


Tapping into Plant–Microbiome Interactions through the Lens of Multi-Omics Techniques

 ,
,  and
and 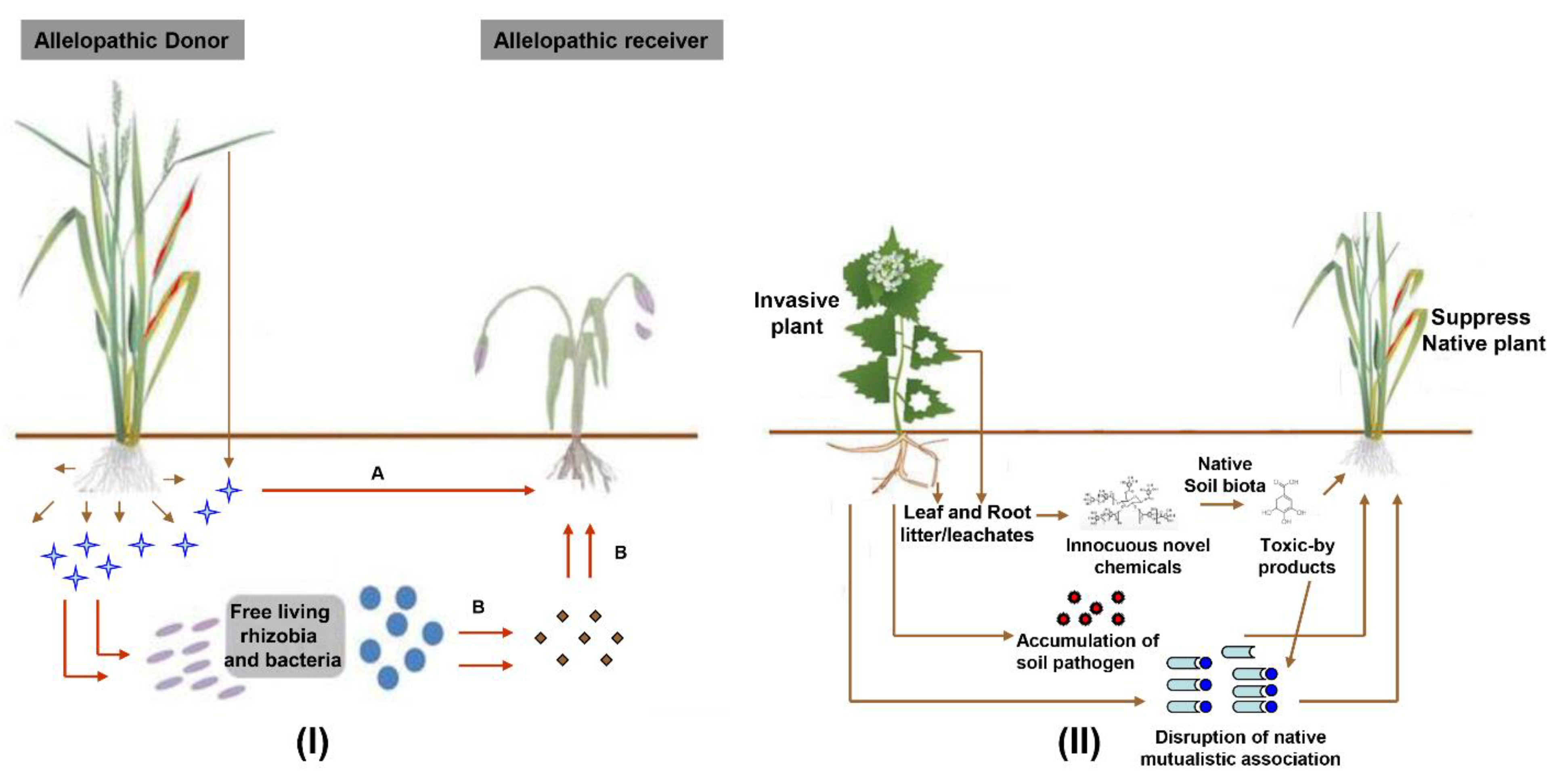{kind=link}
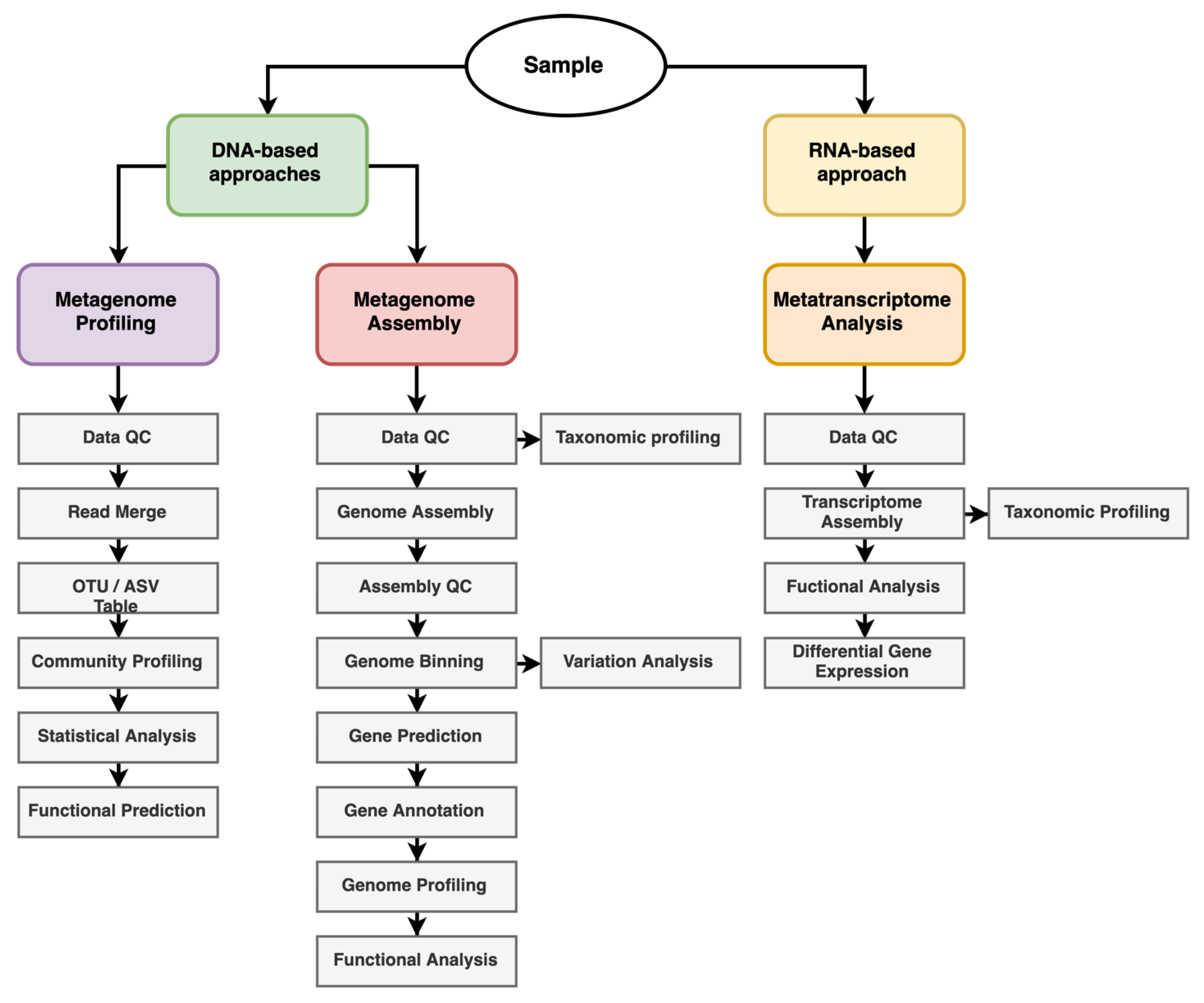{kind=link}
Abstract
1. Introduction
2. Primary Metabolite: Chemical Currency with Multipartite Function
3. Secondary Metabolite: Microbial and Plant Community Modulator Trait
4. Metagenomics and Metatranscriptomics: A Paradigm Shift in Microbiomics
5. Synthetic Microbial Communities (SynComs) Tool: An Exciting Frontier in Rhizosphere Research
6. Genome-Wide Association Study: Tool for Dissecting the Genetic Basis of Secondary Metabolite Variation
7. Metabolomics: The Bridge of Multi-Omics in Metabolite Discovery in Plant–Microbe Interactions
7.1. Instrumentation in Metabolomics
7.2. Instrumental Data Conversion to Meaningful Results
8. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. World Food Situation. FAO Cereal Supply and Demand Brief; FAO: Rome, Italy, 2015. [Google Scholar]
- Damalas, C.A.; Koutroubas, S.D. Farmers’ exposure to pesticides: Toxicity types and ways of prevention. Toxics 2016, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.R.; Gamalero, E. Recent developments in the study of plant microbiomes. Microorganisms 2021, 9, 1533. [Google Scholar] [CrossRef] [PubMed]
- Remus-Emsermann, M.N.; Lücker, S.; Müller, D.B.; Potthoff, E.; Daims, H.; Vorholt, J.A. Spatial distribution analyses of natural phyllosphere-colonizing bacteria on A rabidopsis thaliana revealed by fluorescence in situ hybridization. Environ. Microbiol. 2014, 16, 2329–2340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, H.; Li, M.; Xu, C.; Zhang, Y. Different age-induced changes in rhizosphere microbial composition and function of Panax ginseng in transplantation mode. Front. Plant Sci. 2020, 11, 563240. [Google Scholar] [CrossRef]
- Faddetta, T.; Abbate, L.; Alibrandi, P.; Arancio, W.; Siino, D.; Strati, F.; De Filippo, C.; Fatta Del Bosco, S.; Carimi, F.; Puglia, A.M. The endophytic microbiota of Citrus limon is transmitted from seed to shoot highlighting differences of bacterial and fungal community structures. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Vishwakarma, K.; Kumar, N.; Shandilya, C.; Mohapatra, S.; Bhayana, S.; Varma, A. Revisiting plant–microbe interactions and microbial consortia application for enhancing sustainable agriculture: A review. Front. Microbiol. 2020, 11, 560406. [Google Scholar] [CrossRef]
- Hu, L.; Robert, C.A.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; Van Der Heijden, M.G. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Kong, C.-H.; Xuan, T.D.; Khanh, T.D.; Tran, H.-D.; Trung, N.T. Allelochemicals and signaling chemicals in plants. Molecules 2019, 24, 2737. [Google Scholar] [CrossRef]
- Canarini, A.; Kaiser, C.; Merchant, A.; Richter, A.; Wanek, W. Root exudation of primary metabolites: Mechanisms and their roles in plant responses to environmental stimuli. Front. Plant Sci. 2019, 10, 157. [Google Scholar] [CrossRef]
- Gargallo-Garriga, A.; Sardans, J.; Pérez-Trujillo, M.; Guenther, A.; Llusià, J.; Rico, L.; Terradas, J.; Farré-Armengol, G.; Filella, I.; Parella, T. Shifts in plant foliar and floral metabolomes in response to the suppression of the associated microbiota. BMC Plant Biol. 2016, 16, 1–12. [Google Scholar] [CrossRef]
- Biedrzycki, M.L.; Jilany, T.A.; Dudley, S.A.; Bais, H.P. Root exudates mediate kin recognition in plants. Commun. Integr. Biol. 2010, 3, 28–35. [Google Scholar] [CrossRef]
- Smith, S.E.; Smith, F.A. Roles of arbuscular mycorrhizas in plant nutrition and growth: New paradigms from cellular to ecosystem scales. Annu. Rev. Plant Biol. 2011, 62, 227–250. [Google Scholar] [CrossRef]
- Kuzyakov, Y. Priming effects: Interactions between living and dead organic matter. Soil Biol. Biochem. 2010, 42, 1363–1371. [Google Scholar] [CrossRef]
- Naveed, M.; Brown, L.; Raffan, A.; George, T.S.; Bengough, A.G.; Roose, T.; Sinclair, I.; Koebernick, N.; Cooper, L.; Hackett, C.A. Plant exudates may stabilize or weaken soil depending on species, origin and time. Eur. J. Soil Sci. 2017, 68, 806–816. [Google Scholar] [CrossRef]
- Oleghe, E.; Naveed, M.; Baggs, E.M.; Hallett, P.D. Plant exudates improve the mechanical conditions for root penetration through compacted soils. Plant Soil 2017, 421, 19–30. [Google Scholar] [CrossRef]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef]
- Brady, N.C.; Weil, R.R.; Weil, R.R. The Nature and Properties of Soils; Prentice Hall: Upper Saddle River, NJ, USA, 2008; Volume 13. [Google Scholar]
- Lucas García, J.; Barbas, C.; Probanza, A.; Barrientos, M.; Gutierrez Mañero, F. Low molecular weight organic acids and fatty acids in root exudates of two Lupinus cultivars at flowering and fruiting stages. Phytochem. Anal. Int. J. Plant Chem. Biochem. Tech. 2001, 12, 305–311. [Google Scholar] [CrossRef]
