
Biomechanical Assessment of Macro-Calcification in Human Carotid Atherosclerosis and Its Impact on Smooth Muscle Cell Phenotype

 ,
, 
Abstract
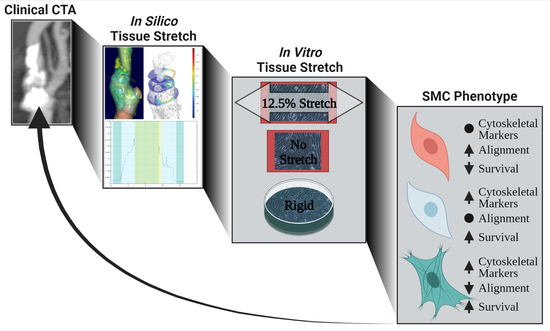
1. Introduction
2. Materials and Methods
2.1. Computed Tomography Angiography Image Analysis
2.2. In Silico Biomechanical Modeling
2.3. In Vitro Cyclic Stretch Assay
2.4. RNA Extraction and Gene Expression Analyses by Quantitative PCR (qPCR)
2.5. Immunofluorescence
2.6. Statistical Analyses
3. Results
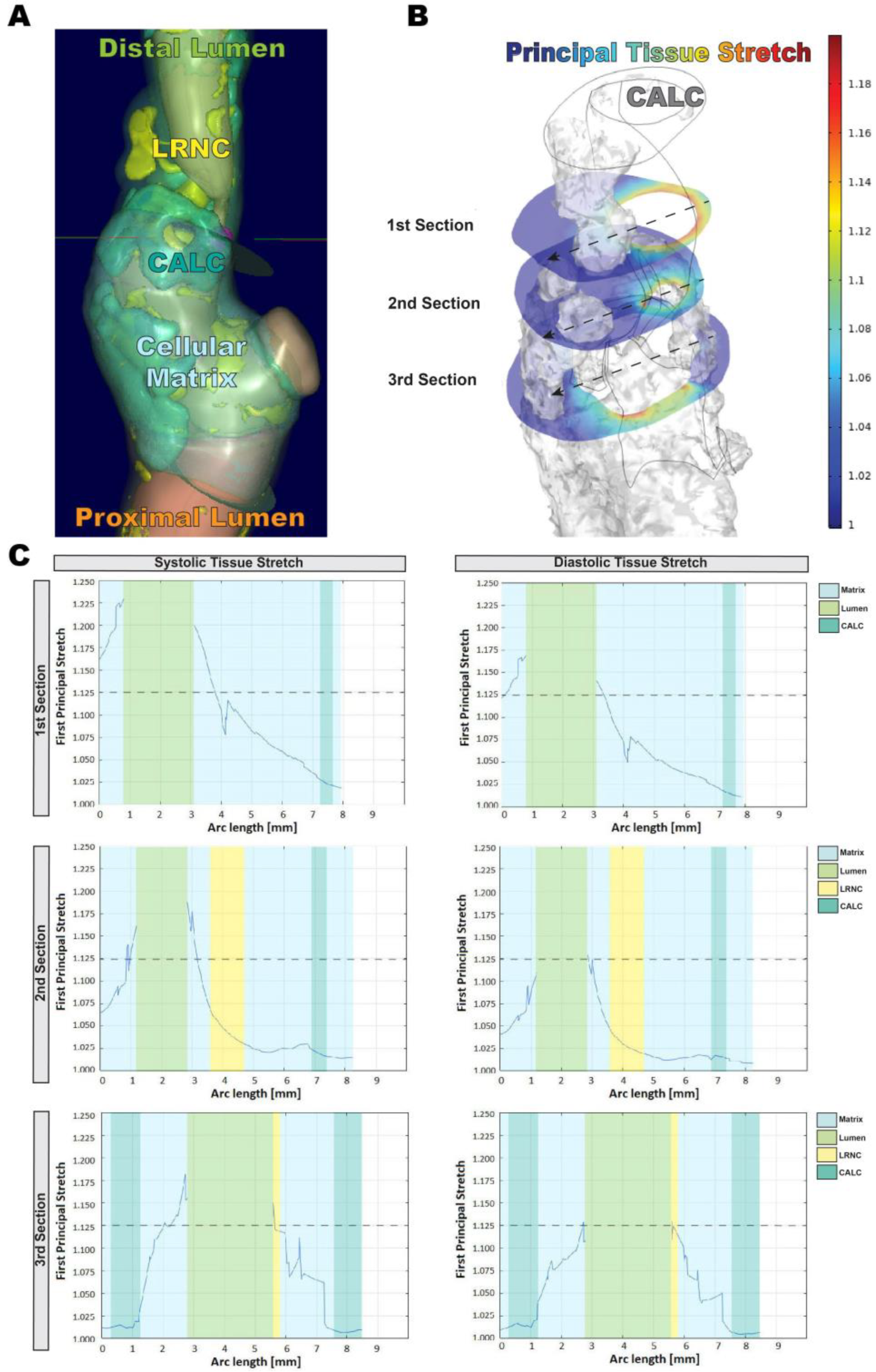
3.1. In Silico Modeling of Biomechanical Stretch Distributions among the Major Plaque Components
3.2. Changed Tissue Biomechanics Alter SMC Alignment and Phenotype In Vitro
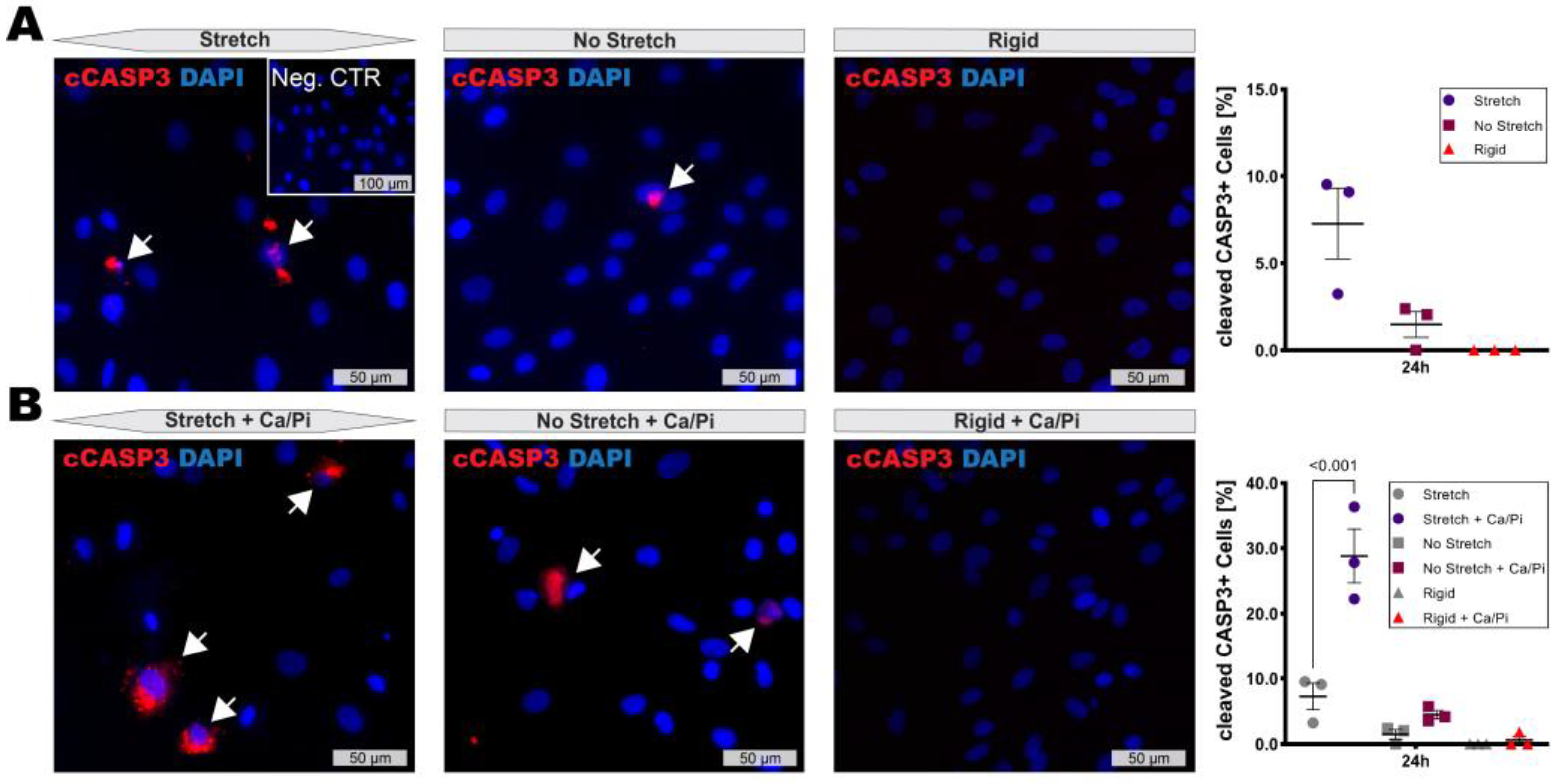
3.3. High Stretch Combined with Calcifying Conditions Induces SMC Apoptosis
4. Discussion
5. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Martinet, W.; Schrijvers, D.M.; De Meyer, G.R. Necrotic cell death in atherosclerosis. Basic Res. Cardiol. 2011, 106, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef]
- Kwee, R.M. Systematic review on the association between calcification in carotid plaques and clinical ischemic symptoms. J. Vasc. Surg. 2010, 51, 1015–1025. [Google Scholar] [CrossRef]
- Perisic, L.; Aldi, S.; Sun, Y.; Folkersen, L.; Razuvaev, A.; Roy, J.; Lengquist, M.; Akesson, S.; Wheelock, C.E.; Maegdefessel, L.; et al. Gene expression signatures, pathways and networks in carotid atherosclerosis. J. Intern. Med. 2016, 279, 293–308. [Google Scholar] [CrossRef]
- Karlöf, E.; Seime, T.; Dias, N.; Lengquist, M.; Witasp, A.; Almqvist, H.; Kronqvist, M.; Gadin, J.R.; Odeberg, J.; Maegdefessel, L.; et al. Correlation of computed tomography with carotid plaque transcriptomes associates calcification with lesion-stabilization. Atherosclerosis 2019, 288, P175–P185. [Google Scholar] [CrossRef] [PubMed]
- Karlöf, E.; Buckler, A.; Liljeqvist, M.L.; Lengquist, M.; Kronqvist, M.; Toonsi, M.A.; Maegdefessel, L.; Matic, L.P.; Hedin, U. Carotid Plaque Phenotyping by Correlating Plaque Morphology from Computed Tomography Angiography with Transcriptional Profiling. Eur. J. Vasc. Endovasc. Surg. 2021, 62, 716–726. [Google Scholar] [CrossRef]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef]
