
Conserved Motifs and Domains in Members of Pospiviroidae


Abstract
1. Introduction
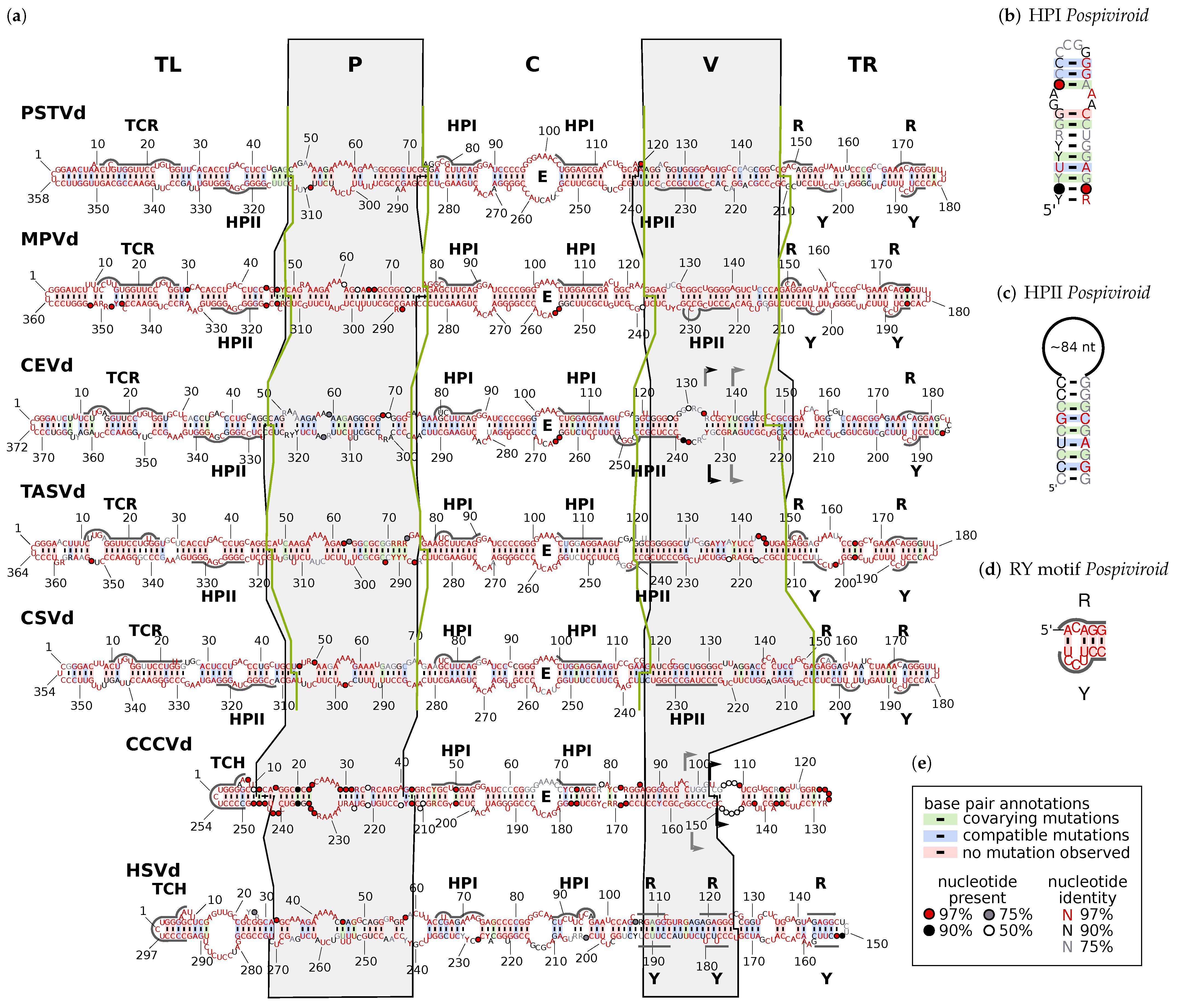
- The terminal left (TL) domain plays an important role in the replication of Pospiviroidae as it contains the starting point for pol II [14,15,16,17]. Furthermore, two subgroups of Pospiviroidae differ by presence and absence, respectively, of two sequence motifs in the TL domain [18,19,20]: members of genus Pospiviroid and some members of Coleviroid contain a terminal conserved region (TCR; see Supplemental Figure S20), while members of genera Cocad- and Hostuviroid contain a terminal conserved hairpin (TCH; Figure S21).
- The pathogenicity (P) domain is associated with symptom severity [21,22,23], but clearly not the only viroid part responsible for virulence (examples can be found in [22,24,25,26,27]). The P domain includes an oligopurine stretch in the upper part and a partly complementary oligopyrimidine stretch in the lower part of the secondary structure. Thus, the region shows alternative structures of low thermodynamic stability, giving rise to the name “premelting region” [28].
- The central (C) domain is the most conserved part between different viroids. It contains a loop E motif (Figure S22b), which shows similarities to loop E of eukaryotic 5Sr RNA, sarcin/ricin loop in 28S rRNA, and loop B of hairpin ribozymes [29,30]: it consists of five non-Watson–Crick basepairs and a bulged nucleotide involved in a triple pair; this unusual conformation allows for the formation of a UV-induced crosslink between two nucleotides of loop E [31,32,33]. The upper part of the C domain can be rearranged into a hairpin (HPI; Figure S22a), which is only present in thermodynamic metastable structures during replication or at biologically non-relevant temperatures [34]. Loop E and HPI are both involved in the processing of linear replication intermediates to mature circles [35,36,37,38,39].