- Dietz, S.; Herz, K.; Gorzolka, K.; Jandt, U.; Bruelheide, H.; Scheel, D. Root exudate composition of grass and forb species in natural grasslands. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Scavo, A.; Abbate, C.; Mauromicale, G. Plant allelochemicals: Agronomic, nutritional and ecological relevance in the soil system. Plant Soil 2019, 442, 23–48. [Google Scholar] [CrossRef]
- Curlango-Rivera, G.; Huskey, D.A.; Mostafa, A.; Kessler, J.O.; Xiong, Z.; Hawes, M.C. Intraspecies variation in cotton border cell production: Rhizosphere microbiome implications. Am. J. Bot. 2013, 100, 1706–1712. [Google Scholar] [CrossRef]
- Berazneva, J.; Conrad, J.M.; Güereña, D.T.; Lehmann, J.; Woolf, D. Agricultural productivity and soil carbon dynamics: A bioeconomic model. Am. J. Agric. Econ. 2019, 101, 1021–1046. [Google Scholar] [CrossRef]
- Jansson, C.; Faiola, C.; Wingler, A.; Zhu, X.-G.; Kravchenko, A.; De Graaff, M.-A.; Ogden, A.J.; Handakumbura, P.P.; Werner, C.; Beckles, D.M. Crops for carbon farming. Front. Plant Sci. 2021, 12, 636709. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Hirte, J.; Bender, S.F.; Mayer, J.; Gattinger, A.; Höschen, C.; Schädler, S.; Iqbal, T.M.; Mueller, C.W. Linking 3D soil structure and plant-microbe-soil carbon transfer in the rhizosphere. Front. Environ. Sci. 2018, 6, 9. [Google Scholar] [CrossRef]
- Bobille, H.; Limami, A.M.; Robins, R.J.; Cukier, C.; Le Floch, G.; Fustec, J. Evolution of the amino acid fingerprint in the unsterilized rhizosphere of a legume in relation to plant maturity. Soil Biol. Biochem. 2016, 101, 226–236. [Google Scholar] [CrossRef]
- Badri, D.V.; Loyola-Vargas, V.M.; Du, J.; Stermitz, F.R.; Broeckling, C.D.; Iglesias-Andreu, L.; Vivanco, J.M. Transcriptome analysis of Arabidopsis roots treated with signaling compounds: A focus on signal transduction, metabolic regulation and secretion. New Phytol. 2008, 179, 209–223. [Google Scholar] [CrossRef]
- Mora-Macías, J.; Ojeda-Rivera, J.O.; Gutiérrez-Alanís, D.; Yong-Villalobos, L.; Oropeza-Aburto, A.; Raya-González, J.; Jiménez-Domínguez, G.; Chávez-Calvillo, G.; Rellán-Álvarez, R.; Herrera-Estrella, L. Malate-dependent Fe accumulation is a critical checkpoint in the root developmental response to low phosphate. Proc. Natl. Acad. Sci. USA 2017, 114, E3563–E3572. [Google Scholar] [CrossRef]
- Manck-Götzenberger, J.; Requena, N. Arbuscular mycorrhiza symbiosis induces a major transcriptional reprogramming of the potato SWEET sugar transporter family. Front. Plant Sci. 2016, 7, 487. [Google Scholar] [CrossRef]
- Pratelli, R.; Voll, L.M.; Horst, R.J.; Frommer, W.B.; Pilot, G. Stimulation of nonselective amino acid export by glutamine dumper proteins. Plant Physiol. 2010, 152, 762–773. [Google Scholar] [CrossRef]
- Yang, H.; Bogner, M.; Stierhof, Y.-D.; Ludewig, U. H+-independent glutamine transport in plant root tips. PLoS ONE 2010, 5, e8917. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Symeonidi, E.; Pang, T.Y.; Denyer, T.; Weidauer, D.; Bezrutczyk, M.; Miras, M.; Zöllner, N.; Hartwig, T.; Wudick, M.M. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell 2021, 33, 511–530. [Google Scholar] [CrossRef]
- Yadav, B.; Jogawat, A.; Rahman, M.S.; Narayan, O.P. Secondary metabolites in the drought stress tolerance of crop plants: A review. Gene Rep. 2021, 23, 101040. [Google Scholar] [CrossRef]
- Piasecka, A.; Jedrzejczak-Rey, N.; Bednarek, P. Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 2015, 206, 948–964. [Google Scholar] [CrossRef]
- Hartman, K.; van der Heijden, M.G.; Roussely-Provent, V.; Walser, J.-C.; Schlaeppi, K. Deciphering composition and function of the root microbiome of a legume plant. Microbiome 2017, 5, 1–13. [Google Scholar] [CrossRef]
- Abdelrahman, M.; El-Sayed, M.A.; Hashem, A.; Abd_Allah, E.F.; Alqarawi, A.A.; Burritt, D.J.; Tran, L.-S.P. Metabolomics and transcriptomics in legumes under phosphate deficiency in relation to nitrogen fixation by root nodules. Front. Plant Sci. 2018, 9, 922. [Google Scholar] [CrossRef]
- Hassan, S.; Mathesius, U. The role of flavonoids in root–rhizosphere signalling: Opportunities and challenges for improving plant–microbe interactions. J. Exp. Bot. 2012, 63, 3429–3444. [Google Scholar] [CrossRef]
- Abdel-Lateif, K.; Bogusz, D.; Hocher, V. The role of flavonoids in the establishment of plant roots endosymbioses with arbuscular mycorrhiza fungi, rhizobia and Frankia bacteria. Plant Signal. Behav. 2012, 7, 636–641. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, B.; Chen, W.; Schlaeppi, K.; Erb, M.; Stirling, E.; Hu, L.; Wang, E.; Zhang, Y.; Zhao, K. Rhizobium symbiotic capacity shapes root-associated microbiomes in soybean. Front. Microbiol. 2021, 12, 709012. [Google Scholar] [CrossRef]