- Schwartz, S.M.; Virmani, R.; Rosenfeld, M.E. The good smooth muscle cells in atherosclerosis. Curr. Atheroscler. Rep. 2000, 2, 422–429. [Google Scholar] [CrossRef]
- Rodríguez, A.I.; Csányi, G.; Ranayhossaini, D.J.; Feck, D.M.; Blose, K.J.; Assatourian, L.; Vorp, D.A.; Pagano, P.J. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2015, 35, 430–438. [Google Scholar] [CrossRef]
- Imoto, K.; Hiro, T.; Fujii, T.; Murashige, A.; Fukumoto, Y.; Hashimoto, G.; Okamura, T.; Yamada, J.; Mori, K.; Matsuzaki, M. Longitudinal structural determinants of atherosclerotic plaque vulnerability: A computational analysis of stress distribution using vessel models and three-dimensional intravascular ultrasound imaging. J. Am. Coll. Cardiol. 2005, 46, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Chow, L.A.; Hsu, J.J.; Perlowski, A.A.; Abedin, M.; Tobis, J.; Tintut, Y.; Mal, A.K.; Klug, W.S.; Demer, L.L. Mechanical stress analysis of a rigid inclusion in distensible material: A model of atherosclerotic calcification and plaque vulnerability. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H802–H810. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Li, Y.; Ding, V.; Jiang, B.; Ball, R.L.; Rodriguez, F.; Fleischmann, D.; Desai, M.; Saloner, D.; Gupta, A.; et al. Semiautomated Characterization of Carotid Artery Plaque Features From Computed Tomography Angiography to Predict Atherosclerotic Cardiovascular Disease Risk Score. J. Comput. Assist. Tomogr. 2019, 43, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, M.; Ma, X.; Paik, D.; Obuchowski, N.A.; St Pierre, S.; Newman, W.P., 3rd; Rae, G.; Perlman, E.S.; Rosol, M.; Keith, J.C., Jr.; et al. Atherosclerotic Plaque Tissue: Noninvasive Quantitative Assessment of Characteristics with Software-aided Measurements from Conventional CT Angiography. Radiology 2018, 286, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Chrencik, M.T.; Khan, A.A.; Luther, L.; Anthony, L.; Yokemick, J.; Patel, J.; Sorkin, J.D.; Sikdar, S.; Lal, B.K. Quantitative assessment of carotid plaque morphology (geometry and tissue composition) using computed tomography angiography. J. Vasc. Surg. 2019, 70, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Uceda, D.E.; Dey, A.K.; Abdelrahman, K.M.; Aksentijevich, M.; Rodante, J.A.; Elnabawi, Y.A.; Reddy, A.; Keel, A.; Erb-Alvarez, J.; et al. Treatment of Psoriasis with Biologic Therapy Is Associated with Improvement of Coronary Artery Plaque Lipid-Rich Necrotic Core: Results from a Prospective, Observational Study. Circ. Cardiovasc. Imaging 2020, 13, e011199. [Google Scholar] [CrossRef]
