
Mechanical Compression of Human Airway Epithelial Cells Induces Release of Extracellular Vesicles Containing Tenascin C

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of Primary Human Bronchial Epithelial HBE Cells
2.2. Application of Mechanical Compression to HBE Cells
2.3. Inhibition of the ERK or TGF-β Receptor Pathway
2.4. RNA-Sequencing
2.5. RT-qPCR
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Western Blot Analysis
2.8. Isolation and Validation of Extracellular Vesicles
2.9. Statistical Analysis
3. Results
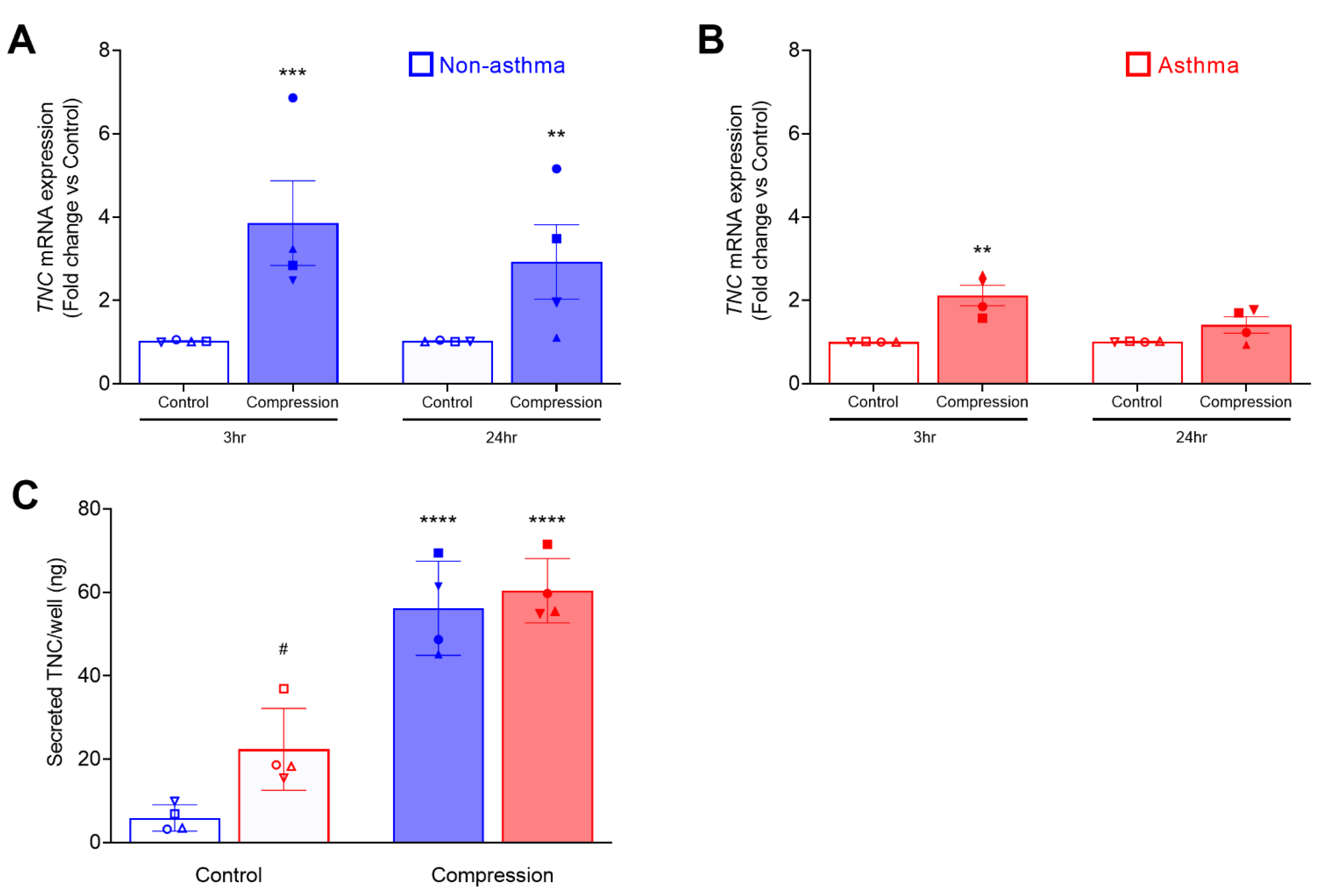
3.1. Mechanical Compression Induces TNC mRNA Expression and Secretion in HBE Cells
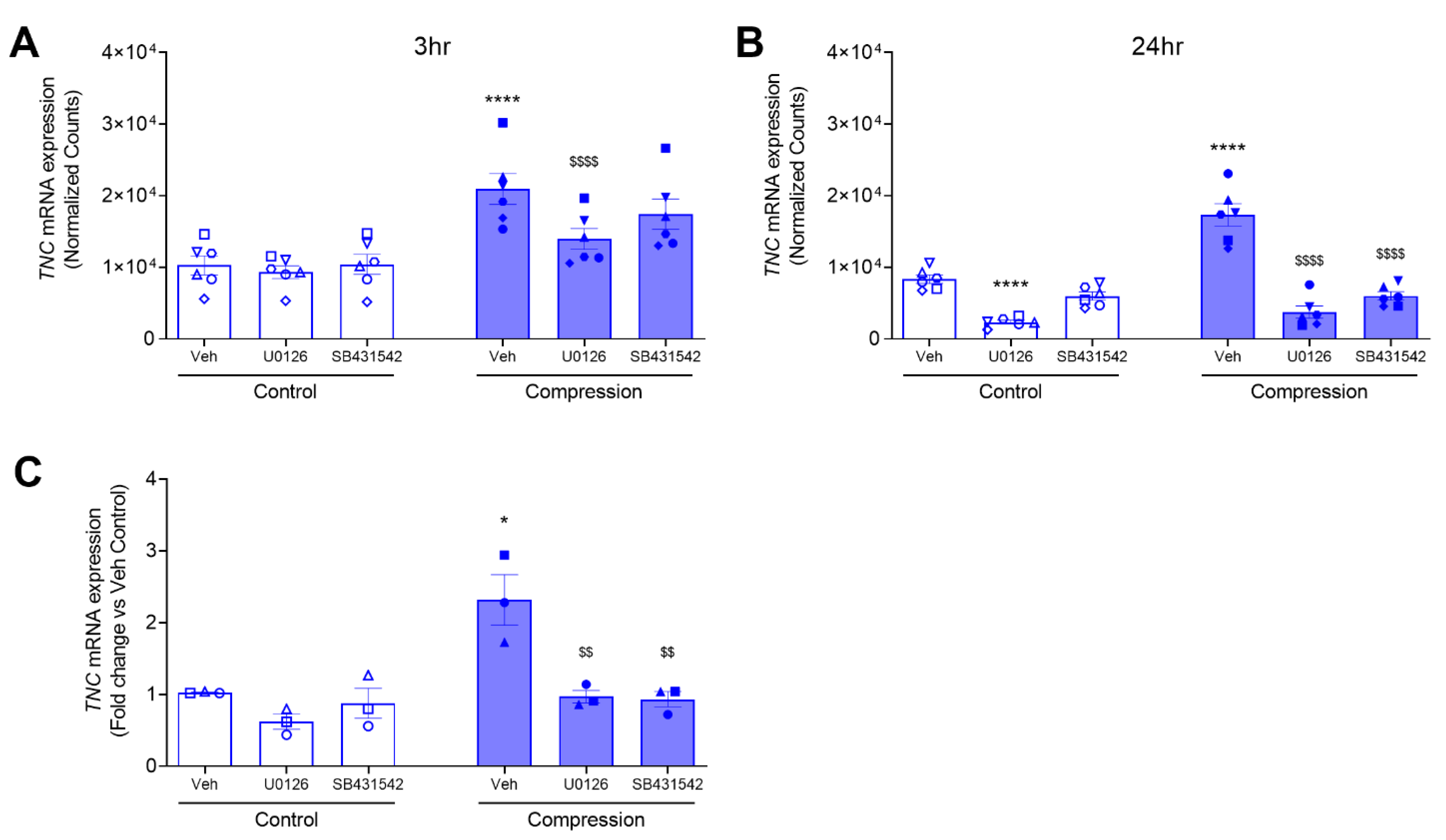
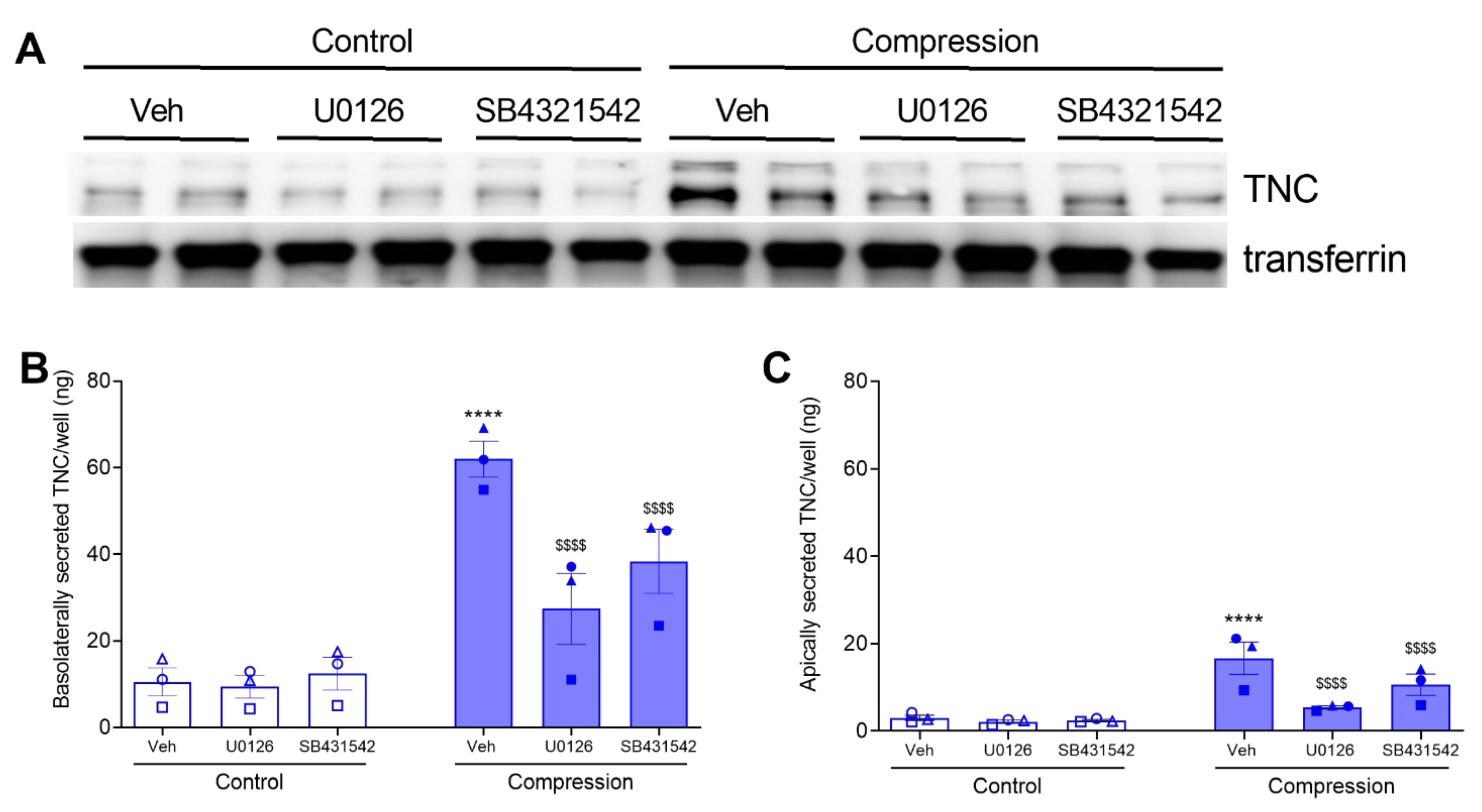
3.2. ERK and TGF-β Receptor Pathways Regulate the Compression-Induced TNC mRNA Expression and Protein Secretion
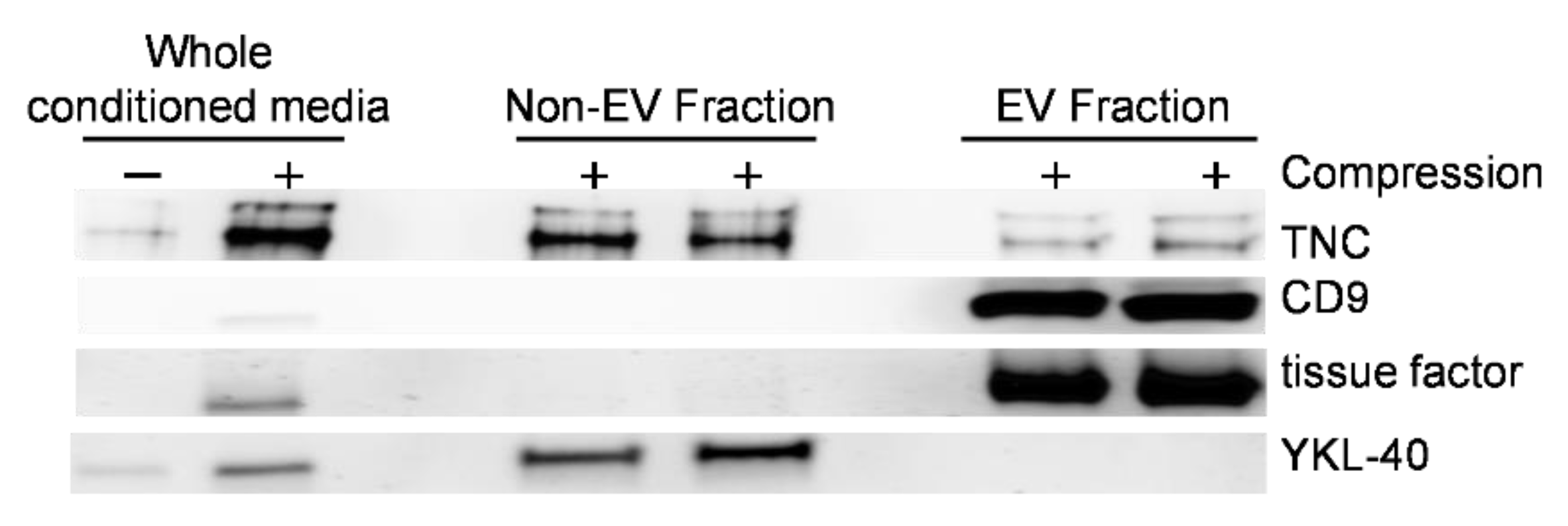
3.3. Mechanical Compression Induces the Release of TNC Protein Contained in EVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fehrenbach, H.; Wagner, C.; Wegmann, M. Airway remodeling in asthma: What really matters. Cell Tissue Res. 2017, 367, 551–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaar, A.L.; Panettieri, R.A., Jr. Is airway remodeling clinically relevant in asthma? Am. J. Med. 2003, 115, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.E. The role of the epithelium in airway remodeling in asthma. Proc. Am. Thorac. Soc. 2009, 6, 678–682. [Google Scholar] [CrossRef] [Green Version]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, B.R.; Hrousis, C.A.; Drazen, J.M.; Kamm, R.D. On the mechanism of mucosal folding in normal and asthmatic airways. J. Appl. Physiol. 1997, 83, 1814–1821. [Google Scholar] [CrossRef] [Green Version]
- Park, J.A.; Fredberg, J.J.; Drazen, J.M. Putting the Squeeze on Airway Epithelia. Physiology 2015, 30, 293–303. [Google Scholar] [CrossRef]
- O’Sullivan, M.J.; Phung, T.N.; Park, J.A. Bronchoconstriction: A potential missing link in airway remodelling. Open Biol. 2020, 10, 200254. [Google Scholar] [CrossRef]
- Kilic, A.; Ameli, A.; Park, J.A.; Kho, A.T.; Tantisira, K.; Santolini, M.; Cheng, F.; Mitchel, J.A.; McGill, M.; O’Sullivan, M.J.; et al. Mechanical forces induce an asthma gene signature in healthy airway epithelial cells. Sci. Rep. 2020, 10, 966. [Google Scholar] [CrossRef]