- Wasson, A.P.; Pellerone, F.I.; Mathesius, U. Silencing the flavonoid pathway in Medicago truncatula inhibits root nodule formation and prevents auxin transport regulation by rhizobia. Plant Cell 2006, 18, 1617–1629. [Google Scholar] [CrossRef]
- White, L.J.; Ge, X.; Brözel, V.S.; Subramanian, S. Root isoflavonoids and hairy root transformation influence key bacterial taxa in the soybean rhizosphere. Environ. Microbiol. 2017, 19, 1391–1406. [Google Scholar] [CrossRef]
- Maxwell, C.A.; Hartwig, U.A.; Joseph, C.M.; Phillips, D.A. A chalcone and two related flavonoids released from alfalfa roots induce nod genes of Rhizobium meliloti. Plant Physiol. 1989, 91, 842–847. [Google Scholar] [CrossRef]
- Popovici, J.; Comte, G.; Bagnarol, É.; Alloisio, N.; Fournier, P.; Bellvert, F.; Bertrand, C.; Fernandez, M.P. Differential effects of rare specific flavonoids on compatible and incompatible strains in the Myrica gale-Frankia actinorhizal symbiosis. Appl. Environ. Microbiol. 2010, 76, 2451–2460. [Google Scholar] [CrossRef]
- Huang, A.C.; Osbourn, A. Plant terpenes that mediate below-ground interactions: Prospects for bioengineering terpenoids for plant protection. Pest Manag. Sci. 2019, 75, 2368–2377. [Google Scholar] [CrossRef]
- Chen, Q.; Jiang, T.; Liu, Y.-X.; Liu, H.; Zhao, T.; Liu, Z.; Gan, X.; Hallab, A.; Wang, X.; He, J. Recently duplicated sesterterpene (C25) gene clusters in Arabidopsis thaliana modulate root microbiota. Sci. China Life Sci. 2019, 62, 947–958. [Google Scholar] [CrossRef]
- Lei, F.; Liu, X.; Huang, H.; Fu, S.; Zou, K.; Zhang, S.; Zhou, L.; Zeng, J.; Liu, H.; Jiang, L. The macleaya cordata symbiont: Revealing the effects of plant niches and alkaloids on the bacterial community. Front. Microbiol. 2021, 12, 1401. [Google Scholar] [CrossRef]
- Bradley, E.L.; Ökmen, B.; Doehlemann, G.; Henrissat, B.; Bradshaw, R.E.; Mesarich, C.H. Secreted glycoside hydrolase (GH) proteins as effectors and invasion patterns of plant-associated fungi and oomycetes. Front. Plant Sci. 2022, 13, 853106. [Google Scholar] [CrossRef]
- Raza, W.; Wei, Z.; Jousset, A.; Shen, Q.; Friman, V.-P. Extended plant metarhizobiome: Understanding volatile organic compound signaling in plant-microbe metapopulation networks. Msystems 2021, 6, e00849-00821. [Google Scholar] [CrossRef] [PubMed]
- Raza, W.; Wang, J.; Jousset, A.; Friman, V.-P.; Mei, X.; Wang, S.; Wei, Z.; Shen, Q. Bacterial community richness shifts the balance between volatile organic compound-mediated microbe–pathogen and microbe–plant interactions. Proc. R. Soc. B 2020, 287, 20200403. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Cheng, Z. Research progress on the use of plant allelopathy in agriculture and the physiological and ecological mechanisms of allelopathy. Front. Plant Sci. 2015, 6, 1020. [Google Scholar] [CrossRef]
- Kong, C.-H.; Zhang, S.-Z.; Li, Y.-H.; Xia, Z.-C.; Yang, X.-F.; Meiners, S.J.; Wang, P. Plant neighbor detection and allelochemical response are driven by root-secreted signaling chemicals. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, N.; Huang, Q.; Raza, W.; Li, R.; Vivanco, J.M.; Shen, Q. Organic acids from root exudates of banana help root colonization of PGPR strain Bacillus amyloliquefaciens NJN-6. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Micallef, S.A.; Channer, S.; Shiaris, M.P.; Colón-Carmona, A. Plant age and genotype impact the progression of bacterial community succession in the Arabidopsis rhizosphere. Plant Signal. Behav. 2009, 4, 777–780. [Google Scholar] [CrossRef]
- Dias, A.C.F.; Dini-Andreote, F.; Hannula, S.E.; Andreote, F.D.; Pereira e Silva, M.d.C.; Salles, J.F.; de Boer, W.; van Veen, J.; van Elsas, J.D. Different selective effects on rhizosphere bacteria exerted by genetically modified versus conventional potato lines. PLoS ONE 2013, 8, e67948. [Google Scholar] [CrossRef]
- Pascale, A.; Proietti, S.; Pantelides, I.S.; Stringlis, I.A. Modulation of the root microbiome by plant molecules: The basis for targeted disease suppression and plant growth promotion. Front. Plant Sci. 2020, 10, 1741. [Google Scholar] [CrossRef]
- Weinert, N.; Piceno, Y.; Ding, G.-C.; Meincke, R.; Heuer, H.; Berg, G.; Schloter, M.; Andersen, G.; Smalla, K. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: Many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 2011, 75, 497–506. [Google Scholar] [CrossRef]
- Haichar, F.e.Z.; Marol, C.; Berge, O.; Rangel-Castro, J.I.; Prosser, J.I.; Balesdent, J.; Heulin, T.; Achouak, W. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008, 2, 1221–1230. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, F. p-Coumaric acid influenced cucumber rhizosphere soil microbial communities and the growth of Fusarium oxysporum f. sp. cucumerinum Owen. PLoS ONE 2012, 7, e48288. [Google Scholar]