- Abdelrahman, K.M.; Chen, M.Y.; Dey, A.K.; Virmani, R.; Finn, A.V.; Khamis, R.Y.; Choi, A.D.; Min, J.K.; Williams, M.C.; Buckler, A.J.; et al. Coronary Computed Tomography Angiography From Clinical Uses to Emerging Technologies: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1226–1243. [Google Scholar] [CrossRef]
- van Assen, M.; Varga-Szemes, A.; Egorova, S.; Johnson, K.; St. Pierre, S.; Zaki, B.; Schoepf, U.J.; Buckler, A.J. Automated plaque analysis for the prognostication of major adverse cardiac events. Eur. Soc. Cardiol. 2019, 116, 8. [Google Scholar] [CrossRef]
- Fang, Q.; Boas, D.A. Tetrahedral mesh generation from volumetric binary and grayscale images. In Proceedings of the IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Boston, MA, USA, 28 June–1 July 2009; pp. 1142–1145. [Google Scholar]
- Heiland, V.M.; Forsell, C.; Roy, J.; Hedin, U.; Gasser, T.C. Identification of carotid plaque tissue properties using an experimental-numerical approach. J. Mech. Behav. Biomed. Mater. 2013, 27, 226–238. [Google Scholar] [CrossRef]
- Chang, Y.T.; Chan, C.K.; Eriksson, I.; Johnson, P.Y.; Cao, X.; Westöö, C.; Norvik, C.; Andersson-Sjöland, A.; Westergren-Thorsson, G.; Johansson, S.; et al. Versican accumulates in vascular lesions in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 347–359. [Google Scholar] [CrossRef]
- Cardoso, L.; Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Weinbaum, S. Effect of tissue properties, shape and orientation of microcalcifications on vulnerable cap stability using different hyperelastic constitutive models. J. Biomech. 2014, 47, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Thavornpattanapong, P.; Cheung, S.C.; Sun, Z.; Tu, J. Effect of calcification on the mechanical stability of plaque based on a three-dimensional carotid bifurcation model. BMC Cardiovasc. Disord. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, M. Mechanical principles in arterial disease. Hypertension 1995, 26, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, S.; Castier, Y.; Tedgui, A. Molecular mechanisms of the vascular responses to haemodynamic forces. J. Intern. Med. 2006, 259, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Su, B.Y.; Shontz, K.M.; Flavahan, N.A.; Nowicki, P.T. The effect of phenotype on mechanical stretch-induced vascular smooth muscle cell apoptosis. J. Vasc. Res. 2006, 43, 229–237. [Google Scholar] [CrossRef]
- Butcher, J.T.; Barrett, B.C.; Nerem, R.M. Equibiaxial strain stimulates fibroblastic phenotype shift in smooth muscle cells in an engineered tissue model of the aortic wall. Biomaterials 2006, 27, 5252–5258. [Google Scholar] [CrossRef] [PubMed]
- Holm Nielsen, S.; Tengryd, C.; Edsfeldt, A.; Brix, S.; Genovese, F.; Bengtsson, E.; Karsdal, M.; Leeming, D.J.; Nilsson, J.; Goncalves, I. Markers of Basement Membrane Remodeling Are Associated with Higher Mortality in Patients with Known Atherosclerosis. J. Am. Heart Assoc. 2018, 7, e009193. [Google Scholar] [CrossRef]
- Röhl, S.; Rykaczewska, U.; Seime, T.; Suur, B.E.; GonzalezDiez, M.; Gådin, J.R.; Gainullina, A.; Sergushichev, A.A.; Wirka, R.; Lengquist, M.; et al. Transcriptomic profiling of experimental arterial injury reveals new mechanisms and temporal dynamics in vascular healing response. J. Vasc. Surg.-Vasc. Sci. 2020, 1, 13–27. [Google Scholar] [CrossRef]
- Cahalane, R.M.; Barrett, H.E.; O’Brien, J.M.; Kavanagh, E.G.; Moloney, M.A.; Walsh, M.T. Relating the mechanical properties of atherosclerotic calcification to radiographic density: A nanoindentation approach. Acta Biomater. 2018, 80, 228–236. [Google Scholar] [CrossRef]
- Standley, P.R.; Cammarata, A.; Nolan, B.P.; Purgason, C.T.; Stanley, M.A.; Camaratta, A. Cyclic stretch induces vascular smooth muscle cell alignment via NO signaling. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1907–H1914. [Google Scholar] [CrossRef]
