
Evolutionary Origin of Insulin-Degrading Enzyme and Its Subcellular Localization and Secretion Mechanism: A Study in Microglial Cells


 , ,
, ,
Abstract
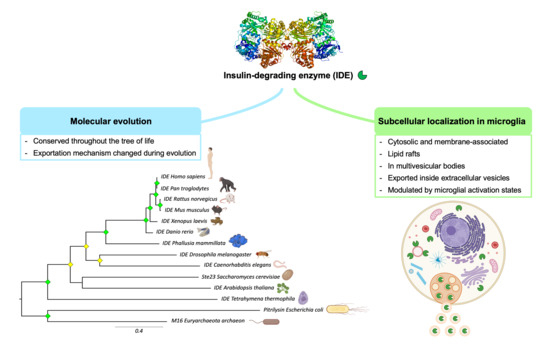
:
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analyses
2.1.1. Sequences Selected for Clan ME Analysis
2.1.2. Searching for IDE Homologous Sequences
2.1.3. Multiple Sequence Alignments (MSA)
2.1.4. Phylogenetic Reconstruction
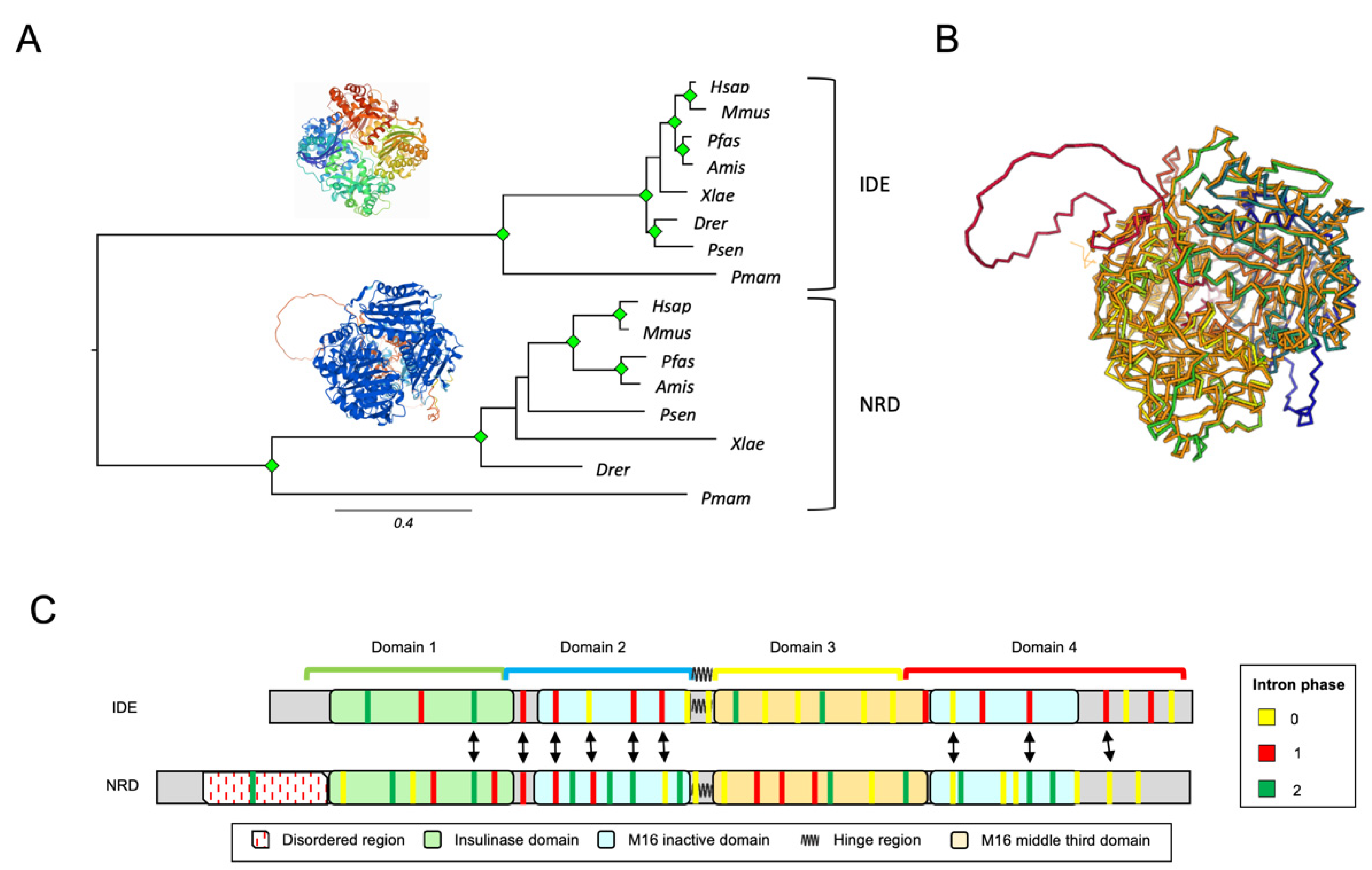
2.1.5. Intronic Features Extraction
2.1.6. Protein Structure Alignments
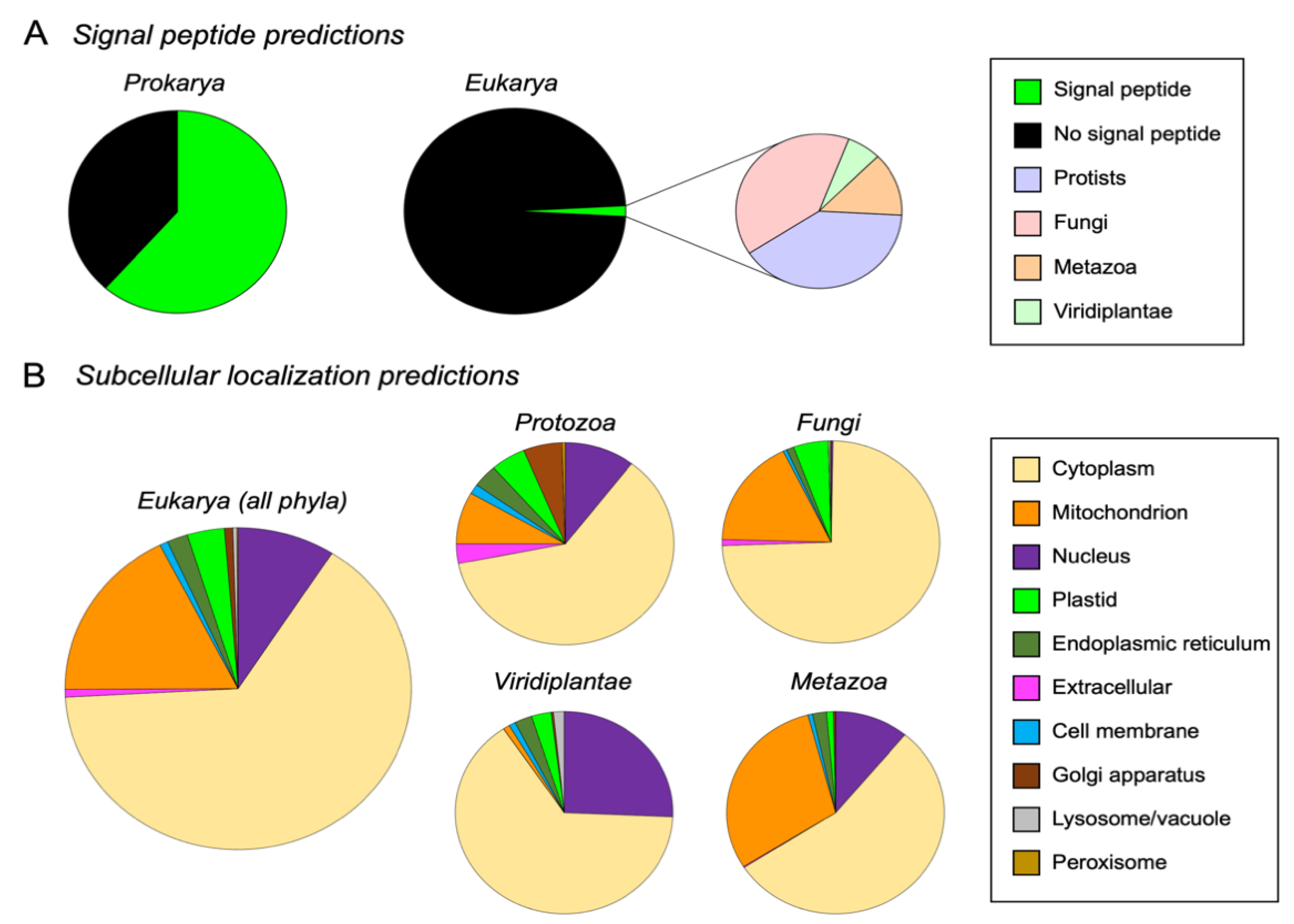