- Cipollini, D.; Rigsby, C.M.; Barto, E.K. Microbes as targets and mediators of allelopathy in plants. J. Chem. Ecol. 2012, 38, 714–727. [Google Scholar] [CrossRef]
- Sanon, A.; Andrianjaka, Z.; Prin, Y.; Bally, R.; Thioulouse, J.; Comte, G.; Duponnois, R. Rhizosphere microbiota interfers with plant-plant interactions. Plant Soil 2009, 321, 259–278. [Google Scholar] [CrossRef]
- Rodríguez-Echeverría, S. Rhizobial hitchhikers from Down Under: Invasional meltdown in a plant–bacteria mutualism? J. Biogeogr. 2010, 37, 1611–1622. [Google Scholar] [CrossRef]
- Elgersma, K.J.; Ehrenfeld, J.G. Linear and non-linear impacts of a non-native plant invasion on soil microbial community structure and function. Biol. Invasions 2011, 13, 757–768. [Google Scholar] [CrossRef]
- Van der Putten, W.H. Impacts of soil microbial communities on exotic plant invasions. Trends Ecol. Evol. 2010, 25, 512–519. [Google Scholar]
- Cantor, A.; Hale, A.; Aaron, J.; Traw, M.B.; Kalisz, S. Low allelochemical concentrations detected in garlic mustard-invaded forest soils inhibit fungal growth and AMF spore germination. Biol. Invasions 2011, 13, 3015–3025. [Google Scholar] [CrossRef]
- Mangla, S.; Inderjit; Callaway, R.M. Exotic invasive plant accumulates native soil pathogens which inhibit native plants. J. Ecol. 2008, 96, 58–67. [Google Scholar] [CrossRef]
- Großkinsky, D.K.; Syaifullah, S.J.; Roitsch, T. Integration of multi-omics techniques and physiological phenotyping within a holistic phenomics approach to study senescence in model and crop plants. J. Exp. Bot. 2018, 69, 825–844. [Google Scholar] [CrossRef]
- Bradbury, J. Isolation and preliminary study of bacteria from plants. Pest Artic. News Summ. 1970, 16, 632–637. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B.; List, F.B.C.A.; Bolchacova, E. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Simmons, T.; Caddell, D.F.; Deng, S.; Coleman-Derr, D. Exploring the root microbiome: Extracting bacterial community data from the soil, rhizosphere, and root endosphere. J. Vis. Exp. 2018, 135, e57561. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Kong, H.H. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Ghannoum, M.A.; Jurevic, R.J.; Mukherjee, P.K.; Cui, F.; Sikaroodi, M.; Naqvi, A.; Gillevet, P.M. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010, 6, e1000713. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.-W.; Yoou, M.-H.; Hong, W.-J.; Kim, Y.-J.; Lee, E.-J.; Jung, K.-H. Re-analysis of 16S amplicon sequencing data reveals soil microbial population shifts in rice fields under drought condition. Rice 2020, 13, 1–7. [Google Scholar] [CrossRef]
- Bolaji, A.J.; Wan, J.C.; Manchur, C.L.; Lawley, Y.; De Kievit, T.R.; Fernando, W.D.; Belmonte, M.F. Microbial community dynamics of soybean (Glycine max) is affected by cropping sequence. Front. Microbiol. 2021, 12, 632280. [Google Scholar] [CrossRef]
- Li, M.; Yang, F.; Wu, X.; Yan, H.; Liu, Y. Effects of continuous cropping of sugar beet (Beta vulgaris L.) on its endophytic and soil bacterial community by high-throughput sequencing. Ann. Microbiol. 2020, 70, 1–12. [Google Scholar] [CrossRef]
- Zhu, B.; Wu, J.; Ji, Q.; Wu, W.; Dong, S.; Yu, J.; Zhang, Q.; Qin, L. Diversity of rhizosphere and endophytic fungi in Atractylodes macrocephala during continuous cropping. PeerJ 2020, 8, e8905. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 47, D94–D99. [Google Scholar] [CrossRef]
- Douglas, G.M.; Beiko, R.G.; Langille, M.G. Predicting the functional potential of the microbiome from marker genes using PICRUSt. In Microbiome Analysis; Springer: Berlin/Heidelberg, Germany, 2018; pp. 169–177. [Google Scholar]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Matsuo, Y.; Komiya, S.; Yasumizu, Y.; Yasuoka, Y.; Mizushima, K.; Takagi, T.; Kryukov, K.; Fukuda, A.; Morimoto, Y.; Naito, Y. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution. BMC Microbiol. 2021, 21, 1–13. [Google Scholar] [CrossRef]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.-T.; Zhang, Z.-F.; Li, W.; Chen, W.; Cai, L. Microbiota in the rhizosphere and seed of rice from China, with reference to their transmission and biogeography. Front. Microbiol. 2020, 11, 995. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sudheer, S.; Usmani, Z.; Rani, R.; Gupta, P. Deciphering the omics of plant-microbe interaction: Perspectives and new insights. Curr. Genom. 2020, 21, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Focus: Microbiome: Metagenomic assembly: Overview, challenges and applications. Yale J. Biol. Med. 2016, 89, 353. [Google Scholar]
- Knief, C. Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 2014, 5, 216. [Google Scholar] [CrossRef]