- Park, J.A.; Tschumperlin, D.J. Chronic intermittent mechanical stress increases MUC5AC protein expression. Am. J. Respir. Cell Mol. Biol. 2009, 41, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Swartz, M.A.; Tschumperlin, D.J.; Kamm, R.D.; Drazen, J.M. Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proc. Natl. Acad. Sci. USA 2001, 98, 6180–6185. [Google Scholar] [CrossRef] [Green Version]
- Lan, B.; Mitchel, J.A.; O’Sullivan, M.J.; Park, C.Y.; Kim, J.H.; Cole, W.C.; Butler, J.P.; Park, J.A. Airway epithelial compression promotes airway smooth muscle proliferation and contraction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L645–L652. [Google Scholar] [CrossRef]
- Kim, S.H.; Mitchel, J.A.; McGill, M.; Cremona, T.P.; Baek, J.W.; Kasahara, D.I.; Anathy, V.; Israel, E.; Park, J.A. Increased extracellular maspin levels after mechanical compression in vitro or allergen challenge in vivo. J. Allergy Clin. Immunol. 2019, 144, 1116–1118.e1114. [Google Scholar] [CrossRef] [Green Version]
- Park, J.A.; Sharif, A.S.; Tschumperlin, D.J.; Lau, L.; Limbrey, R.; Howarth, P.; Drazen, J.M. Tissue factor-bearing exosome secretion from human mechanically stimulated bronchial epithelial cells in vitro and in vivo. J. Allergy Clin. Immunol. 2012, 130, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschumperlin, D.J.; Shively, J.D.; Kikuchi, T.; Drazen, J.M. Mechanical stress triggers selective release of fibrotic mediators from bronchial epithelium. Am. J. Respir. Cell Mol. Biol. 2003, 28, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschumperlin, D.J.; Shively, J.D.; Swartz, M.A.; Silverman, E.S.; Haley, K.J.; Raab, G.; Drazen, J.M. Bronchial epithelial compression regulates MAP kinase signaling and HB-EGF-like growth factor expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L904–L911. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Drazen, J.M.; Tschumperlin, D.J. The chitinase-like protein YKL-40 is secreted by airway epithelial cells at base line and in response to compressive mechanical stress. J. Biol. Chem. 2010, 285, 29817–29825. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Hsia, H.C.; Schwarzbauer, J.E. Meet the tenascins: Multifunctional and mysterious. J. Biol. Chem. 2005, 280, 26641–26644. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.L.; Jones, F.S. Tenascin-C in development and disease: Gene regulation and cell function. Matrix Biol. 2000, 19, 581–596. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Mackie, E.J.; Pearson, C.A.; Sakakura, T. Tenascin: An extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell 1986, 47, 131–139. [Google Scholar] [CrossRef]
- Roth-Kleiner, M.; Hirsch, E.; Schittny, J.C. Fetal lungs of tenascin-C-deficient mice grow well, but branch poorly in organ culture. Am. J. Respir. Cell Mol. Biol. 2004, 30, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Mund, S.I.; Schittny, J.C. Tenascin-C deficiency impairs alveolarization and microvascular maturation during postnatal lung development. J. Appl. Physiol. 2020, 128, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Gremlich, S.; Roth-Kleiner, M.; Equey, L.; Fytianos, K.; Schittny, J.C.; Cremona, T.P. Tenascin-C inactivation impacts lung structure and function beyond lung development. Sci. Rep. 2020, 10, 5118. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estany, S.; Vicens-Zygmunt, V.; Llatjos, R.; Montes, A.; Penin, R.; Escobar, I.; Xaubet, A.; Santos, S.; Manresa, F.; Dorca, J.; et al. Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFbeta1. BMC Pulm. Med. 2014, 14, 120. [Google Scholar] [CrossRef] [Green Version]
- Kaarteenaho-Wiik, R.; Kinnula, V.L.; Herva, R.; Soini, Y.; Pollanen, R.; Paakko, P. Tenascin-C is highly expressed in respiratory distress syndrome and bronchopulmonary dysplasia. J. Histochem. Cytochem. 2002, 50, 423–431. [Google Scholar] [CrossRef]
- Lofdahl, M.; Kaarteenaho, R.; Lappi-Blanco, E.; Tornling, G.; Skold, M.C. Tenascin-C and alpha-smooth muscle actin positive cells are increased in the large airways in patients with COPD. Respir. Res. 2011, 12, 48. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, A.; Altraja, A.; Kampe, M.; Linden, M.; Virtanen, I.; Laitinen, L.A. Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am. J. Respir. Crit. Care Med. 1997, 156, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Karjalainen, E.M.; Lindqvist, A.; Laitinen, L.A.; Kava, T.; Altraja, A.; Halme, M.; Laitinen, A. Airway inflammation and basement membrane tenascin in newly diagnosed atopic and nonatopic asthma. Respir. Med. 2003, 97, 1045–1051. [Google Scholar] [CrossRef] [Green Version]
- Kariyawasam, H.H.; Aizen, M.; Barkans, J.; Robinson, D.S.; Kay, A.B. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, L.A.; Laitinen, A.; Altraja, A.; Virtanen, I.; Kampe, M.; Simonsson, B.G.; Karlsson, S.E.; Hakansson, L.; Venge, P.; Sillastu, H. Bronchial biopsy findings in intermittent or “early” asthma. J. Allergy Clin. Immunol. 1996, 98, S3–S6, discussion S33–S40. [Google Scholar] [CrossRef]
- Yasuda, M.; Harada, N.; Harada, S.; Ishimori, A.; Katsura, Y.; Itoigawa, Y.; Matsuno, K.; Makino, F.; Ito, J.; Ono, J.; et al. Characterization of tenascin-C as a novel biomarker for asthma: Utility of tenascin-C in combination with periostin or immunoglobulin E. Allergy Asthma Clin. Immunol. 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Good, J.; Rollins, D.; Verma, M.; Chu, H.; Pham, T.H.; Martin, R.J. Airway and serum biochemical correlates of refractory neutrophilic asthma. J. Allergy Clin. Immunol. 2017, 140, 1004–1014.e1013. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, H.; Gabazza, E.C.; Fujimoto, H.; Nishii, Y.; D’Alessandro-Gabazza, C.N.; Bruno, N.E.; Takagi, T.; Hayashi, T.; Maruyama, J.; Maruyama, K.; et al. Deficiency of tenascin C attenuates allergen-induced bronchial asthma in the mouse. Eur. J. Immunol. 2006, 36, 3334–3345. [Google Scholar] [CrossRef] [PubMed]
- Fluck, M.; Mund, S.I.; Schittny, J.C.; Klossner, S.; Durieux, A.C.; Giraud, M.N. Mechano-regulated tenascin-C orchestrates muscle repair. Proc. Natl. Acad. Sci. USA 2008, 105, 13662–13667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanaka-Yoshida, K.; Aoki, H. Tenascin-C and mechanotransduction in the development and diseases of cardiovascular system. Front. Physiol. 