2.1.7. Bioinformatic Prediction of Subcellular Localization
2.2. Animals, Cell Cultures and Treatments
2.3. Crude Membrane Preparations
2.4. Isolation of Lipid Rafts by Sucrose Gradient Centrifugation
2.5. Immunoblot Analysis
2.6. Immunocytochemistry
2.7. Electron Microscopy
2.8. Extracellular Media Processing and Extracellular Vesicles Isolation
2.9. Statistical Analysis
3. Results
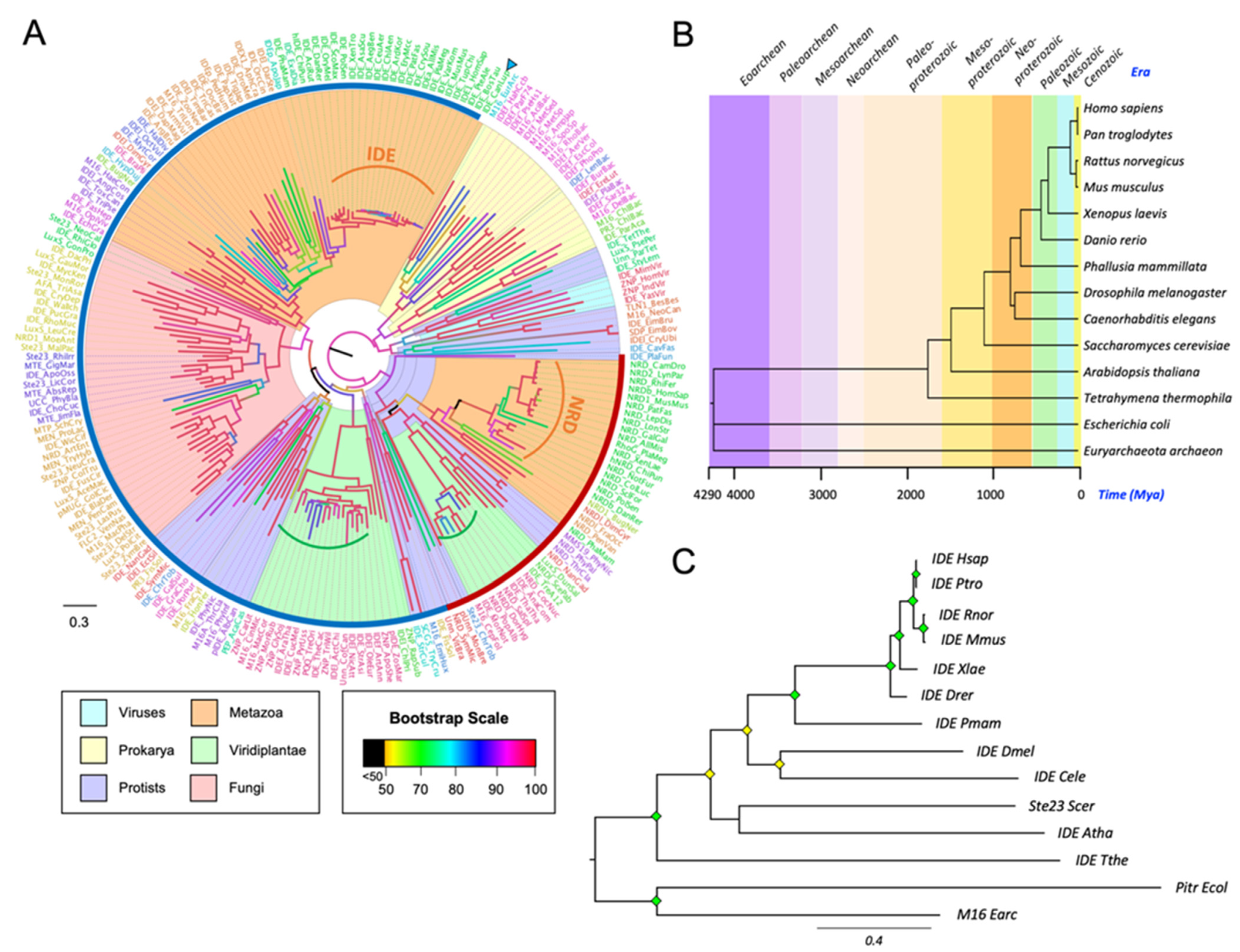
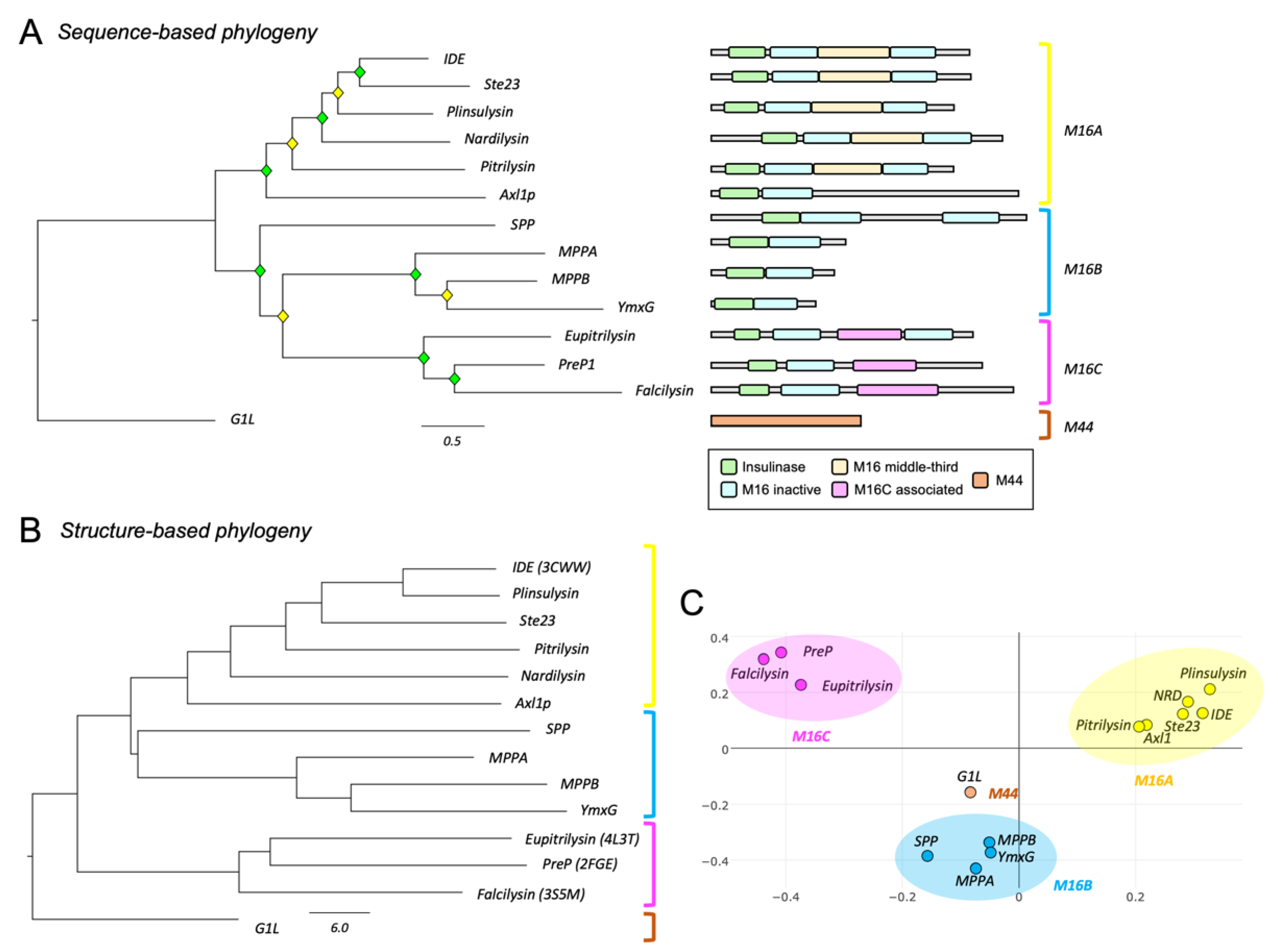
3.1. Phylogeny of Metalloendopeptidases Belonging to Clan ME