- The variable (V) domain has the lowest sequence similarity even between closely related species [11] but contains one part of hairpin II (HPII; Figure S25), which is a metastable structural element critical for the transcription of -stranded replication intermediates in pospiviroids [40,41]. The 3’ part of HPII is located in the lower part of the TL domain.
- NucAln is no longer available, but its algorithm is still part of ClustalΩ [48] for the fast production of pairwise alignments. Thus, we modified ClustalΩ to output not only scores but also aligned positions from the fast alignment step (for details see Supplement and Figure S1). In the following, this modified ClustalΩ is called NucAln.
- vmatch [51] determines maximal substring matches between two sequences.

2. Materials and Methods
2.1. Consensus Sequence Compilation
2.2. NucAln
2.3. JAli
2.4. vmatch
- -
- d: report (only) direct matches;
- -
- l 50: report matches of length ;
- -
- e 20: edit distance—only report matches that contain at most 20 mismatches, insertions or deletions;
- -
- leastscore 50: only report matches if their score is ;
- -
- seedlength 4: length of exact seeds.
2.5. Pairwise Sequence Identity
3. Results and Discussion
3.1. Can We Retrace the Results of Keese and Symons?
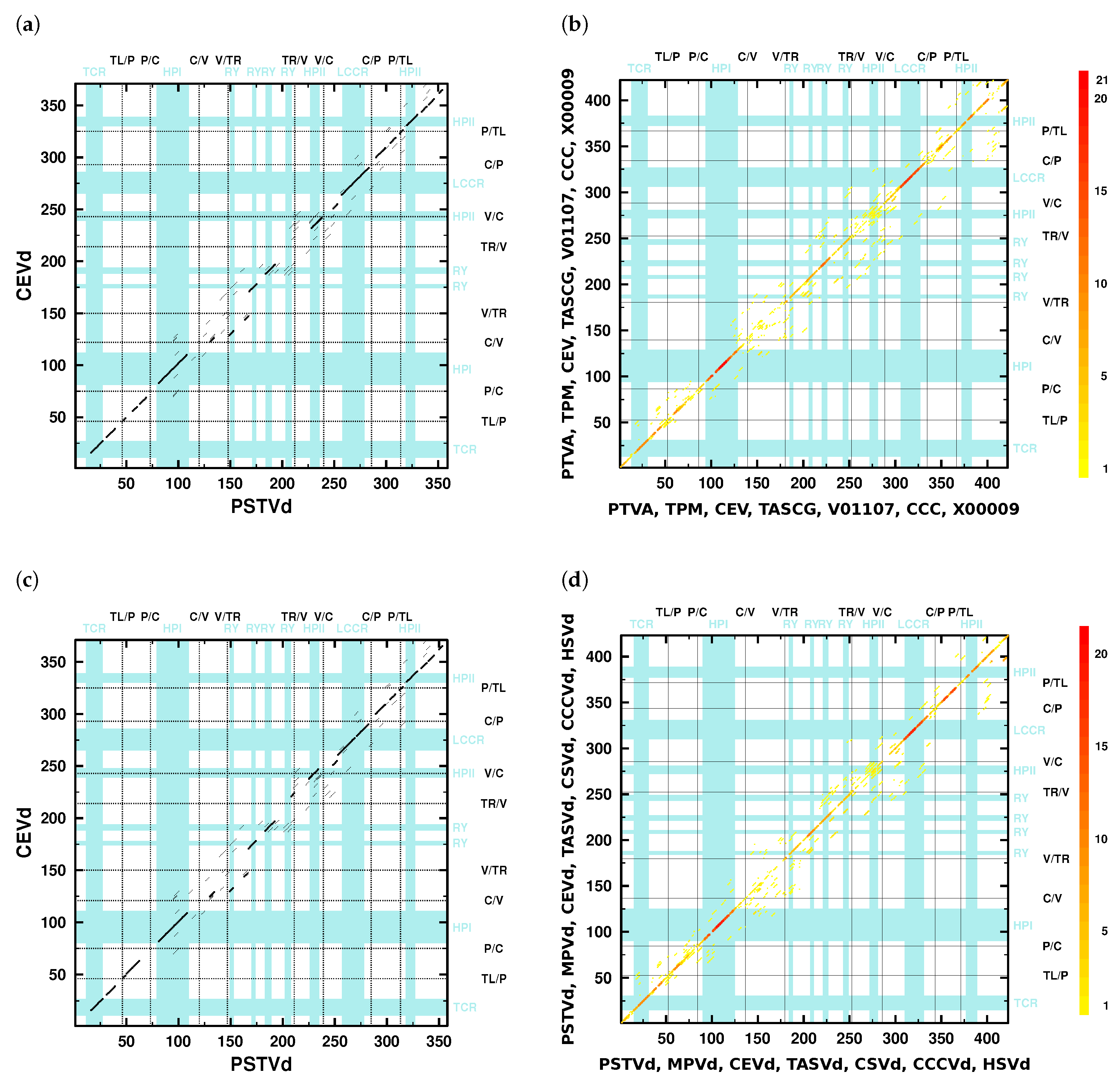
- The V domain shows the “greatest variability between closely related viroids” [12] and “is the most variable region … between otherwise closely related viroids, such as between TASV[d] and CEV[d] or TPMV [MPVd] and PSTV[d]” [11].Indeed, only a few diagonals show up in the dotplots (Figure 2) in the V domain; minor similarities in the lower part of the V domain are due to the 3’ part of HPII. The average pairwise sequence identity (APSI) in a mafft X-INS-i alignment of consensus sequences of species used in [11] is 64%, while the APSI of the V domain in the same alignment is only around 49% (Table S4b).
