly depolarized RMP of astrocytes [
14], it is also possible that β-COP also regulates the surface expression of Kir4.1 channels in hippocampal astrocytes. In addition, it is also worthwhile to examine the specific roles of K2P channels and Kir4.1 channels in various region-specific astrocytes.
In conclusion, we reveal for the first time that an important characteristic of astrocytes, passive conductance, is regulated through the protein–protein interactions with β-COP. β-COP is an important binding protein capable of modulating passive conductance mediated by the TWIK1/TREK1 heterodimeric channel. Therefore, these data suggest a new molecular structural mechanism for passive conductance. This research advanced our understanding of the mechanisms that regulate K+ clearance in astrocytes and the various regulatory pathways of TWIK1/TREK1 heterodimeric channels promoted by their binding proteins.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