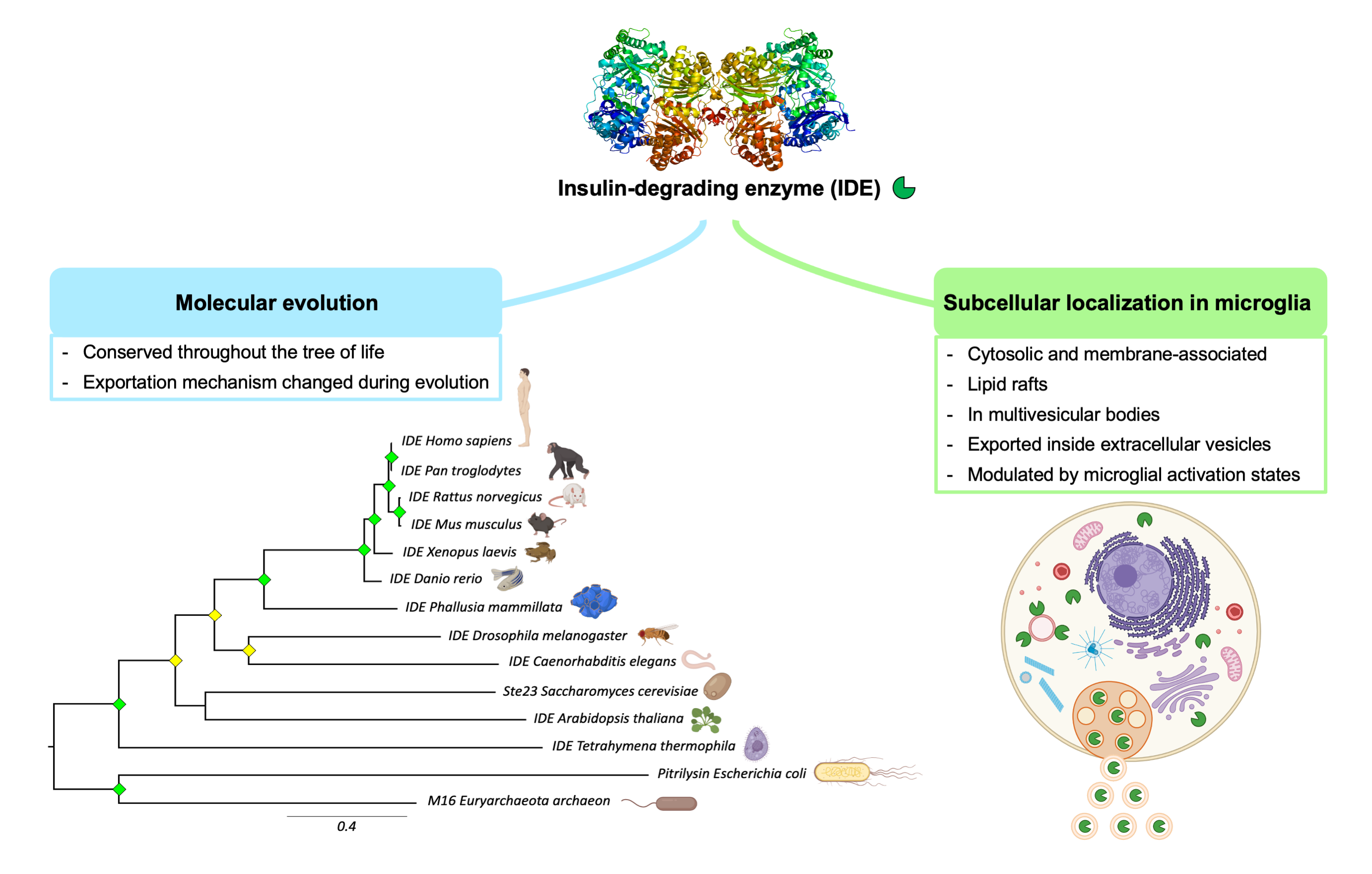
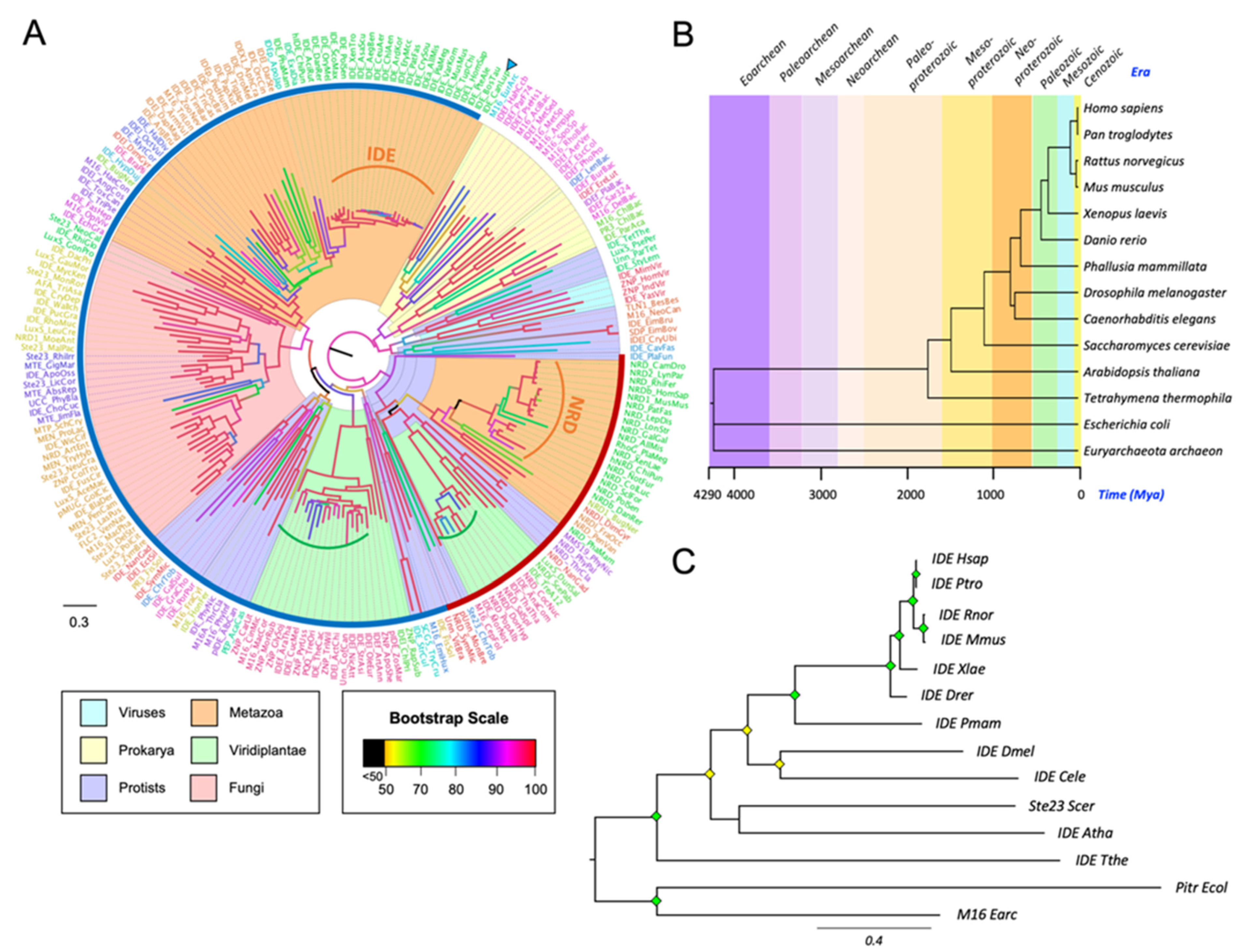
3.2. The M16A Family Is Highly Conserved across Evolution
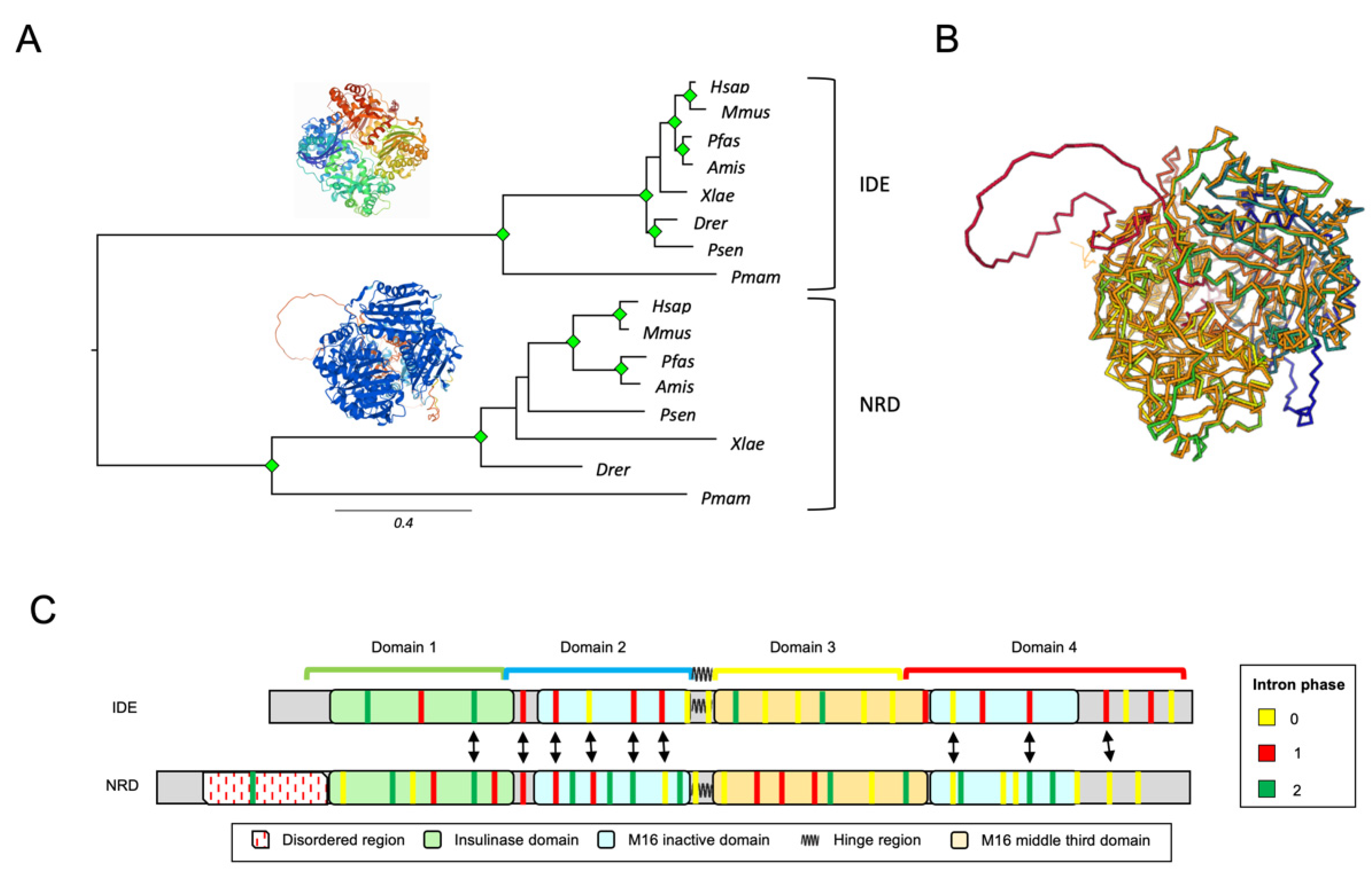