- “In pairwise sequence comparisons of viroids containing highly homologous C domains … there is significantly less sequence homology in the P and V regions, occurring about 5–9 residues 5’ and about 7–15 residues 3’ of the inverted repeat” [11].The number of kmer matches in the C domain regions is higher than in P and V regions; compare the dot colors of these domains in the overlay dotplots (Figure 2b,d).
- “…in comparisons of PSTV[d], TPMV [MPVd], TASV[d], and CSV[d], a change from low homology in the V domain to high homology in the T2 [TR] domain defines the boundary for these two domains” [11].The overlay dotplots clearly show a higher number of kmer matches in the TR domain regions than in the V domain regions (Figure 2b,d). The TR domains have %, while the V domains have % (Table S4b). Only the mentioned viroids have much larger differences in their individual PSI values with % and % (see values in bold font in columns V and TR of Table 2). Other viroid pairs have more similar PSI values of their V and TR domains, which is not sufficient to define their V/TR borders.
- “The P domain, with a conserved oligo(A) sequence flanked by regions with greater variability, has its borders based on homologies between the P region of HSV[d] and other viroids such as PSTV[d] and by certain pairwise comparisons such as CEV[d]-A and TASV[d] in which there is significant change from relatively low sequence homology in the P region to higher homology in the adjacent T1 [TL] and C domains” [11].Pairwise dotplots produced by NucAlN of HSVd with PSTVd and CEVd with TASVd, mentioned in the above sentence, are shown in Figure S5f,h. Here, as well as in further dotplots (f. e. Figure 2), the oligo-purine sequence in the upper and the partially complementary oligo-pyrimidine sequence in the lower P domain regions give rise to long kmer diagonals. In alignments (f. e. Figures S4 and S7), these sequences are also easily detectable.
- “In addition, sequence data show that three viroids (TASV[d], TPMV [MPVd], and CCCV[d]) exhibit unusual relationships with respect to their terminal sequences. For example, TASV[d] shares 73% overall sequence homology with CEV[d]-A but the T2 [TR] domains are only 46% homologous … In contrast, TASV[d] shares less overall sequence homology with PSTV[d] (64%) but the T2 [TR] domains are highly homologous (90%). Therefore, TASV[d] appears to be a recombinant between the T2 [TR] domain of a PSTV[d]-like viroid and all but the T2 [TR] domain of a CEV[d]-like viroid” [11].Clearly, several viroid sequences show PSI values of individual domains largely deviating (>20%) from the PSI values of their full sequences (Tables S2 and S3). One of the examples is the relation between TASVd, CEVd, and PSTVd (or MPVd), mentioned above: the full-sequence PSI values of pairs TASVd/CEVd, TASVd/PSTVd, and TASVd/MPVd are around 80%, 71%, and 76%, respectively; in contrast, the TR PSI values are around 57%, 91%, and 97%, respectively. That is, despite the highest full-sequences PSI of TASVd/CEVd, their TR PSI is much lower, and the opposite is true for the TASVd/PSTVd and TASVd/MPVd pairs, which led Keese and Symons to suggest that TASVd is recombinant of a CEVd-like sequence and a TR domain of a PSTVd- (or MPVd-)like sequence.
- “TPMV [MPVd] shares 76% overall sequence homology with PSTV[d] but the T1 [TL] domains are less homologous (67%). In contrast, TPMV [MPVd] shares less overall sequence homology with CEV[d]-A (60%) but the T1 [TL] domains are more homologous (80%). Thus, TPMV [MPVd] appears to be a recombinant between the T1 [TL] domain of a CEV[d]-like viroid and all but the T1 [TL] domain of a PSTV[d]-like viroid” [11].According to alignments by with NucAln , (Table S2) or , (Table S3), and by mafft X-INS-i (Table 2), the PSI values of full-sequence alignments for MPVd and PSTVd are around 77%, 82%, and 80%, respectively, and for MPVd and CEVd, are around 66%, 67%, and 66%, respectively; the PSI values of TL domains are lower with around 68%, 75%, and 77% for MPVd and PSTVd, and higher with around 75%, 77%, and 76% for MPVd and CEVd. Despite the difference between our values and the values from [11] (see below), all values support the given conclusion by Keese and Symons.