- Perisic Matic, L.; Rykaczewska, U.; Razuvaev, A.; Sabater-Lleal, M.; Lengquist, M.; Miller, C.L.; Ericsson, I.; Rohl, S.; Kronqvist, M.; Aldi, S.; et al. Phenotypic Modulation of Smooth Muscle Cells in Atherosclerosis Is Associated with Downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arter. Thromb. Vasc. Biol. 2016, 36, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- von Kleeck, R.; Castagnino, P.; Roberts, E.; Talwar, S.; Ferrari, G.; Assoian, R.K. Decreased vascular smooth muscle contractility in Hutchinson-Gilford Progeria Syndrome linked to defective smooth muscle myosin heavy chain expression. Sci. Rep. 2021, 11, 10625. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Kinsella, M.G.; Wight, T.N.; Hedin, U.; Clowes, A.W. Heparan sulfate proteoglycans mediate a potent inhibitory signal for migration of vascular smooth muscle cells. Circ. Res. 1998, 83, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.K.; Tran-Lundmark, K.; Soininen, R.; Tryggvason, K.; Thyberg, J.; Hedin, U. Increased intimal hyperplasia and smooth muscle cell proliferation in transgenic mice with heparan sulfate-deficient perlecan. Circ. Res. 2004, 94, 550–558. [Google Scholar] [CrossRef]
- Hedin, U.; Roy, J.; Tran, P.K. Control of smooth muscle cell proliferation in vascular disease. Curr. Opin. Lipidol. 2004, 15, 559–565. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Yahagi, K.; Mori, H.; Romero, M.E.; Trout, H.H.; Finn, A.V.; Virmani, R. High-risk carotid plaque: Lessons learned from histopathology. Semin. Vasc. Surg. 2017, 30, 31–43. [Google Scholar] [CrossRef]
- McEvoy, J.W.; Blaha, M.J.; Defilippis, A.P.; Budoff, M.J.; Nasir, K.; Blumenthal, R.S.; Jones, S.R. Coronary artery calcium progression: An important clinical measurement? A review of published reports. J. Am. Coll. Cardiol. 2010, 56, 1613–1622. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef]
- Genge, B.R.; Wu, L.N.; Wuthier, R.E. In vitro modeling of matrix vesicle nucleation: Synergistic stimulation of mineral formation by annexin A5 and phosphatidylserine. J. Biol. Chem. 2007, 282, 26035–26045. [Google Scholar] [CrossRef]
- Raggi, P.; Genest, J.; Giles, J.T.; Rayner, K.J.; Dwivedi, G.; Beanlands, R.S.; Gupta, M. Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis 2018, 276, 98–108. [Google Scholar] [CrossRef]
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular Vesicles As Mediators of Cardiovascular Calcification. Front. Cardiovasc. Med. 2017, 4, 78. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.P.; Quignard, J.F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Lifshitz, L.M.; Tuft, R.A.; Fogarty, K.E.; Singer, J.J. Visualization of Ca2+ entry through single stretch-activated cation channels. Proc. Natl. Acad. Sci. USA 2002, 99, 6404–6409. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Whole Cohort, n |