2014, 5, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiquet, M.; Sarasa-Renedo, A.; Tunc-Civelek, V. Induction of tenascin-C by cyclic tensile strain versus growth factors: Distinct contributions by Rho/ROCK and MAPK signaling pathways. Biochim. Biophys. Acta 2004, 1693, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Yuyama, N.; Davies, D.E.; Akaiwa, M.; Matsui, K.; Hamasaki, Y.; Suminami, Y.; Yoshida, N.L.; Maeda, M.; Pandit, A.; Lordan, J.L.; et al. Analysis of novel disease-related genes in bronchial asthma. Cytokine 2002, 19, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kaminski, N.; Dolganov, G.; Grunig, G.; Koth, L.; Solomon, C.; Erle, D.J.; Sheppard, D. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am. J. Respir. Cell Mol. Biol. 2001, 25, 474–485. [Google Scholar] [CrossRef]
- Mills, J.T.; Schwenzer, A.; Marsh, E.K.; Edwards, M.R.; Sabroe, I.; Midwood, K.S.; Parker, L.C. Airway Epithelial Cells Generate Pro-inflammatory Tenascin-C and Small Extracellular Vesicles in Response to TLR3 Stimuli and Rhinovirus Infection. Front. Immunol. 2019, 10, 1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marzio, M.; Kilic, A.; Maiorino, E.; Mitchel, J.A.; Mwase, C.; O’Sullivan, M.J.; McGill, M.; Chase, R.; Fredberg, J.J.; Park, J.A.; et al. Genomic signatures of the unjamming transition in compressed human bronchial epithelial cells. Sci. Adv. 2021, 7, eabf1088. [Google Scholar] [CrossRef]
- Albacete-Albacete, L.; Sanchez-Alvarez, M.; Del Pozo, M.A. Extracellular Vesicles: An Emerging Mechanism Governing the Secretion and Biological Roles of Tenascin-C. Front. Immunol. 2021, 12, 671485. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.J.; Kim, O.Y.; Gho, Y.S. Extracellular vesicles as emerging intercellular communicasomes. BMB Rep. 2014, 47, 531–539. [Google Scholar] [CrossRef]
- Mitchel, J.A.; Das, A.; O’Sullivan, M.J.; Stancil, I.T.; DeCamp, S.J.; Koehler, S.; Ocana, O.H.; Butler, J.P.; Fredberg, J.J.; Nieto, M.A.; et al. In primary airway epithelial cells, the unjamming transition is distinct from the epithelial-to-mesenchymal transition. Nat. Commun. 2020, 11, 5053. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, M.J.; Mitchel, J.A.; Mwase, C.; McGill, M.; Kanki, P.; Park, J.A. In well-differentiated primary human bronchial epithelial cells, TGF-beta1 and TGF-beta2 induce expression of furin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L246–L253. [Google Scholar] [CrossRef]