3.2. Consistent Domain Borders of Pospiviroid Members
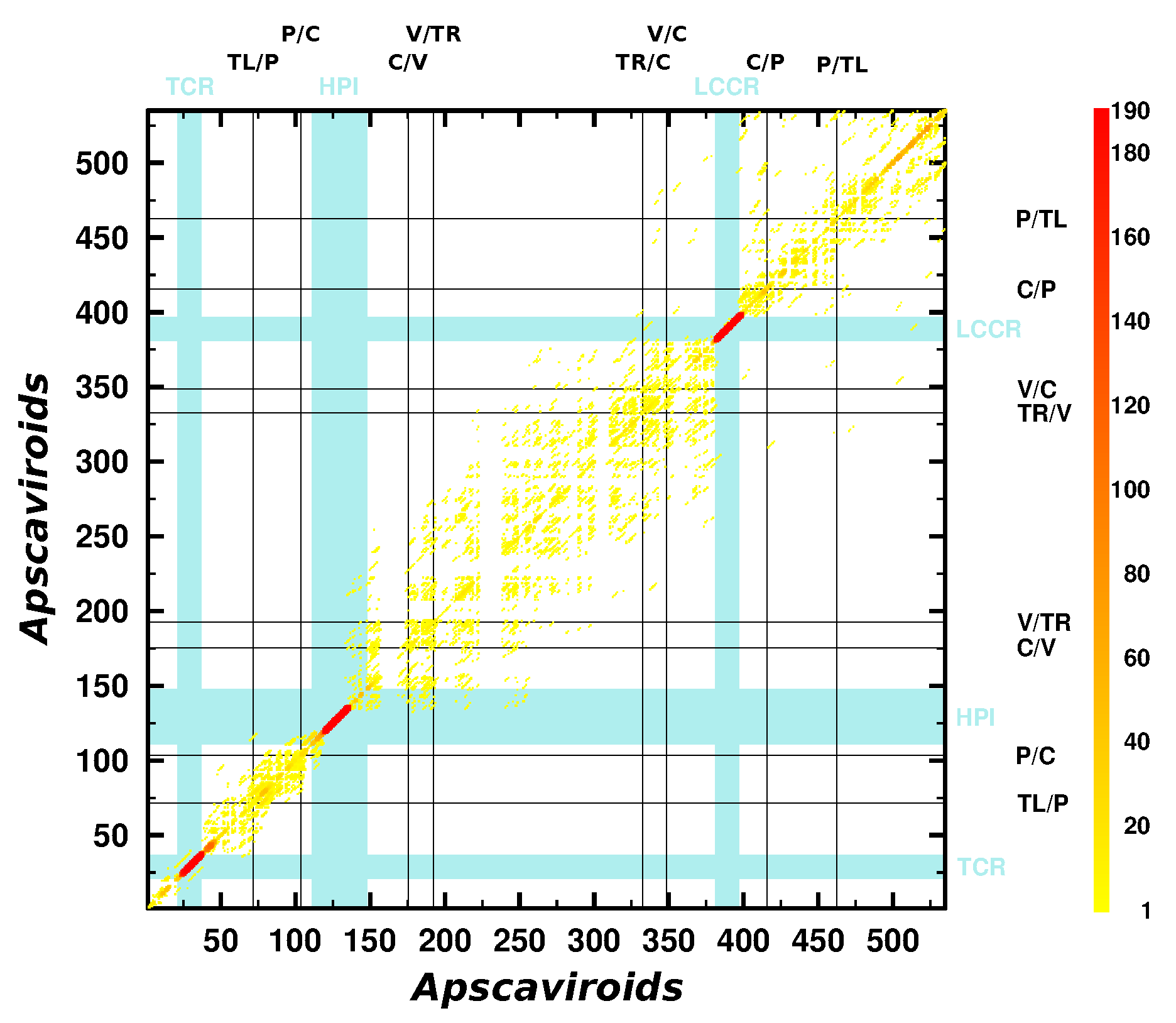
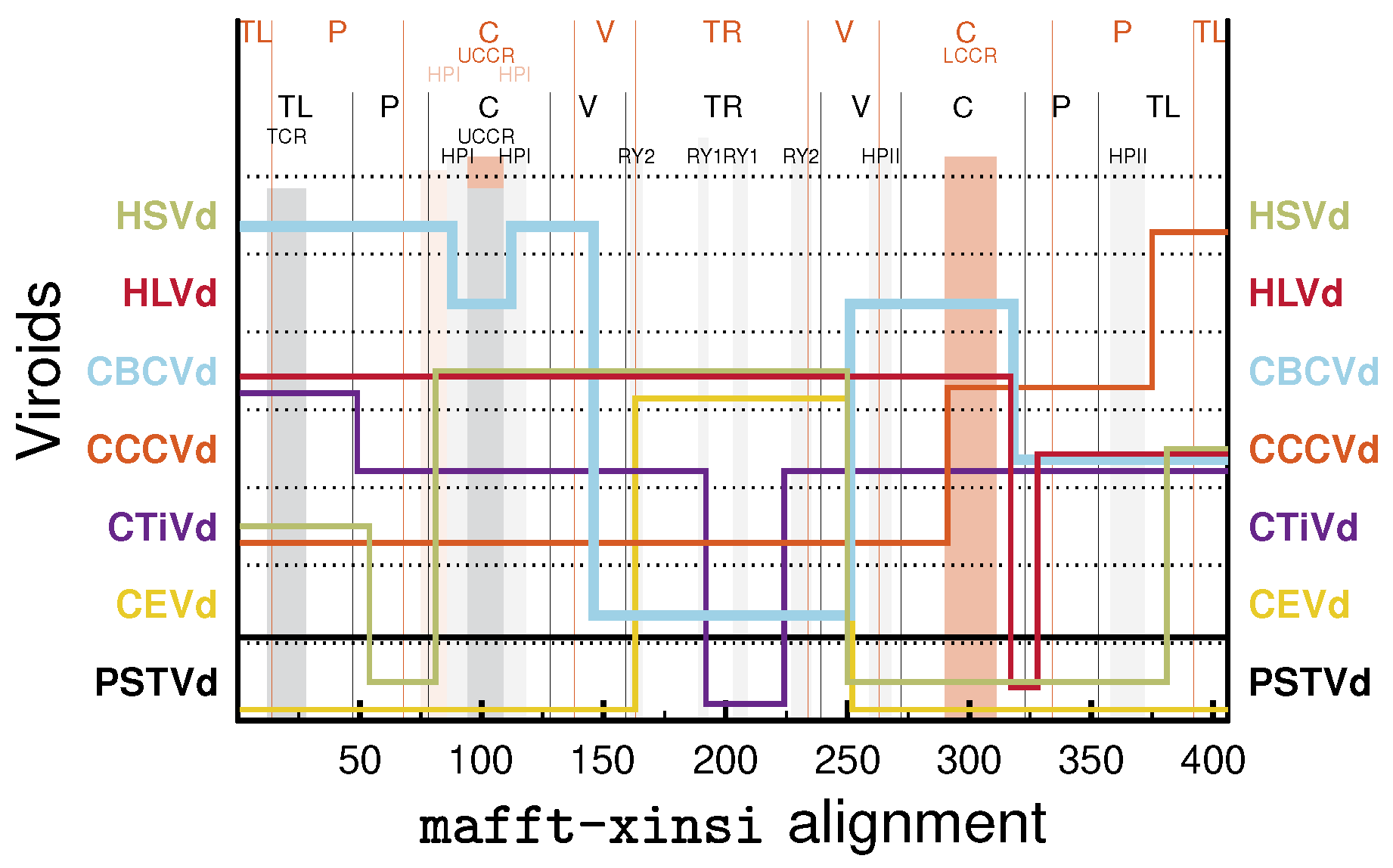
3.3. Can We Extend the Domain Hypothesis to the Other Genera of Pospiviroidae?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sänger, H.; Klotz, G.; Riesner, D.; Gross, H.; Kleinschmidt, A. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Kaeseberg, P.; Diener, T. Potato spindle tuber viroid. XII. An investigation of viroid RNA as a messenger for protein synthesis. Virology 1974, 61, 281–286. [Google Scholar] [CrossRef]
- Hadidi, A.; Randles, J.; Flores, R.; Palukaitis, P. (Eds.) Viroids and Satellites; Academic Press, Elsevier: Cambridge, MA, USA, 2017. [Google Scholar]
- Harders, J.; Lukács, N.; Robert-Nicoud, M.; Jovin, T.; Riesner, D. Imaging of viroids in nuclei from tomato leaf tissue by in situ hybridization and confocal laser scanning microscopy. EMBO J. 1989, 8, 3941–3949. [Google Scholar] [CrossRef]
- Schindler, I.M.; Mühlbach, H.P. Involvement of nuclear DNA-dependent RNA polymerases in potato spindle tuber viroid replication: A reevaluation. Plant Sci. 1992, 84, 221–229. [Google Scholar] [CrossRef]
- Qi, Y.; Ding, B. Differential subnuclear localization of RNA strands of opposite polarity derived from an autonomously replicating viroid. Plant Cell 2003, 15, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Dissanayaka Mudiyanselage, S.; Qu, J.; Tian, N.; Jiang, J.; Wang, Y. Potato spindle tuber viroid RNA-templated transcription: Factors and regulation. Viruses 2018, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.; Perreault, J.P. Structure and associated biological functions of viroids. Adv. Virus Res. 2016, 94, 141–172. [Google Scholar] [CrossRef]
- Giguère, T.; Adkar-Purushothama, C.; Perreault, J. Comprehensive secondary structure elucidation of four genera of the family Pospiviroidae. PLoS ONE 2014, 9, e98655. [Google Scholar] [CrossRef]
- López-Carrasco, A.; Flores, R. Dissecting the secondary structure of the circular RNA of a nuclear viroid in vivo: A “naked” rod-like conformation similar but not identical to that observed in vitro. RNA Biol. 2017, 14, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Keese, P.; Symons, R. Domains in viroids: Evidence of intermolecular RNA rearrangement and their contribution to viroid evolution. Proc. Natl. Acad. Sci. USA 1985, 82, 4582–4586. [Google Scholar] [CrossRef]
- McInnes, J.; Symons, R. Comparative structure of viroids and their rapid detection using radioactive and nonradioactive nucleic acid probes. In Viroids and Satellites: Molecular Parasites at the Frontier of Life; Maramorosch, K., Ed.; CRC Press: Boca Raton, FL, USA, 1991; pp. 21–58. [Google Scholar]
- Koltunow, A.; Rezaian, M. A scheme for viroid classification. Intervirology 1989, 30, 194–201. [Google Scholar] [CrossRef]
- Goodman, T.; Nagel, L.; Rappold, W.; Klotz, G.; Riesner, D. Viroid replication: Equilibrium association constant and comparative activity measurements for the viroid-polymerase interactions. Nucleic Acids Res. 1984, 12, 6231–6246. [Google Scholar] [CrossRef] [PubMed]
