
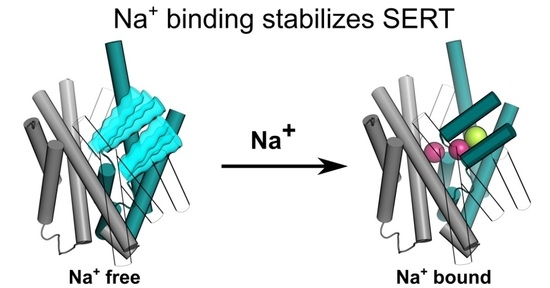
Sodium Binding Stabilizes the Outward-Open State of SERT by Limiting Bundle Domain Motions



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. System Creation and Molecular Dynamics Simulations
2.2. Data and Statistical Analysis
3. Results
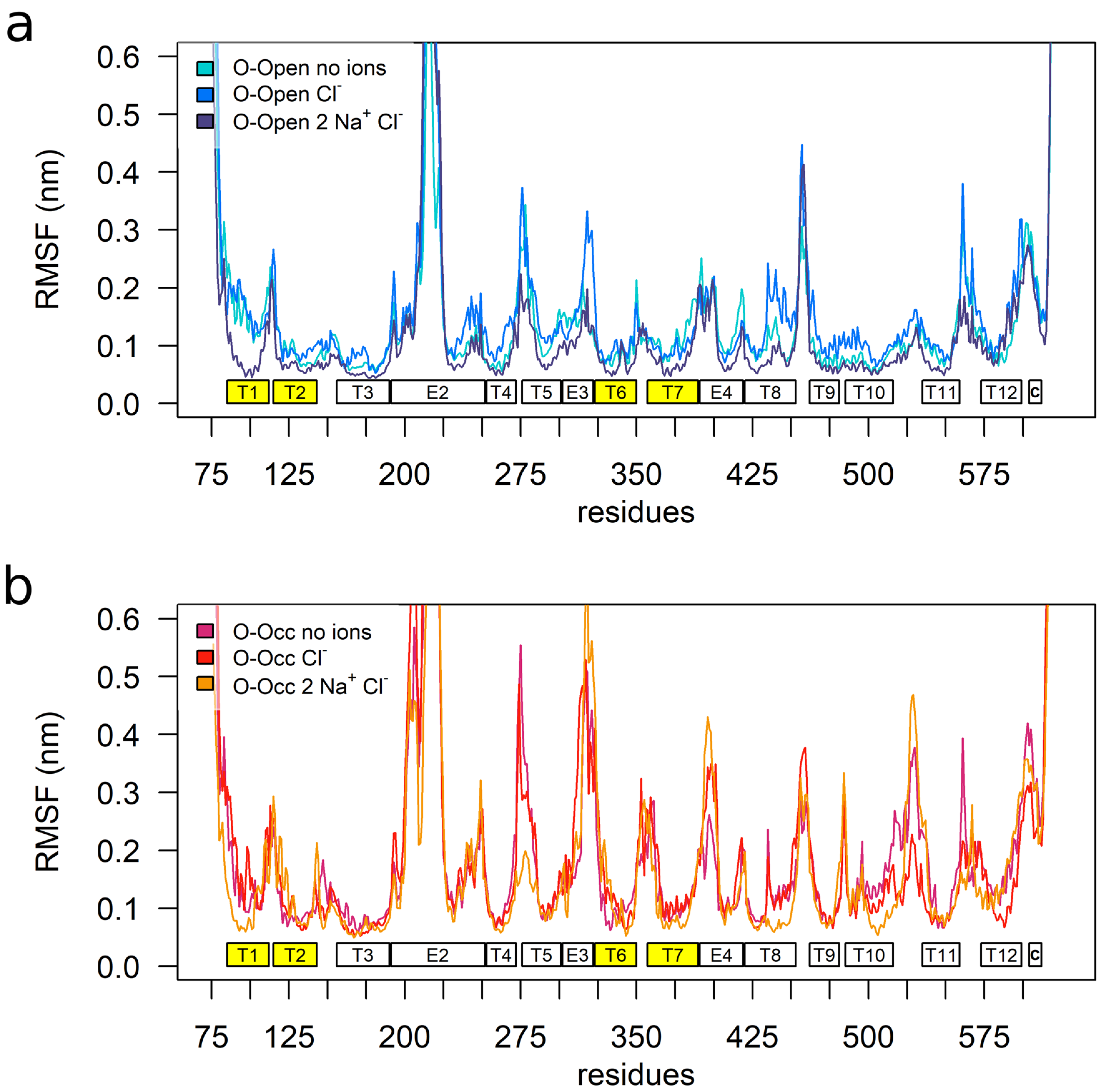
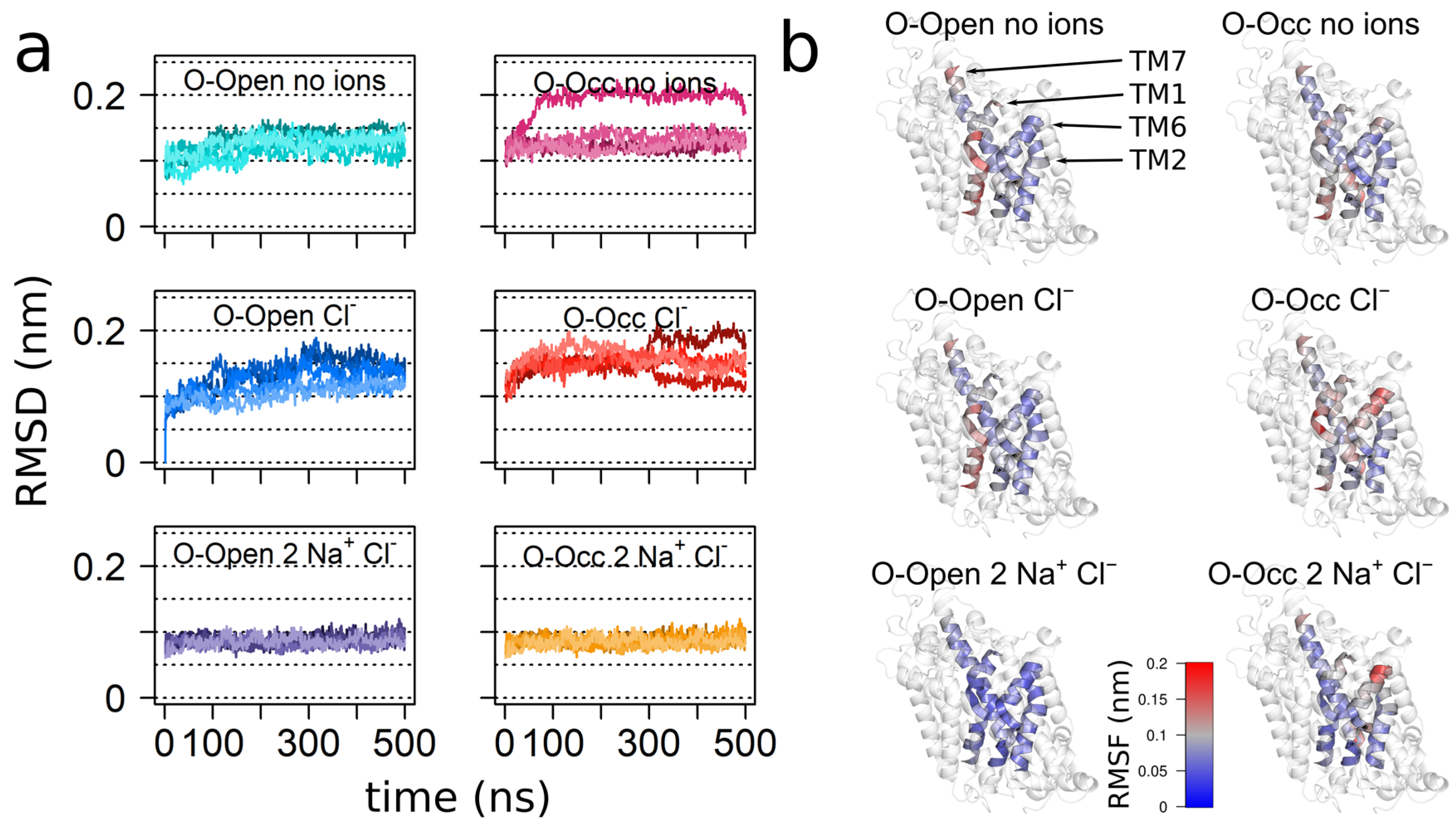
3.1. Overall Transporter Dynamics and Mobility of the Bundle Domain
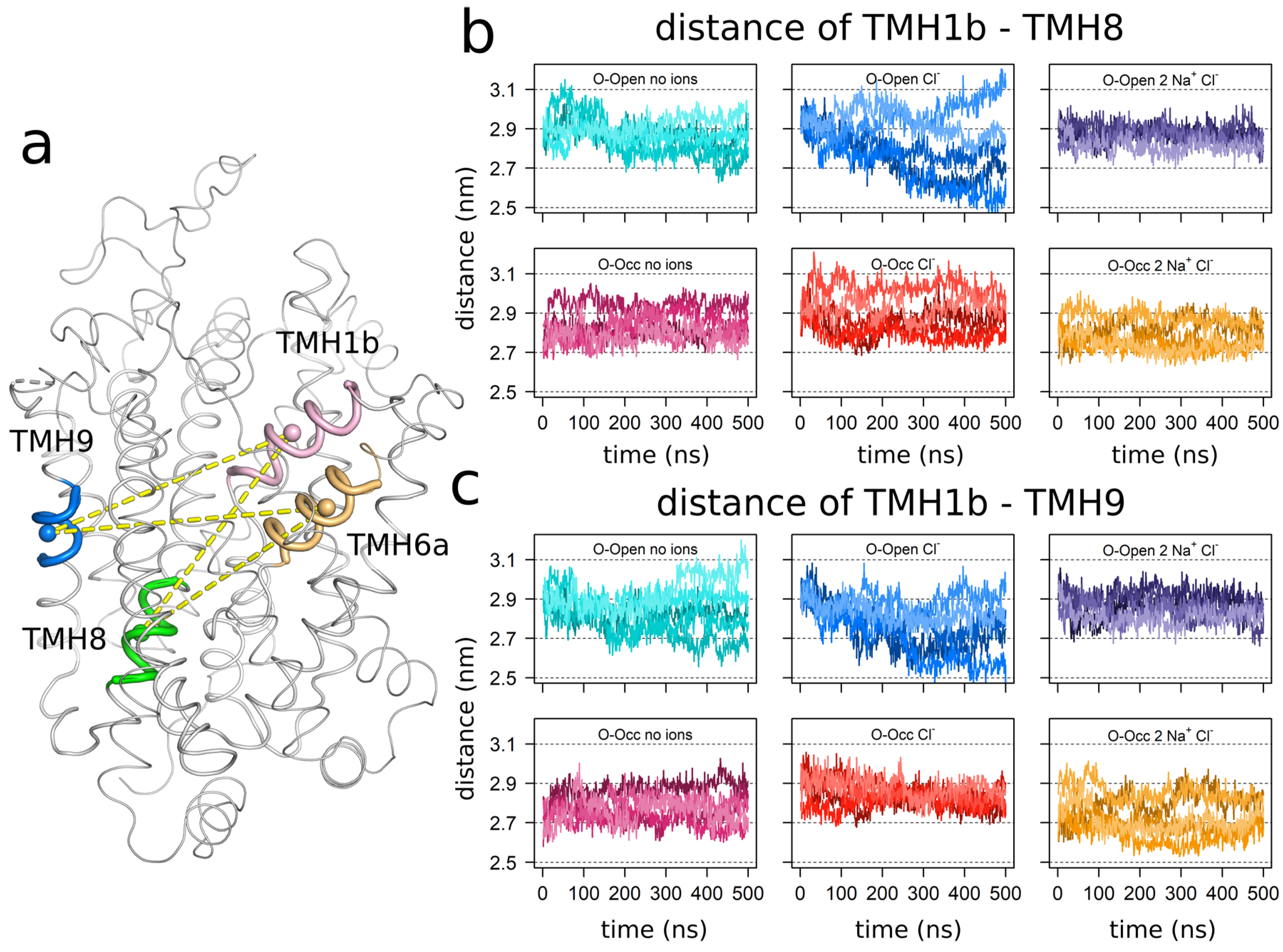
3.2. The Bound Sodium Ions Stabilize TMH1b
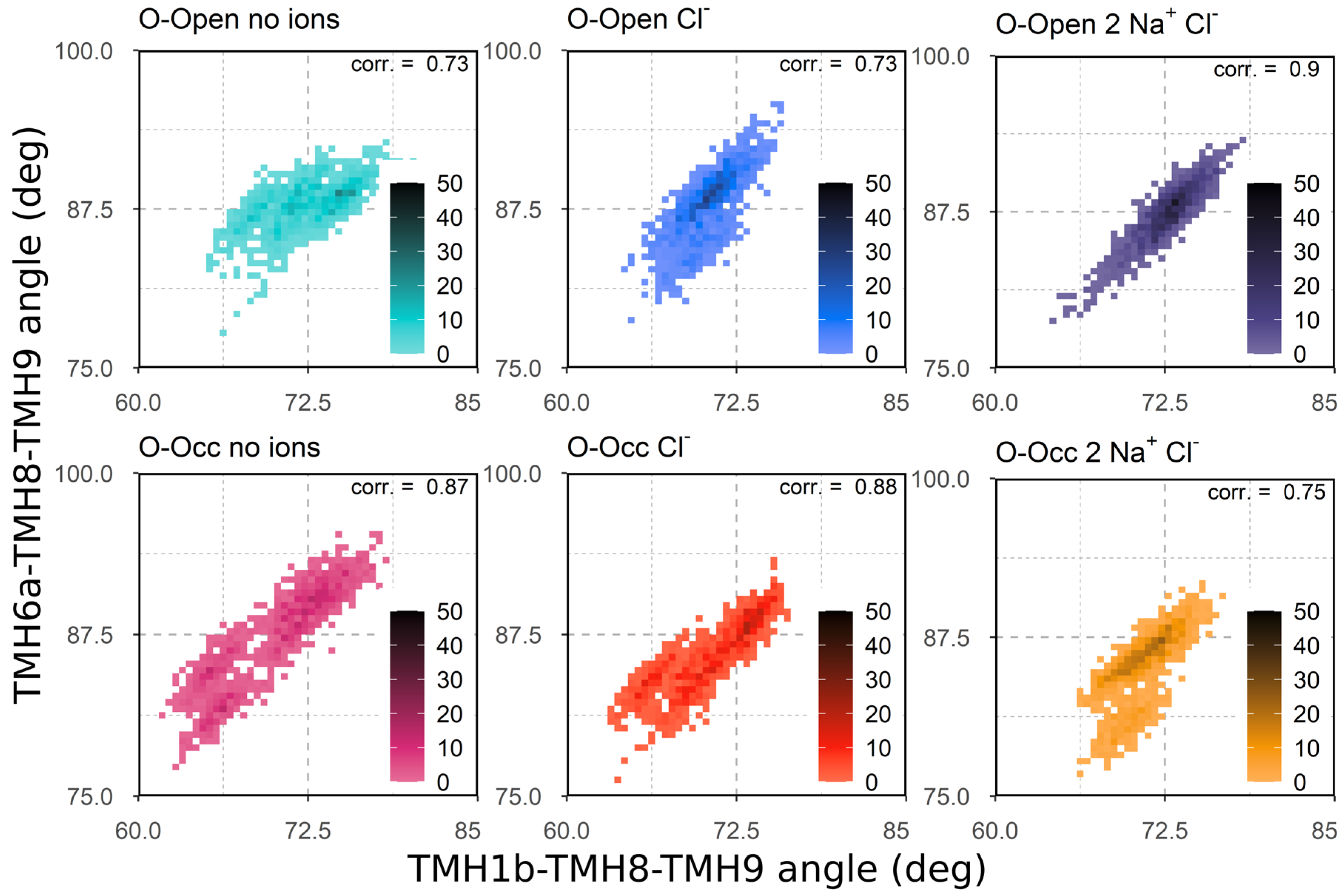
3.3. Correlation of TMH1b and TMH6a Movements
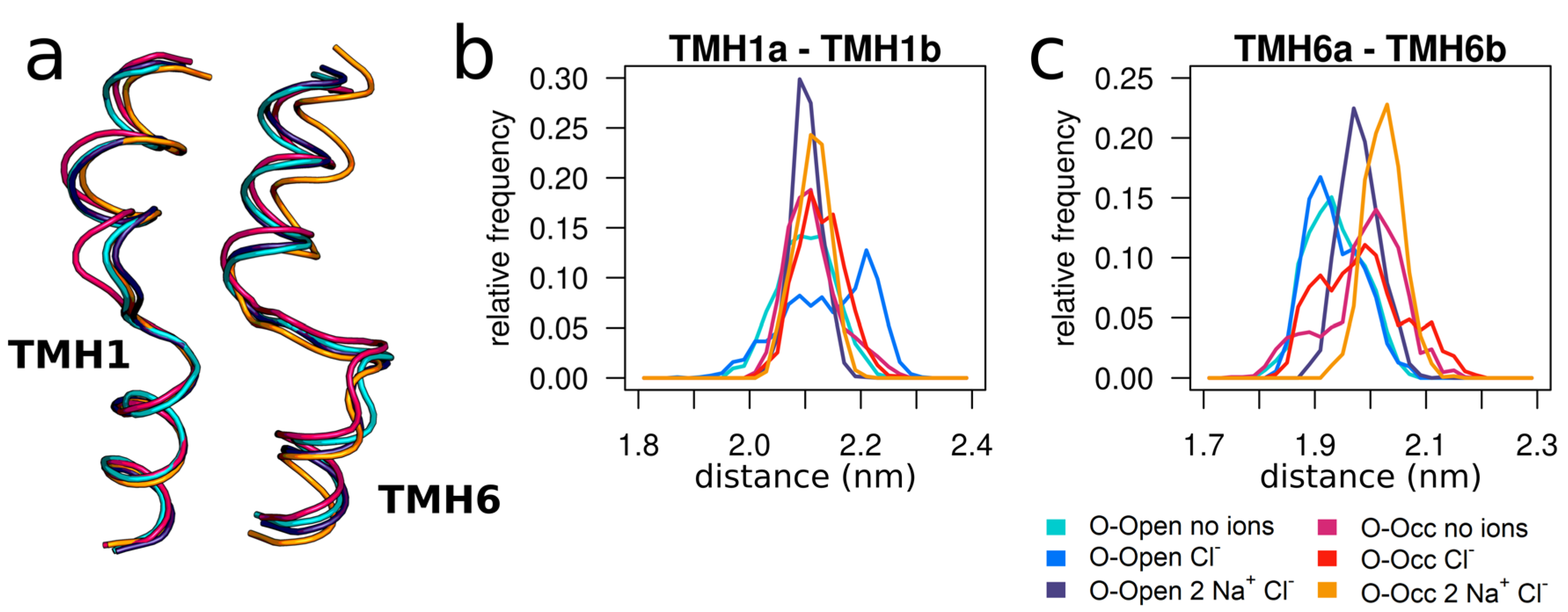
3.4. Sodium Binding Reduces the Internal Dynamics of TMH1 and TMH6
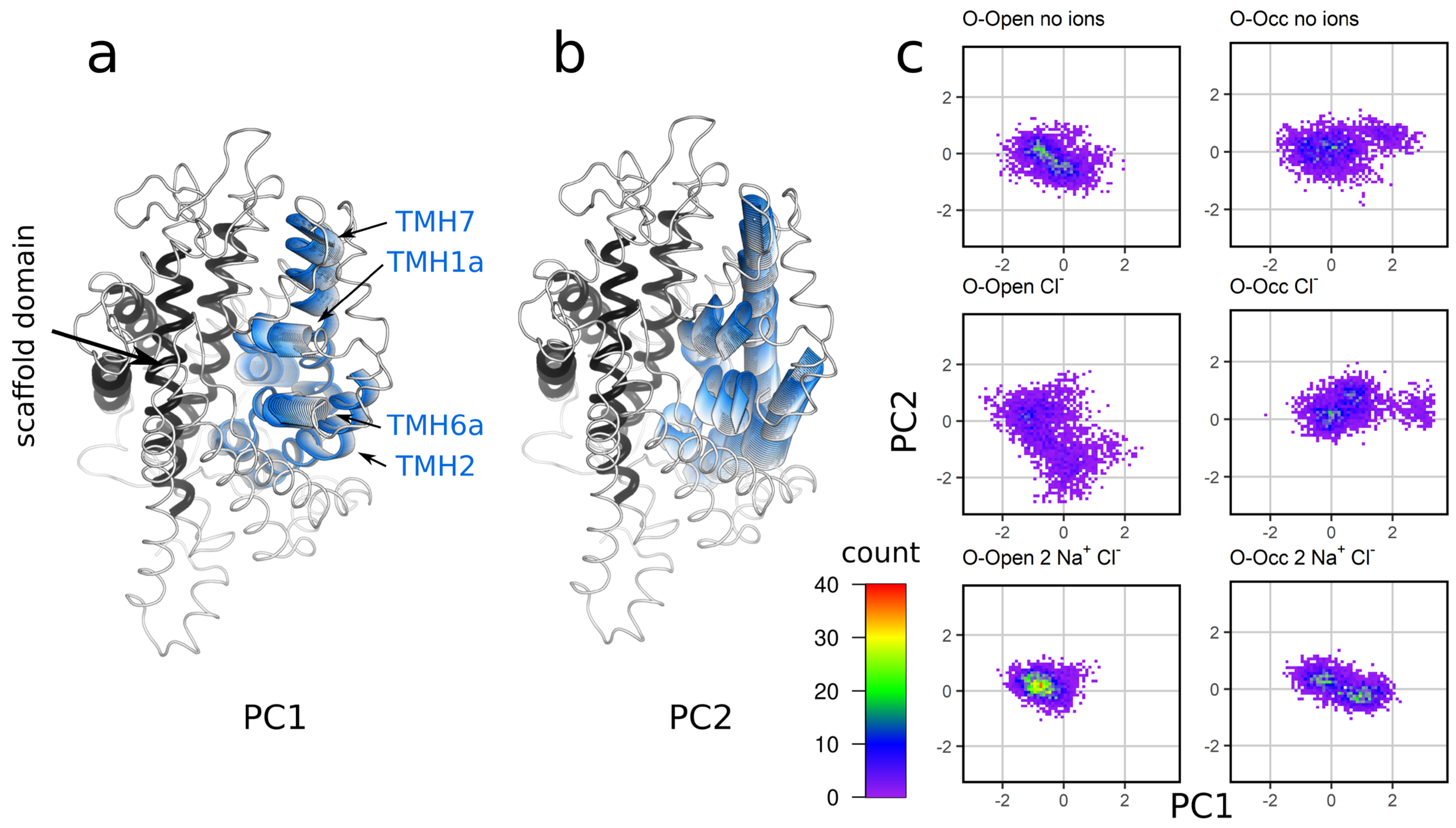
3.5. Sodium Binding Decreases the Intradomain Mobility of the Bundle Domain of the O-Open Conformation
3.6. Analysis of the Global Motions of the Bundle Domain Shows That Ion Binding Restrains the Overall Bundle Domain Motions
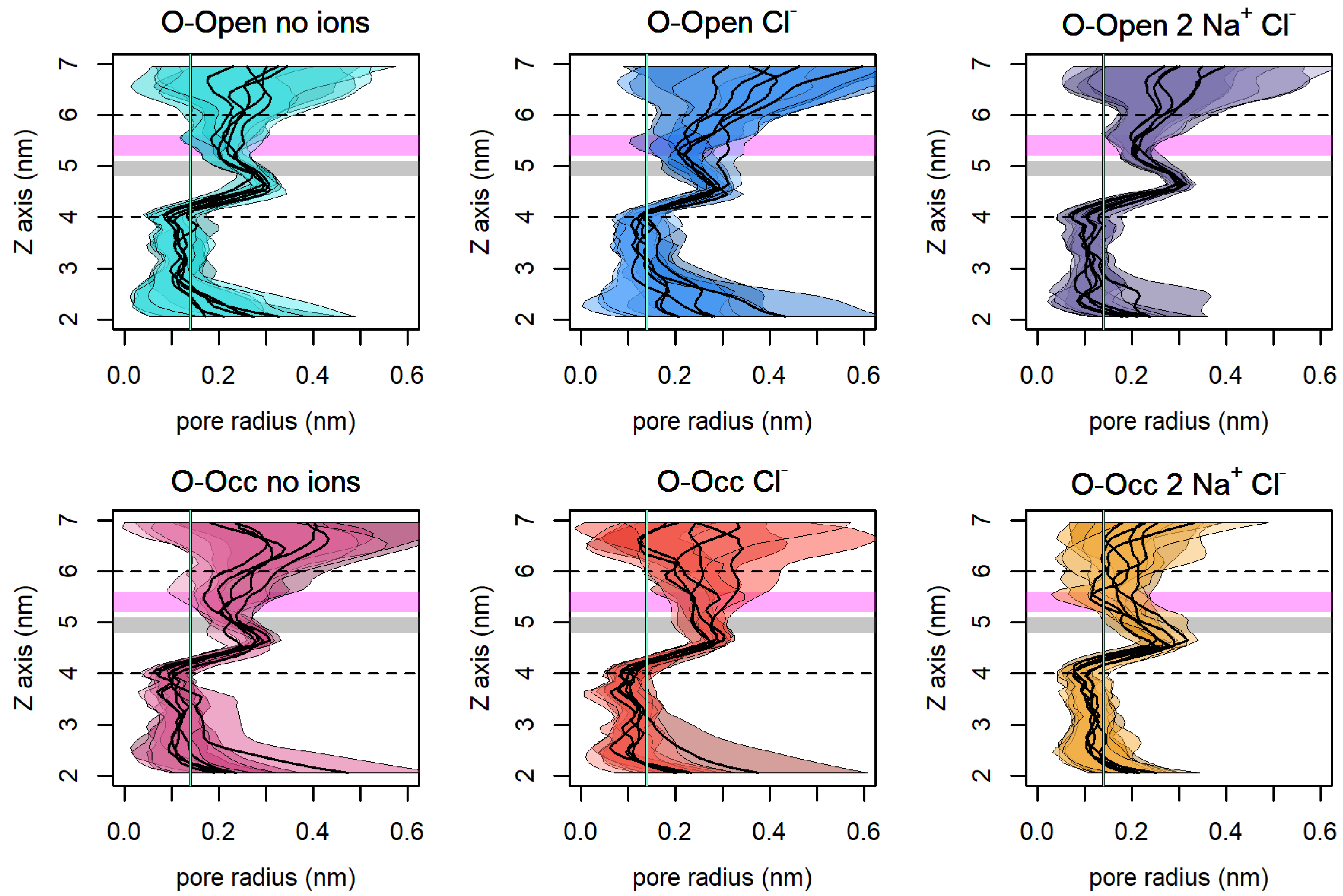