- Park, J.A.; Kim, J.H.; Bi, D.; Mitchel, J.A.; Qazvini, N.T.; Tantisira, K.; Park, C.Y.; McGill, M.; Kim, S.H.; Gweon, B.; et al. Unjamming and cell shape in the asthmatic airway epithelium. Nat. Mater. 2015, 14, 1040–1048. [Google Scholar] [CrossRef]
- Mitchel, J.A.; Antoniak, S.; Lee, J.H.; Kim, S.H.; McGill, M.; Kasahara, D.I.; Randell, S.H.; Israel, E.; Shore, S.A.; Mackman, N.; et al. IL-13 Augments Compressive Stress-Induced Tissue Factor Expression in Human Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2016, 54, 524–531. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.J.; Dailey, L.A.; Brighton, L.E.; Devlin, R.B. Transcriptional profiling of mucociliary differentiation in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2007, 37, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005, 107, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Gajos, K.; Kaminska, A.; Awsiuk, K.; Bajor, A.; Gruszczynski, K.; Pawlak, A.; Zadlo, A.; Kowalik, A.; Budkowski, A.; Stepien, E. Immobilization and detection of platelet-derived extracellular vesicles on functionalized silicon substrate: Cytometric and spectrometric approach. Anal. Bioanal. Chem. 2017, 409, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Dai, G.; Maly, I.V.; Kikuchi, T.; Laiho, L.H.; McVittie, A.K.; Haley, K.J.; Lilly, C.M.; So, P.T.; Lauffenburger, D.A.; et al. Mechanotransduction through growth-factor shedding into the extracellular space. Nature 2004, 429, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Zerah, F.; Harf, A.; Perlemuter, L.; Lorino, H.; Lorino, A.M.; Atlan, G. Effects of obesity on respiratory resistance. Chest 1993, 103, 1470–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutting, S.; Mahadev, S.; Tonga, K.O.; Bailey, D.L.; Dame Carroll, J.R.; Farrow, C.E.; Thamrin, C.; Chapman, D.G.; King, G.G. Obesity alters the topographical distribution of ventilation and the regional response to bronchoconstriction. J. Appl. Physiol. 2020, 128, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Badyda, A.J.; Dabrowiecki, P.; Czechowski, P.O.; Majewski, G. Risk of bronchi obstruction among non-smokers—Review of environmental factors affecting bronchoconstriction. Respir. Physiol. Neurobiol. 