- Kolonko, N.; Bannach, O.; Aschermann, K.; Hu, K.H.; Moors, M.; Schmitz, M.; Steger, G.; Riesner, D. Transcription of potato spindle tuber viroid by RNA polymerase II starts in the left terminal loop. Virology 2006, 347, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Bojić, T.; Beeharry, Y.; Zhang, D.; Pelchat, M. Tomato RNA polymerase II interacts with the rod-like conformation of the left terminal domain of the potato spindle tuber viroid positive RNA genome. J. Gen. Virol. 2012, 93, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Dissanayaka Mudiyanselage, S.; Wang, Y. Evidence Supporting That RNA Polymerase II Catalyzes De Novo Transcription Using Potato Spindle Tuber Viroid Circular RNA Templates. Viruses 2020, 12, 371. [Google Scholar] [CrossRef]
- Koltunow, A.; Rezaian, M. Grapevine yellow speckle viroid: Structural features of a new viroid group. Nucleic Acids Res. 1988, 16, 849–864. [Google Scholar] [CrossRef]
- Puchta, H.; Ramm, K.; Sänger, H. The molecular structure of hop latent viroid (HLV), a new viroid occurring worldwide in hops. Nucleic Acids Res. 1988, 16, 4197–4216. [Google Scholar] [CrossRef]
- Di Serio, F.; Li, S.F.; Palls, V.; Pwens, R.; Randles, J.; Sano, T.; Verhoeven, J.; Vidalakis, G.; Flores, R. Viroid taxonomy. In Viroids and Satellites; Hadidi, A., Randles, J., Flores, R., Palukaitis, P., Eds.; Academic Press, Elsevier: Cambridge, MA, USA, 2017; pp. 135–146. [Google Scholar]
- Schnölzer, M.; Haas, B.; Ramm, K.; Hofmann, H.; Sänger, H. Correlation between structure and pathogenicity of potato spindle tuber viroid (PSTV). EMBO J. 1985, 4, 2181–2190. [Google Scholar] [CrossRef]
- Visvader, J.; Symons, R. Eleven new sequence variants of citrus exocortis viroid and the correlation of sequence with pathogenicity. Nucleic Acids Res. 1985, 13, 2907–2920. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, S.; Tsushima, D.; Adkar-Purushothama, C.; Sano, T. Identification and Molecular Mechanisms of Key Nucleotides Causing Attenuation in Pathogenicity of Dahlia Isolate of Potato Spindle Tuber Viroid. Int. J. Mol. Sci. 2020, 21, 7352. [Google Scholar] [CrossRef]
- Sano, T.; Candresse, T.; Hammond, R.; Diener, T.; Owens, R. Identification of multiple structural domains regulating viroid pathogenicity. Proc. Natl. Acad. Sci. USA 1992, 89, 10104–10108. [Google Scholar] [CrossRef]
- Rodriguez, M.; Randles, J. Coconut cadang-cadang viroid (CCCVd) mutants associated with severe disease vary in both the pathogenicity domain and the central conserved region. Nucleic Acids Res. 1993, 21, 2771. [Google Scholar] [CrossRef] [PubMed]
- Chaffai, M.; Serra, P.; Gandía, M.; Hernández, C.; Duran-Vila, N. Molecular characterization of CEVd strains that induce different phenotypes in Gynura aurantiaca: Structure-pathogenicity relationships. Arch. Virol. 2007, 152, 1283–1294. [Google Scholar] [CrossRef]
- Li, R.; Padmanabhan, C.; Ling, K. A single base pair in the right terminal domain of tomato planta macho viroid is a virulence determinant factor on tomato. Virology 2016, 500, 238–246. [Google Scholar] [CrossRef]
- Steger, G.; Hofmann, H.; Förtsch, J.; Gross, H.; Randles, J.; Sänger, H.; Riesner, D. Conformational transitions in viroids and virusoids: Comparison of results from energy minimization algorithm and from experimental data. J. Biomol. Struct. Dyn. 1984, 2, 543–571. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Cai, Z.; Tinoco, I. RNA structure at high resolution. FASEB J. 1995, 9, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Leontis, N.; Qian, S.; Itaya, A.; Qi, Y.; Boris-Lawrie, K.; Ding, B. Tertiary structural and functional analyses of a viroid RNA motif by isostericity matrix and mutagenesis reveal its essential role in replication. J. Virol. 2006, 80, 8566–8581. [Google Scholar] [CrossRef] [PubMed]