- Xie, H.; Yang, C.; Sun, Y.; Igarashi, Y.; Jin, T.; Luo, F. PacBio long reads improve metagenomic assemblies, gene catalogs, and genome binning. Front. Genet. 2020, 11, 516269. [Google Scholar] [CrossRef]
- Adedayo, A.A.; Fadiji, A.E.; Babalola, O.O. The effects of plant health status on the community structure and metabolic pathways of rhizosphere microbial communities associated with solanum lycopersicum. Horticulturae 2022, 8, 404. [Google Scholar] [CrossRef]
- Mukherjee, A.; Reddy, M.S. Metatranscriptomics: An approach for retrieving novel eukaryotic genes from polluted and related environments. 3 Biotech 2020, 10, 1–19. [Google Scholar] [CrossRef]
- Turner, T.R.; Ramakrishnan, K.; Walshaw, J.; Heavens, D.; Alston, M.; Swarbreck, D.; Osbourn, A.; Grant, A.; Poole, P.S. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 2013, 7, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.L.; Chen, Y.; Bruns, T.; Peay, K.; Taylor, J.; Branco, S.; Talbot, J.; Vilgalys, R. Metatranscriptomic analysis of ectomycorrhizal roots reveals genes associated with P iloderma–P inus symbiosis: Improved methodologies for assessing gene expression in situ. Environ. Microbiol. 2014, 16, 3730–3742. [Google Scholar] [CrossRef] [PubMed]
- Haveman, N.J.; Khodadad, C.L.; Dixit, A.R.; Louyakis, A.S.; Massa, G.D.; Venkateswaran, K.; Foster, J.S. Evaluating the lettuce metatranscriptome with MinION sequencing for future spaceflight food production applications. npj Microgravity 2021, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Trivedi, P. Microbiome and the future for food and nutrient security. Microb. Biotechnol. 2017, 10, 50. [Google Scholar] [CrossRef]
- Sessitsch, A.; Pfaffenbichler, N.; Mitter, B. Microbiome applications from lab to field: Facing complexity. Trends Plant Sci. 2019, 24, 194–198. [Google Scholar] [CrossRef]
- McCarty, N.S.; Ledesma-Amaro, R. Synthetic biology tools to engineer microbial communities for biotechnology. Trends Biotechnol. 2019, 37, 181–197. [Google Scholar] [CrossRef]
- Armanhi, J.S.L.; De Souza, R.S.C.; Damasceno, N.d.B.; De Araujo, L.M.; Imperial, J.; Arruda, P. A community-based culture collection for targeting novel plant growth-promoting bacteria from the sugarcane microbiome. Front. Plant Sci. 2018, 8, 2191. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Zhuang, L.; Yu, Y.; Liu, J.; Zhang, L.; Gao, Z.; Wu, Y.; Gao, W.; Ding, G.-c. A rhizosphere-derived consortium of Bacillus subtilis and Trichoderma harzianum suppresses common scab of potato and increases yield. Comput. Struct. Biotechnol. J. 2019, 17, 645–653. [Google Scholar] [CrossRef]
- Andrade-Domínguez, A.; Salazar, E.; del Carmen Vargas-Lagunas, M.; Kolter, R.; Encarnacion, S. Eco-evolutionary feedbacks drive species interactions. ISME J. 2014, 8, 1041–1054. [Google Scholar] [CrossRef]
- Dolinšek, J.; Goldschmidt, F.; Johnson, D.R. Synthetic microbial ecology and the dynamic interplay between microbial genotypes. FEMS Microbiol. Rev. 2016, 40, 961–979. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef]
- Voges, M.J.; Bai, Y.; Schulze-Lefert, P.; Sattely, E.S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 12558–12565. [Google Scholar] [CrossRef]
- Niu, B.; Paulson, J.N.; Zheng, X.; Kolter, R. Simplified and representative bacterial community of maize roots. Proc. Natl. Acad. Sci. USA 2017, 114, E2450–E2459. [Google Scholar] [CrossRef]
- Stanley, C.E.; Shrivastava, J.; Brugman, R.; Heinzelmann, E.; van Swaay, D.; Grossmann, G. Dual-flow-RootChip reveals local adaptations of roots towards environmental asymmetry at the physiological and genetic levels. New Phytol. 2018, 217, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Carreno-Quintero, N.; Bouwmeester, H.J.; Keurentjes, J.J. Genetic analysis of metabolome–phenotype interactions: From model to crop species. Trends Genet. 2013, 29, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, F.; Nakabayashi, R.; Yang, Z.; Okazaki, Y.; Yonemaru, J.i.; Ebana, K.; Yano, M.; Saito, K. Metabolome-genome-wide association study dissects genetic architecture for generating natural variation in rice secondary metabolism. Plant J. 2015, 81, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Gemmer, M.R.; Richter, C.; Schmutzer, T.; Raorane, M.L.; Junker, B.; Pillen, K.; Maurer, A. Genome-wide association study on metabolite accumulation in a wild barley NAM population reveals natural variation in sugar metabolism. PLoS ONE 2021, 16, e0246510. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Li, D.; Li, X.; Gao, Y.; Li, W.; Li, H.; Liu, J.; Liu, H.; Chen, W.; Luo, J. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mönchgesang, S.; Strehmel, N.; Schmidt, S.; Westphal, L.; Taruttis, F.; Müller, E.; Herklotz, S.; Neumann, S.; Scheel, D. Natural variation of root exudates in Arabidopsis thaliana-linking metabolomic and genomic data. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Förster, N.; Ulrichs, C.; Schreiner, M.; Arndt, N.; Schmidt, R.; Mewis, I. Ecotype variability in growth and secondary metabolite profile in Moringa oleifera: Impact of sulfur and water availability. J. Agric. Food Chem. 2015, 63, 2852–2861. [Google Scholar] [CrossRef] [PubMed]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 1–8. [Google Scholar] [CrossRef]