2015, 209, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Tucker, R.P.; Chiquet-Ehrismann, R. The regulation of tenascin expression by tissue microenvironments. Biochim. Biophys. Acta 2009, 1793, 888–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Jinnin, M.; Ihn, H.; Asano, Y.; Yamane, K.; Trojanowska, M.; Tamaki, K. Tenascin-C upregulation by transforming growth factor-beta in human dermal fibroblasts involves Smad3, Sp1, and Ets1. Oncogene 2004, 23, 1656–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainge, C.L.; Lau, L.C.; Ward, J.A.; Dulay, V.; Lahiff, G.; Wilson, S.; Holgate, S.; Davies, D.E.; Howarth, P.H. Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 2011, 364, 2006–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Radicioni, G.; Abdelwahab, S.; Dang, H.; Carpenter, J.; Chua, M.; Mieczkowski, P.A.; Sheridan, J.T.; Randell, S.H.; Kesimer, M. Intercellular Communication between Airway Epithelial Cells Is Mediated by Exosome-Like Vesicles. Am. J. Respir. Cell Mol. Biol. 2019, 60, 209–220. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Ji, H.; Greening, D.W.; Barnes, T.W.; Lim, J.W.; Tauro, B.J.; Rai, A.; Xu, R.; Adda, C.; Mathivanan, S.; Zhao, W.; et al. Proteome profiling of exosomes derived from human primary and metastatic colorectal cancer cells reveal differential expression of key metastatic factors and signal transduction components. Proteomics 2013, 13, 1672–1686. [Google Scholar] [CrossRef] [PubMed]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adh. Migr. 2015, 9, 48–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Hernandez, J.M.; Doussot, A.; Bojmar, L.; Zambirinis, C.P.; Costa-Silva, B.; van Beek, E.; Mark, M.T.; Molina, H.; Askan, G.; et al. Extracellular matrix proteins and carcinoembryonic antigen-related cell adhesion molecules characterize pancreatic duct fluid exosomes in patients with pancreatic cancer. HPB 2018, 20, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Sur, S.; Khatun, M.; Steele, R.; Isbell, T.S.; Ray, R.; Ray, R.B. Exosomes from COVID-19 Patients Carry Tenascin-C and Fibrinogen-beta in Triggering Inflammatory Signals in Cells of Distant Organ. Int. J. Mol. Sci. 2021, 22, 3184. [Google Scholar] [CrossRef] [PubMed]