- Branch, A.; Benenfeld, B.; Robertson, H. Ultraviolet light-induced crosslinking reveals a unique region of local tertiary structure in potato spindle tuber viroid and HeLa 5 S RNA. Proc. Natl. Acad. Sci. USA 1985, 82, 6590–6594. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, X.; Itaya, A.; Ding, B. Evidence for the existence of the loop E motif of Potato spindle tuber viroid in vivo. J. Virol. 2007, 81, 2074–2077. [Google Scholar] [CrossRef]
- Eiras, M.; Kitajima, E.; Flores, R.; Darós, J. Existence in vivo of the loop E motif in potato spindle tuber viroid RNA. Arch. Virol. 2007, 152, 1389–1393. [Google Scholar] [CrossRef]
- Henco, K.; Sänger, H.; Riesner, D. Fine structure melting of viroids as studied by kinetic methods. Nucleic Acids Res. 1979, 6, 3041–3059. [Google Scholar] [CrossRef]
- Diener, T. Viroid processing: A model involving the central conserved region and hairpin I. Proc. Natl. Acad. Sci. USA 1986, 83, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.; Tabler, M.; Brüggemann, W.; Colpan, M.; Klotz, G.; Sänger, H.; Riesner, D. Structure of viroid replicative intermediates: Physico-chemical studies on SP6 transcripts of cloned oligomeric potato spindle tuber viroid. Nucleic Acids Res. 1986, 14, 9613–9630. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baumstark, T.; Schröder, A.; Riesner, D. Viroid processing: Switch from cleavage to ligation is driven by a change from a tetraloop to a loop E conformation. EMBO J. 1997, 16, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Schrader, O.; Baumstark, T.; Riesner, D. A mini-RNA containing the tetraloop, wobble-pair and loop E motifs of the central conserved region of potato spindle tuber viroid is processed into a minicircle. Nucleic Acids Res. 2003, 31, 988–998. [Google Scholar] [CrossRef]
- Gas, M.; Hernández, C.; Flores, R.; Daròs, J. Processing of nuclear viroids in vivo: An interplay between RNA conformations. PLoS Pathog. 2007, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Loss, P.; Schmitz, M.; Steger, G.; Riesner, D. Formation of a thermodynamically metastable structure containing hairpin II is critical for infectivity of potato spindle tuber viroid RNA. EMBO J. 1991, 10, 719–727. [Google Scholar] [CrossRef]
- Qu, F.; Heinrich, C.; Loss, P.; Steger, G.; Tien, P.; Riesner, D. Multiple pathways of reversion in viroids for conservation of structural elements. EMBO J. 1993, 12, 2129–2139. [Google Scholar] [CrossRef]
- Gozmanova, M.; Denti, M.; Minkov, I.; Tsagris, M.; Tabler, M. Characterization of the RNA motif responsible for the specific interaction of potato spindle tuber viroid RNA (PSTVd) and the tomato protein Virp1. Nucleic Acids Res. 2003, 31, 5534–5543. [Google Scholar] [CrossRef]
- Martínez de Alba, A.; Sägesser, R.; Tabler, M.; Tsagris, M. A bromodomain-containing protein from tomato specifically binds potato spindle tuber viroid RNA in vitro and in vivo. J. Virol. 2003, 77, 9685–9694. [Google Scholar] [CrossRef]
- Steger, G. Modelling the three-dimensional structure of the right-terminal domain of pospiviroids. Sci. Rep. 2017, 7, 711. [Google Scholar] [CrossRef] [PubMed]
- Kalantidis, K.; Denti, M.; Tzortzakaki, S.; Marinou, E.; Tabler, M.; Tsagris, M. Virp1 is a host protein with a major role in Potato spindle tuber viroid infection in Nicotiana plants. J. Virol. 2007, 81, 12872–12880. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Kalantidis, K.; Rao, A. A bromodomain-containing host protein mediates the nuclear importation of a satellite RNA of Cucumber mosaic virus. J. Virol. 2013, 88, 1890–1896. [Google Scholar] [CrossRef]