3.7. The Size of the Vestibule Indicates That Na+ Stabilizes an Open Access Path to the S1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freissmuth, M.; Stockner, T.; Sucic, S. SLC6 Transporter Folding Diseases and Pharmacochaperoning. In Targeting Trafficking in Drug Development. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 249–270. [Google Scholar]
- Sitte, H.H.; Freissmuth, M. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 2015, 36, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.A.; Yang, D.; Zhao, Z.; Wen, P.-C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin transporter–ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef]
- Hasenhuetl, P.S.; Freissmuth, M.; Sandtner, W. Electrogenic Binding of Intracellular Cations Defines a Kinetic Decision Point in the Transport Cycle of the Human Serotonin Transporter. J. Biol. Chem. 2016, 291, 25864–25876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.J.; Rudnick, G. Coupling between platelet 5-hydroxytryptamine and potassium transport. J. Biol. Chem. 1979, 254, 10084–10089. [Google Scholar] [CrossRef]
- Zhao, Y.; Terry, D.S.; Shi, L.; Quick, M.; Weinstein, H.; Blanchard, S.C.; Javitch, J.A. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature 2011, 474, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schicker, K.; Uzelac, Z.; Gesmonde, J.; Bulling, S.; Stockner, T.; Freissmuth, M.; Boehm, S.; Rudnick, G.; Sitte, H.H.; Sandtner, W. Unifying concept of serotonin transporter-associated currents. J. Biol. Chem. 2012, 287, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Talvenheimo, J.; Fishkes, H.; Nelson, P.J.; Rudnick, G. The serotonin transporter-imipramine “receptor”. J. Biol. Chem. 1983, 258, 6115–6119. [Google Scholar] [CrossRef]
- Lingjaerde, O. Uptake of serotonin in blood platelets: Dependence on sodium and chloride, and inhibition by choline. FEBS Lett. 1969, 3, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Androutsellis-Theotokis, A.; Rudnick, G. Accessibility and Conformational Coupling in Serotonin Transporter Predicted Internal Domains. J. Neurosci. 2002, 22, 8370–8378. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- de Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Ingólfsson, H.I.; Böckmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.A.; Pluhackova, K.; Böckmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.G.; Hoefling, M.; Aponte-Santamaría, C.; Grubmüller, H.; Groenhof, G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31, 2169–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jämbeck, J.P.M.M.; Lyubartsev, A.P. Another piece of the membrane puzzle: Extending slipids further. J. Chem. Theory Comput. 2013, 9, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Jämbeck, J.P.M.; Lyubartsev, A.P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Smart, O.S.; Neduvelil, J.G.; Wang, X.; Wallace, B.A.; Sansom, M.S.P. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger LLC: New York, NY, USA, 2002. [Google Scholar]
- Nielsen, A.K.; Möller, I.R.; Wang, Y.; Rasmussen, S.G.F.; Lindorff-Larsen, K.; Rand, K.D.; Loland, C.J. Substrate-induced conformational dynamics of the dopamine transporter. Nat. Commun. 2019, 10, 2714. [Google Scholar] [CrossRef]
- Hasenhuetl, P.S.; Bhat, S.; Mayer, F.P.; Sitte, H.H.; Freissmuth, M.; Sandtner, W. A kinetic account for amphetamine-induced monoamine release. J. Gen. Physiol. 2018, 150, 431–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grouleff, J.; Ladefoged, L.K.; Koldsø, H.; Schiøtt, B. Monoamine transporters: Insights from molecular dynamics simulations. Front. Pharmacol. 2015, 6, 235. [Google Scholar] [CrossRef] [Green Version]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat. Struct. Mol. Biol. 2015, 22, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Möller, I.R.; Slivacka, M.; Nielsen, A.K.; Rasmussen, S.G.F.; Gether, U.; Loland, C.J.; Rand, K.D. Conformational dynamics of the human serotonin transporter during substrate and drug binding. Nat. Commun. 2019, 10, 1687. [Google Scholar] [CrossRef]
- Mitchell, S.M.; Lee, E.; Garcia, M.L.; Stephan, M.M. Structure and Function of Extracellular Loop 4 of the Serotonin Transporter as Revealed by Cysteine-scanning Mutagenesis. J. Biol. Chem. 2004, 279, 24089–24099. [Google Scholar] [CrossRef] [Green Version]