|---|---|
| N | 4 |
| Age (year, mean) | 68 |
| Sex (female/male) | 1/3 |
| BMI (mean) | 29 |
| Smoking | |
| Present | 1 |
| Former | 1 |
| Never | 2 |
| Comorbidities | |
| previous myocardial infarction | 1 |
| angina pectoris | 2 |
| diabetes | 0 |
| Therapy | |
| lipid lowering (ezetimib, HMG-CoA reductase inhibitors) | 4 |
| antidiabetics | 0 |
| antihypertensives (ACE inhibitors, beta-blockers, diuretics, angiotensine II blockers) | 3 |
| Symptoms | |
| Amaurosis fugax | 1 |
| TIA | 1 |
| Minor Stroke | 0 |
| Asymptomatic | 2 |
| MATX | CALC | LRNC |
|---|---|---|
| c1 = 2.35 104 | c1 = 3.02 105 | c1 = 2.96 104 |
| c2 = 1.26 105 | c2 = −2.28 105 | c2 = −3.32 104 |
| c3 = 1.12 105 | c3 = 2.61 105 | c3 = 1.29 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seime, T.; van Wanrooij, M.; Karlöf, E.; Kronqvist, M.; Johansson, S.; Matic, L.; Gasser, T.C.; Hedin, U. Biomechanical Assessment of Macro-Calcification in Human Carotid Atherosclerosis and Its Impact on Smooth Muscle Cell Phenotype. Cells 2022, 11, 3279. https://doi.org/10.3390/cells11203279
Seime T, van Wanrooij M, Karlöf E, Kronqvist M, Johansson S, Matic L, Gasser TC, Hedin U. Biomechanical Assessment of Macro-Calcification in Human Carotid Atherosclerosis and Its Impact on Smooth Muscle Cell Phenotype. Cells. 2022; 11(20):3279. https://doi.org/10.3390/cells11203279
Chicago/Turabian StyleSeime, Till, Max van Wanrooij, Eva Karlöf, Malin Kronqvist, Staffan Johansson, Ljubica Matic, T. Christian Gasser, and Ulf Hedin. 2022. "Biomechanical Assessment of Macro-Calcification in Human Carotid Atherosclerosis and Its Impact on Smooth Muscle Cell Phenotype" Cells 11, no. 20: 3279. https://doi.org/10.3390/cells11203279
APA StyleSeime, T., van Wanrooij, M., Karlöf, E., Kronqvist, M., Johansson, S., Matic, L., Gasser, T. C., & Hedin, U. (2022). Biomechanical Assessment of Macro-Calcification in Human Carotid Atherosclerosis and Its Impact on Smooth Muscle Cell Phenotype. Cells, 11(20), 3279. https://doi.org/10.3390/cells11203279