- Tadesse, W.; Ogbonnaya, F.; Jighly, A.; Sanchez-Garcia, M.; Sohail, Q.; Rajaram, S.; Baum, M. Genome-wide association mapping of yield and grain quality traits in winter wheat genotypes. PLoS ONE 2015, 10, e0141339. [Google Scholar] [CrossRef]
- Patil, G.; Do, T.; Vuong, T.D.; Valliyodan, B.; Lee, J.-D.; Chaudhary, J.; Shannon, J.G.; Nguyen, H.T. Genomic-assisted haplotype analysis and the development of high-throughput SNP markers for salinity tolerance in soybean. Sci. Rep. 2016, 6, 1–13. [Google Scholar]
- Olukolu, B.A.; Tracy, W.F.; Wisser, R.; De Vries, B.; Balint-Kurti, P.J. A genome-wide association study for partial resistance to maize common rust. Phytopathology 2016, 106, 745–751. [Google Scholar] [CrossRef]
- Zhang, T.; Yu, L.-X.; Zheng, P.; Li, Y.; Rivera, M.; Main, D.; Greene, S.L. Identification of loci associated with drought resistance traits in heterozygous autotetraploid alfalfa (Medicago sativa L.) using genome-wide association studies with genotyping by sequencing. PLoS ONE 2015, 10, e0138931. [Google Scholar] [CrossRef]
- Horton, M.W.; Bodenhausen, N.; Beilsmith, K.; Meng, D.; Muegge, B.D.; Subramanian, S.; Vetter, M.M.; Vilhjálmsson, B.J.; Nordborg, M.; Gordon, J.I. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Lisec, J.; Czedik-Eysenberg, A.; Sulpice, R.; Flis, A.; Grieder, C.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. USA 2012, 109, 8872–8877. [Google Scholar] [CrossRef]
- Turner, M.F.; Heuberger, A.L.; Kirkwood, J.S.; Collins, C.C.; Wolfrum, E.J.; Broeckling, C.D.; Prenni, J.E.; Jahn, C.E. Non-targeted metabolomics in diverse sorghum breeding lines indicates primary and secondary metabolite profiles are associated with plant biomass accumulation and photosynthesis. Front. Plant Sci. 2016, 7, 953. [Google Scholar] [CrossRef]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for plant improvement: Status and prospects. Front. Plant Sci. 2017, 8, 1302. [Google Scholar] [CrossRef]
- Kusano, M.; Saito, K. Role of metabolomics in crop improvement. J. Plant Biochem. Biotechnol. 2012, 21, 24–31. [Google Scholar] [CrossRef]
- Sakurai, N. Recent applications of metabolomics in plant breeding. Breed. Sci. 2022, 72, 21065. [Google Scholar] [CrossRef]
- Fritsche-Guenther, R.; Gloaguen, Y.; Bauer, A.; Opialla, T.; Kempa, S.; Fleming, C.A.; Redmond, H.P.; Kirwan, J.A. Optimized workflow for on-line derivatization for targeted metabolomics approach by gas chromatography-mass spectrometry. Metabolites 2021, 11, 888. [Google Scholar] [CrossRef]
- Mushtaq, M.Y.; Choi, Y.H.; Verpoorte, R.; Wilson, E.G. Extraction for metabolomics: Access to the metabolome. Phytochem. Anal. 2014, 25, 291–306. [Google Scholar] [CrossRef]
- ElNaker, N.A.; Daou, M.; Ochsenkühn, M.A.; Amin, S.A.; Yousef, A.F.; Yousef, L.F. A metabolomics approach to evaluate the effect of lyophilization versus oven drying on the chemical composition of plant extracts. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Ellis, D.I.; Goodacre, R. Metabolic fingerprinting in disease diagnosis: Biomedical applications of infrared and Raman spectroscopy. Analyst 2006, 131, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Kamnev, A.A. FTIR spectroscopic studies of bacterial cellular responses to environmental factors, plant-bacterial interactions and signalling. Spectroscopy 2008, 22, 83–95. [Google Scholar] [CrossRef]
- Cao, Z.; Wang, Z.; Shang, Z.; Zhao, J. Classification and identification of Rhodobryum roseum Limpr. and its adulterants based on fourier-transform infrared spectroscopy (FTIR) and chemometrics. PLoS ONE 2017, 12, e0172359. [Google Scholar] [CrossRef] [PubMed]
- Naumann, A.; Heine, G.; Rauber, R. Efficient discrimination of oat and pea roots by cluster analysis of Fourier transform infrared (FTIR) spectra. Field Crops Res. 2010, 119, 78–84. [Google Scholar] [CrossRef]
- Legner, N.; Meinen, C.; Rauber, R. Root differentiation of agricultural plant cultivars and proveniences using FTIR spectroscopy. Front. Plant Sci. 2018, 9, 748. [Google Scholar] [CrossRef]
- El Zemrany, H.; Czarnes, S.; Hallett, P.D.; Alamercery, S.; Bally, R.; Jocteur Monrozier, L. Early changes in root characteristics of maize (Zea mays) following seed inoculation with the PGPR Azospirillum lipoferum CRT1. Plant Soil 2007, 291, 109–118. [Google Scholar] [CrossRef]