- Wilbur, W.; Lipman, D. Rapid similarity searches of nucleic acid and protein data banks. Proc. Natl. Acad. Sci. USA 1983, 80, 726–730. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Spang, R.; Rehmsmeier, M.; Stoye, J. Sequence database search using jumping alignments. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2000, 8, 367–375. [Google Scholar]
- Spang, R.; Rehmsmeier, M.; Stoye, J. A novel approach to remote homology detection: Jumping alignments. J. Comput. Biol. 2002, 9, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Abouelhoda, M.; Kurtz, S.; Ohlebusch, E. Replacing suffix trees with enhanced suffix arrays. J. Discr. Algorithms 2004, 2, 53–86. [Google Scholar] [CrossRef]
- Keese, P.; Symons, R. Molecular structure (primary and secondary). In The Viroids; Diener, T., Ed.; Plenum Press: New York, NY, USA, 1987; pp. 37–62. [Google Scholar]
- Katoh, K.; Toh, H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinform. 2008, 9, 212. [Google Scholar] [CrossRef]
- Wilm, A.; Linnenbrink, K.; Steger, G. ConStruct: Improved construction of RNA consensus structures. BMC Bioinform. 2008, 9, 219. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, Z.; Breaker, R. R2R—Software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinform. 2011, 12, 3. [Google Scholar] [CrossRef]
- Weinberg, Z.; Barrick, J.; Yao, Z.; Roth, A.; Kim, J.; Gore, J.; Wang, J.; Lee, E.; Block, K.; Sudarsan, N.; et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007, 35, 4809–4819. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.; Clements, J.; Eddy, S. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 2017, 14, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Semancik, J.; Szychowski, J.; Rakowski, A.; Symons, R. Isolates of citrus exocortis viroid recovered by host and tissue selection. J. Gen. Virol. 1993, 74, 2427–2436. [Google Scholar] [CrossRef] [PubMed]
- Semancik, J.; Szychowski, J.; Rakowski, A.; Symons, R. A stable 463 nucleotide variant of citrus exocortis viroid produced by terminal repeats. J. Gen. Virol. 1994, 75, 727–732. [Google Scholar] [CrossRef]
- Fadda, Z.; Darós, J.; Flores, R.; Duran-Vila, N. Identification in eggplant of a variant of citrus exocortis viroid (CEVd) with a 96 nucleotide duplication in the right terminal region of the rod-like secondary structure. Virus Res. 2003, 97, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Haseloff, J.; Mohamed, N.; Symons, R. Viroid RNAs of cadang-cadang disease of coconuts. Nature 1982, 299, 316–321. [Google Scholar] [CrossRef]
- Vadamalai, G.; Hanold, D.; Rezaian, M.; Randles, J. Variants of Coconut cadang-cadang viroid isolated from an African oil palm (Elaies guineensis Jacq.) in Malaysia. Arch. Virol. 2006, 151, 1447–1456. [Google Scholar] [CrossRef]
- Boratyn, G.; Camacho, C.; Cooper, P.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.; Matten, W.; McGinnis, S.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef]
- Graphics Layout Engine. Available online: http://glx.sourceforge.net/ (accessed on 1 November 2021).
- Katoh, K.; Kuma, K.; Miyata, T.; Toh, H. Improvement in the accuracy of multiple sequence alignment program MAFFT. Genome Inform. Ser. 2005, 16, 22–33. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.; Hofacker, I. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Viral Genome Browser (Pospiviroidae). Available online: https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=185751 (accessed on 1 November 2021).