- Jardetzky, O. Simple Allosteric Model for Membrane Pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Khafizov, K.; Staritzbichler, R.; Stamm, M.; Forrest, L.R. A Study of the Evolution of Inverted-Topology Repeats from LeuT-Fold Transporters Using AlignMe. Biochemistry 2010, 49, 10702–10713. [Google Scholar] [CrossRef] [PubMed]
- Claxton, D.P.; Quick, M.; Shi, L.; de Carvalho, F.D.; Weinstein, H.; Javitch, J.A.; Mchaourab, H.S. Ion/substrate-dependent conformational dynamics of a bacterial homolog of Neurotransmitter: Sodium symporters. Nat. Struct. Mol. Biol. 2010, 17, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.J.; Rudnick, G. The role of chloride ion in platelet serotonin transport. J. Biol. Chem. 1982, 257, 6151–6155. [Google Scholar] [CrossRef]
- Henry, L.K.; Iwamoto, H.; Field, J.R.; Kaufmann, K.; Dawson, E.S.; Jacobs, M.T.; Adams, C.; Felts, B.; Zdravkovic, I.; Armstrong, V.; et al. A Conserved Asparagine Residue in Transmembrane Segment 1 (TM1) of Serotonin Transporter Dictates Chloride-coupled Neurotransmitter Transport. J. Biol. Chem. 2011, 286, 30823–30836. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, Z.; Tajkhorshid, E. Locking Two Rigid-body Bundles in an Outward-Facing Conformation: The Ion-coupling Mechanism in a LeuT-fold Transporter. Sci. Rep. 2019, 9, 19479. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Stolzenberg, S.; Gracia, L.; Weinstein, H.; Noskov, S.; Shi, L. Ion-Controlled Conformational Dynamics in the Outward-Open Transition from an Occluded State of LeuT. Biophys. J. 2012, 103, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Stolzenberg, S.; Quick, M.; Zhao, C.; Gotfryd, K.; Khelashvili, G.; Gether, U.; Loland, C.J.; Javitch, J.A.; Noskov, S.; Weinstein, H.; et al. Mechanism of the Association between Na+ Binding and Conformations at the Intracellular Gate in Neurotransmitter:Sodium Symporters. J. Biol. Chem. 2015, 290, 13992–14003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomot, E.; Gur, M.; Bahar, I. Microseconds Simulations Reveal a New Sodium-binding Site and the Mechanism of Sodium-coupled Substrate Uptake by LeuT. J. Biol. Chem. 2015, 290, 544–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, C.J.; Wall, S.C.; Rudnick, G. ligand Binding to the Serotonin Transporter: Equilibria, Kinetics, and Ion Dependence. Biochemistry 1994, 33, 9118–9125. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szöllősi, D.; Stockner, T. Sodium Binding Stabilizes the Outward-Open State of SERT by Limiting Bundle Domain Motions. Cells 2022, 11, 255. https://doi.org/10.3390/cells11020255
Szöllősi D, Stockner T. Sodium Binding Stabilizes the Outward-Open State of SERT by Limiting Bundle Domain Motions. Cells. 2022; 11(2):255. https://doi.org/10.3390/cells11020255
Chicago/Turabian StyleSzöllősi, Dániel, and Thomas Stockner. 2022. "Sodium Binding Stabilizes the Outward-Open State of SERT by Limiting Bundle Domain Motions" Cells 11, no. 2: 255. https://doi.org/10.3390/cells11020255
APA StyleSzöllősi, D., & Stockner, T. (2022). Sodium Binding Stabilizes the Outward-Open State of SERT by Limiting Bundle Domain Motions. Cells, 11(2), 255. https://doi.org/10.3390/cells11020255