- Rewald, B.; Meinen, C.; Trockenbrodt, M.; Ephrath, J.E.; Rachmilevitch, S. Root taxa identification in plant mixtures–current techniques and future challenges. Plant Soil 2012, 359, 165–182. [Google Scholar] [CrossRef]
- Mal, T.K.; Tian, Y.; Patterson, A.D. Sample preparation and data analysis for NMR-based metabolomics. In Translational Bioinformatics for Therapeutic Development; Springer: Berlin/Heidelberg, Germany, 2021; pp. 301–313. [Google Scholar]
- Fan, T.W.-M.; Lane, A.N. Structure-based profiling of metabolites and isotopomers by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2008, 2, 69–117. [Google Scholar] [CrossRef]
- Gao, X. Recent advances in computational methods for nuclear magnetic resonance data processing. Genom. Proteom. Bioinform. 2013, 11, 29–33. [Google Scholar] [CrossRef]
- Arany, A.M.; De Jong, T.; Kim, H.; Van Dam, N.; Choi, Y.; Verpoorte, R.; Van der Meijden, E. Glucosinolates and other metabolites in the leaves of Arabidopsis thaliana from natural populations and their effects on a generalist and a specialist herbivore. Chemoecology 2008, 18, 65–71. [Google Scholar] [CrossRef]
- Ali, K.; Maltese, F.; Zyprian, E.; Rex, M.; Choi, Y.H.; Verpoorte, R. NMR metabolic fingerprinting based identification of grapevine metabolites associated with downy mildew resistance. J. Agric. Food Chem. 2009, 57, 9599–9606. [Google Scholar] [CrossRef]
- Padhi, E.M.; Maharaj, N.; Lin, S.-Y.; Mishchuk, D.O.; Chin, E.; Godfrey, K.; Foster, E.; Polek, M.; Leveau, J.H.; Slupsky, C.M. Metabolome and microbiome signatures in the roots of citrus affected by huanglongbing. Phytopathology 2019, 109, 2022–2032. [Google Scholar] [CrossRef]
- Nemadodzi, L.E.; Vervoort, J.; Prinsloo, G. NMR-based metabolomic analysis and microbial composition of soil supporting burkea africana growth. Metabolites 2020, 10, 402. [Google Scholar] [CrossRef]
- Ren, J.-L.; Zhang, A.-H.; Kong, L.; Wang, X.-J. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC advances 2018, 8, 22335–22350. [Google Scholar] [CrossRef]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Liebal, U.W.; Phan, A.N.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine learning applications for mass spectrometry-based metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Q.; Zhang, X.; Chen, J.; Liang, X.; Kettrup, A. High-performance liquid chromatography–tandem mass spectrometry for identification of isoflavones and description of the biotransformation of kudzu root. Anal. Bioanal. Chem. 2005, 383, 787–796. [Google Scholar] [CrossRef]
- Kisiala, A.; Laffont, C.; Emery, R.N.; Frugier, F. Bioactive cytokinins are selectively secreted by Sinorhizobium meliloti nodulating and nonnodulating strains. Mol. Plant-Microbe Interact. 2013, 26, 1225–1231. [Google Scholar] [CrossRef]
- Nebbioso, A.; De Martino, A.; Eltlbany, N.; Smalla, K.; Piccolo, A. Phytochemical profiling of tomato roots following treatments with different microbial inoculants as revealed by IT-TOF mass spectrometry. Chem. Biol. Technol. Agric. 2016, 3, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Shi, H.; Xu, P.; Yu, D. Metabolomics and microbiome reveal potential root microbiota affecting the alkaloidal metabolome in Aconitum vilmorinianum Kom. BMC Microbiol. 2022, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rampler, E.; Abiead, Y.E.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent topics in mass spectrometry-based metabolomics and lipidomics—Standardization, coverage, and throughput. Anal. Chem. 2020, 93, 519–545. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.T.; Guillarme, D.; Rudaz, S.; Veuthey, J.L. Fast analysis in liquid chromatography using small particle size and high pressure. J. Sep. Sci. 2006, 29, 1836–1848. [Google Scholar] [CrossRef]
- Lopez, S.H.; Dias, J.; Mol, H.; de Kok, A. Selective multiresidue determination of highly polar anionic pesticides in plant-based milk, wine and beer using hydrophilic interaction liquid chromatography combined with tandem mass spectrometry. J. Chromatogr. A 2020, 1625, 461226. [Google Scholar] [CrossRef]
- Lee, L.-S.; Choi, J.H.; Son, N.; Kim, S.-H.; Park, J.-D.; Jang, D.-J.; Jeong, Y.; Kim, H.-J. Metabolomic analysis of the effect of shade treatment on the nutritional and sensory qualities of green tea. J. Agric. Food Chem. 2013, 61, 332–338. [Google Scholar] [CrossRef]
- Zhou, L.; Sun, S.; Zhang, W.; He, Y.-W. Ultra-performance liquid chromatography/mass spectrometry for the detection and quantification of diffusible signal factor (DSF) family quorum-sensing signals. In Quorum Sensing; Springer: Berlin/Heidelberg, Germany, 2018; pp. 97–105. [Google Scholar]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry–based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef]