- Brister, J.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Eddy, S. RSEARCH: Finding homologs of single structured RNA sequences. BMC Bioinform. 2003, 4, 44. [Google Scholar] [CrossRef]
- Keese, P.; Osorio-Keese, M.; Symons, R. Coconut tinangaja viroid: Sequence homology with coconut cadang-cadang viroid and other potato spindle tuber viroid related RNAs. Virology 1988, 162, 508–510. [Google Scholar] [CrossRef]
- Koltunow, A.; Rezaian, M. Grapevine viroid 1B, a new member of the apple scar skin viroid group contains the left terminal region of tomato planta macho viroid. Virology 1989, 170, 575–578. [Google Scholar] [CrossRef]
- Stasys, R.; Dry, I.; Rezaian, M. The termini of a new citrus viroid contain duplications of the central conserved regions from two viroid groups. FEBS Lett. 1995, 358, 182–184. [Google Scholar] [CrossRef][Green Version]
- Puchta, H.; Ramm, K.; Luckinger, R.; Hadas, R.; Bar-Joseph, M.; Sänger, H. Primary and secondary structure of citrus viroid IV (CVd IV), a new chimeric viroid present in dwarfed grapefruit in Israel. Nucleic Acids Res. 1991, 19, 6640. [Google Scholar] [CrossRef]
- Verhoeven, J.; Meekes, E.; Roenhorst, J.; Flores, R.; Serra, P. Dahlia latent viroid: A recombinant new species of the family Pospiviroidae posing intriguing questions about its origin and classification. J. Gen. Virol. 2012, 94, 711–719. [Google Scholar] [CrossRef] [PubMed][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | Version | URL | Ref. |
|---|---|---|---|
| BLASTn | 2.9.0 | https://blast.ncbi.nlm.nih.gov/Blast.cgi | [63] |
| ClustalΩ | 1.2.4 | http://www.clustal.org/omega/#Download | [48] |
| ConStruct | 3.2.2 | http://www.biophys.uni-duesseldorf.de/html/local/construct3/ | [54] |
| gle | 4.2.5 | https://sourceforge.net/projects/glx/ | [64] |
| JAli | 1.3 | https://bibiserv.cebitec.uni-bielefeld.de/jali | [49,50] |
| mafft X-INS-i, L-INS-I | 7.402 | https://mafft.cbrc.jp/alignment/software/ | [53,65,66] |
| R2R | 1.0.2 | https://sourceforge.net/projects/weinberg-r2r/ | [55] |
| RNAfold | 2.4.6 | https://www.tbi.univie.ac.at/RNA/ | [67] |
| R-scape | 1.2.3 | http://eddylab.org/R-scape/ | [57] |
| vmatch | 2.3.0 | http://www.vmatch.de/ | [51] |
| Viroids Usedfor PairwiseComparisons | % Sequence Homology | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Domains | ||||||||||||
| TL | P | C | V | TR | |||||||||
| 1 | 2 | KS | WS | KS | WS | KS | S | KS | WS | KS | WS | KS | WS |
| TASVd | CEVd | 73 | 79.4 | 91 | 93.4 | 54 | 63.3 | 99 | 97.9 | 49 | 59.2 | 46 | 61.9 |
| PSTVd | 64 | 69.9 | 67 | 78.1 | 59 | 69.1 | 65 | 74.2 | 30 | 44.9 | 90 | 92.1 | |
| MPVd | PSTVd | 76 | 80.2 | 67 | 76.6 | 73 | 82.1 | 94 | 96.8 | 42 | 42.3 | 95 | 95.2 |
| CEVd | 60 | 66.1 | 80 | 76.1 | 70 | 62.5 | 69 | 78.3 | 29 | 42.3 | 37 | 61.9 | |
| CCCVd | PSTVd | 38 | 59.3 | 25 | 52.2 | 14 | 47.5 | 70 | 71.6 | 37 | 57.1 | 27 | 53.7 |
| HSVd | 39 | 56.5 | 52 | 82.6 | 33 | 39.0 | 42 | 63.2 | 31 | 46.4 | 50 | 58.5 | |
| HSVd | PSTVd | 35 | 53.9 | 23 | 49.1 | 58 | 71.2 | 35 | 52.6 | 37 | 43.2 | 28 | 51.1 |
| CSVd | CEVd | 59 | 67.4 | 77 | 80.9 | 42 | 57.4 | 82 | 91.2 | 28 | 36.7 | 38 | 60.4 |
| PSTVd | 61 | 69.7 | 69 | 71.9 | 49 | 64.8 | 71 | 78.9 | 31 | 46.6 | 81 | 92.5 | |
| CEVd | PSTVd | 55 | 64.6 | 62 | 70.0 | 71 | 67.9 | 65 | 74.2 | 31 | 43.4 | 38 | 60.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wüsthoff, K.-P.; Steger, G. Conserved Motifs and Domains in Members of Pospiviroidae. Cells 2022, 11, 230. https://doi.org/10.3390/cells11020230
Wüsthoff K-P, Steger G. Conserved Motifs and Domains in Members of Pospiviroidae. Cells. 2022; 11(2):230. https://doi.org/10.3390/cells11020230
Chicago/Turabian StyleWüsthoff, Kevin-Phil, and Gerhard Steger. 2022. "Conserved Motifs and Domains in Members of Pospiviroidae" Cells 11, no. 2: 230. https://doi.org/10.3390/cells11020230
APA StyleWüsthoff, K.-P., & Steger, G. (2022). Conserved Motifs and Domains in Members of Pospiviroidae. Cells, 11(2), 230. https://doi.org/10.3390/cells11020230