- Albacete-Albacete, L.; Navarro-Lerida, I.; Lopez, J.A.; Martin-Padura, I.; Astudillo, A.M.; Ferrarini, A.; Van-Der-Heyden, M.; Balsinde, J.; Orend, G.; Vazquez, J.; et al. ECM deposition is driven by caveolin-1-dependent regulation of exosomal biogenesis and cargo sorting. J. Cell Biol. 2020, 219, e202006178. [Google Scholar] [CrossRef]
- Campos, A.; Salomon, C.; Bustos, R.; Diaz, J.; Martinez, S.; Silva, V.; Reyes, C.; Diaz-Valdivia, N.; Varas-Godoy, M.; Lobos-Gonzalez, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine 2018, 13, 2597–2609. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences |
|---|---|
| GAPDH | FW: 5′-TGGGCTACACTGAGCACCAG-3′ RV: 5′-GGGTGTCGCTGTTGAAGTCA-3′ |
| TNC | FW: 5′-TCCCAGTGTTCGGTGGATCT-3′ RV: 5′-TTGATGCGATGTGTGAAGACA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwase, C.; Phung, T.-K.N.; O’Sullivan, M.J.; Mitchel, J.A.; De Marzio, M.; Kılıç, A.; Weiss, S.T.; Fredberg, J.J.; Park, J.-A. Mechanical Compression of Human Airway Epithelial Cells Induces Release of Extracellular Vesicles Containing Tenascin C. Cells 2022, 11, 256. https://doi.org/10.3390/cells11020256
Mwase C, Phung T-KN, O’Sullivan MJ, Mitchel JA, De Marzio M, Kılıç A, Weiss ST, Fredberg JJ, Park J-A. Mechanical Compression of Human Airway Epithelial Cells Induces Release of Extracellular Vesicles Containing Tenascin C. Cells. 2022; 11(2):256. https://doi.org/10.3390/cells11020256
Chicago/Turabian StyleMwase, Chimwemwe, Thien-Khoi N. Phung, Michael J. O’Sullivan, Jennifer A. Mitchel, Margherita De Marzio, Ayşe Kılıç, Scott T. Weiss, Jeffrey J. Fredberg, and Jin-Ah Park. 2022. "Mechanical Compression of Human Airway Epithelial Cells Induces Release of Extracellular Vesicles Containing Tenascin C" Cells 11, no. 2: 256. https://doi.org/10.3390/cells11020256
APA StyleMwase, C., Phung, T.-K. N., O’Sullivan, M. J., Mitchel, J. A., De Marzio, M., Kılıç, A., Weiss, S. T., Fredberg, J. J., & Park, J.-A. (2022). Mechanical Compression of Human Airway Epithelial Cells Induces Release of Extracellular Vesicles Containing Tenascin C. Cells, 11(2), 256. https://doi.org/10.3390/cells11020256