- Roessner, U.; Wagner, C.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Simultaneous analysis of metabolites in potato tuber by gas chromatography–mass spectrometry. Plant J. 2000, 23, 131–142. [Google Scholar] [CrossRef]
- De Vos, R.C.; Moco, S.; Lommen, A.; Keurentjes, J.J.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef]
- Patel, M.K.; Pandey, S.; Kumar, M.; Haque, M.I.; Pal, S.; Yadav, N.S. Plants metabolome study: Emerging tools and techniques. Plants 2021, 10, 2409. [Google Scholar] [CrossRef]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS-and GC/MS-based metabolomics: State of the field and future prospects. Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef]
- Cody, R.B.; Fouquet, T.N.; Takei, C. Thermal desorption and pyrolysis direct analysis in real time mass spectrometry for qualitative characterization of polymers and polymer additives. Rapid Commun. Mass Spectrom. 2020, 34, e8687. [Google Scholar] [CrossRef]
- Mallouchos, A.; Mikrou, T.; Gardeli, C. Gas Chromatography–Mass Spectrometry-Based Metabolite Profiling for the Assessment of Freshness in Gilthead Sea Bream (Sparus aurata). Foods 2020, 9, 464. [Google Scholar] [CrossRef]
- Arora, S.; Saini, M. Gas chromatography mass spectrometry profiling in methanolic and ethyl-acetate root and stem extract of Corbichonia decumbens (Forssk.) exell from Thar Desert of Rajasthan, India. Pharmacogn. Res. 2017, 9, S48. [Google Scholar] [CrossRef]
- Tayade, A.B.; Dhar, P.; Kumar, J.; Sharma, M.; Chauhan, R.S.; Chaurasia, O.P.; Srivastava, R.B. Chemometric profile of root extracts of Rhodiola imbricata Edgew. with hyphenated gas chromatography mass spectrometric technique. PLoS ONE 2013, 8, e52797. [Google Scholar] [CrossRef]
- Tapfuma, K.I.; Nchabeleng, E.K.; Adebo, O.A.; Hussan, R.; Williams, R.D.; Ravuluvulu, A.B.; Ndinteh, D.T.; Gan, R.-Y.; Habimana, O.; Niemann, N. Antibacterial activity and gas chromatography mass spectrometry (GC–MS)-based metabolite profiles of Celtis africana and its endophytic extracts. Ind. Crops Prod. 2020, 157, 112933. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Huang, L.; Fang, M.; Cupp-Sutton, K.A.; Wang, Z.; Smith, K.; Wu, S. Spray-capillary-based capillary electrophoresis mass spectrometry for metabolite analysis in single cells. Anal. Chem. 2021, 93, 4479–4487. [Google Scholar] [CrossRef]
- Maia, M.; Figueiredo, A.; Cordeiro, C.; Sousa Silva, M. FT-ICR-MS-based metabolomics: A deep dive into plant metabolism. Mass Spectrom. Rev. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, G.; Guo, J.; Wiedenhoeft, A.C.; Liu, C.C.; Yin, Y. Timber species identification from chemical fingerprints using direct analysis in real time (DART) coupled to Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS): Comparison of wood samples subjected to different treatments. Holzforschung 2019, 73, 975–985. [Google Scholar] [CrossRef]
- Dekermanjian, J.; Labeikovsky, W.; Ghosh, D.; Kechris, K. MSCAT: A machine learning assisted catalog of metabolomics software tools. Metabolites 2021, 11, 678. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Vorst, O.; De Vos, C.; Lommen, A.; Staps, R.; Visser, R.; Bino, R.; Hall, R. A non-directed approach to the differential analysis of multiple LC–MS-derived metabolic profiles. Metabolomics 2005, 1, 169–180. [Google Scholar] [CrossRef]
- Eylem, C.C.; Nemutlu, E.; Dogan, A.; Acik, V.; Matyar, S.; Gezercan, Y.; Altintas, S.; Okten, A.I.; Akduman, N.E.B. High-Throughput Single-Step plasma sample extraction optimization strategies with experimental design for LC-MS and GC–MS integrated metabolomics and lipidomics analysis. Microchem. J. 2022, 179, 107525. [Google Scholar] [CrossRef]
- Griffiths, W.J. Metabolomics, Metabonomics and Metabolite Profiling; Royal Society of Chemistry: London, UK, 2007. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Sudalaimuthuasari, N.; Hazzouri, K.M.; Saeed, E.E.; Shah, I.; Amiri, K.M.A. Tapping into Plant–Microbiome Interactions through the Lens of Multi-Omics Techniques. Cells 2022, 11, 3254. https://doi.org/10.3390/cells11203254
Mishra AK, Sudalaimuthuasari N, Hazzouri KM, Saeed EE, Shah I, Amiri KMA. Tapping into Plant–Microbiome Interactions through the Lens of Multi-Omics Techniques. Cells. 2022; 11(20):3254. https://doi.org/10.3390/cells11203254
Chicago/Turabian StyleMishra, Ajay Kumar, Naganeeswaran Sudalaimuthuasari, Khaled M. Hazzouri, Esam Eldin Saeed, Iltaf Shah, and Khaled M. A. Amiri. 2022. "Tapping into Plant–Microbiome Interactions through the Lens of Multi-Omics Techniques" Cells 11, no. 20: 3254. https://doi.org/10.3390/cells11203254
APA StyleMishra, A. K., Sudalaimuthuasari, N., Hazzouri, K. M., Saeed, E. E., Shah, I., & Amiri, K. M. A. (2022). Tapping into Plant–Microbiome Interactions through the Lens of Multi-Omics Techniques. Cells, 11(20), 3254. https://doi.org/10.3390/cells11203254