3.3. IDE and NRD Are Paralogous Proteins with Different Gene Structure
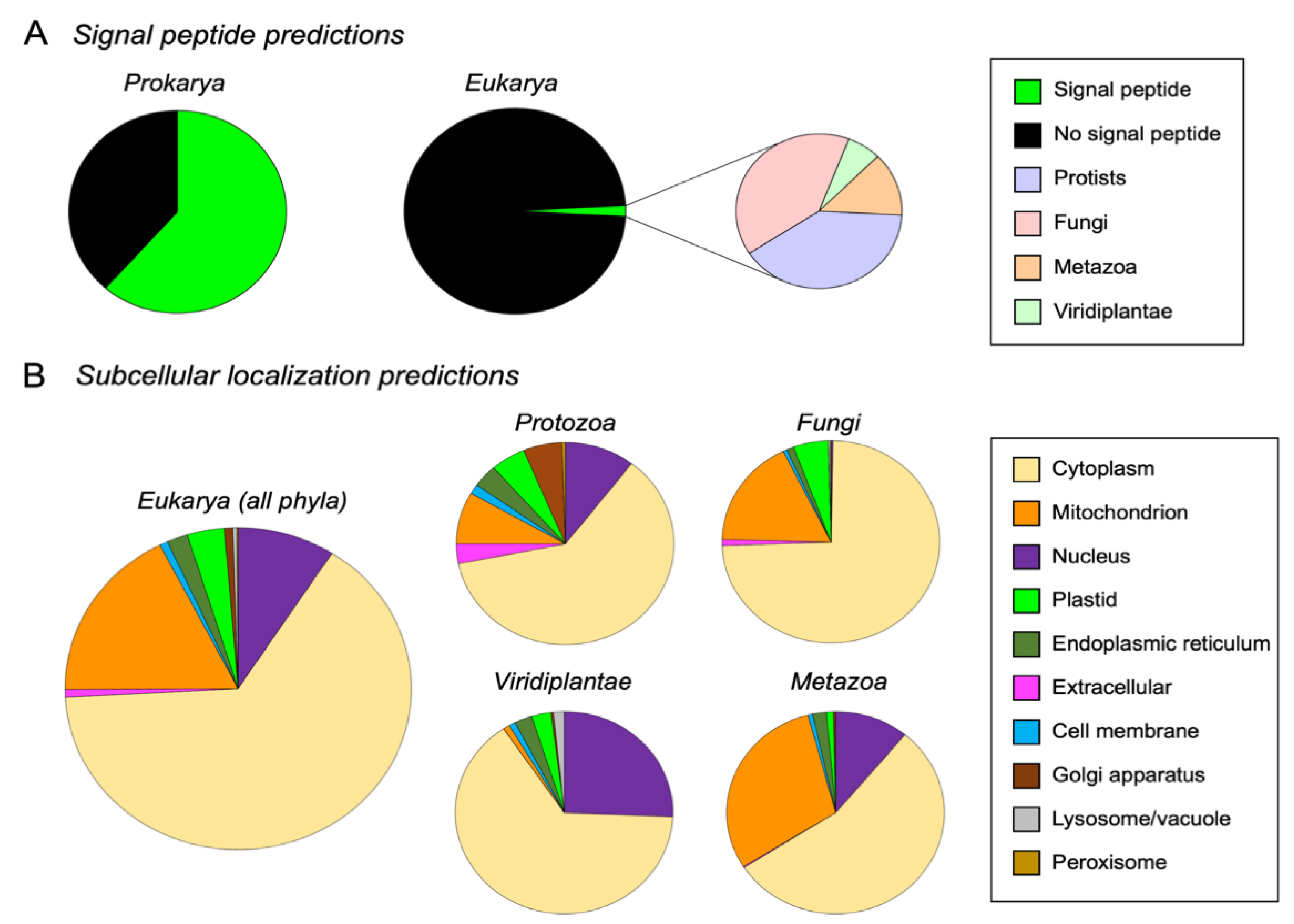
3.4. M16A Proteins Topogenesis Changed during Evolution
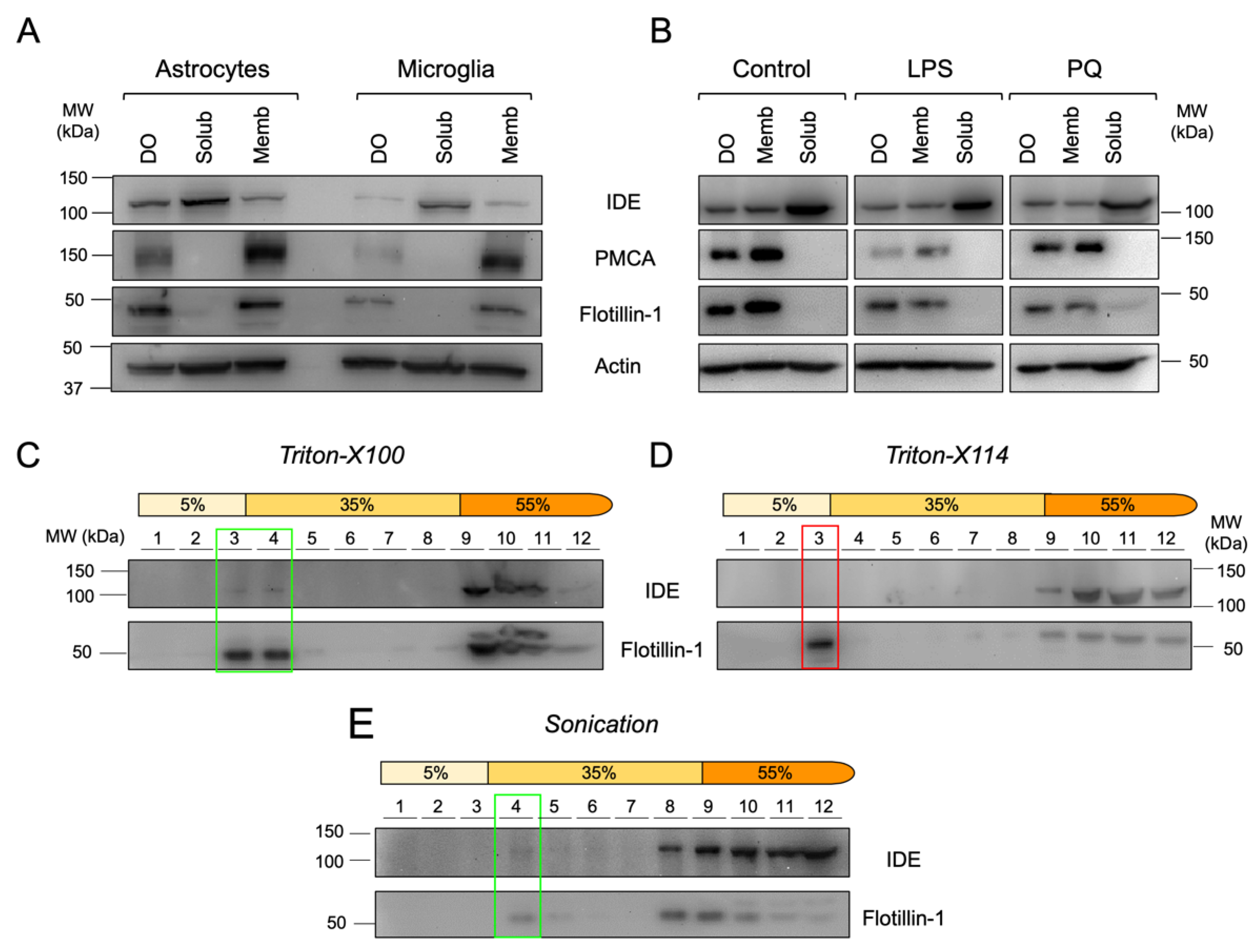
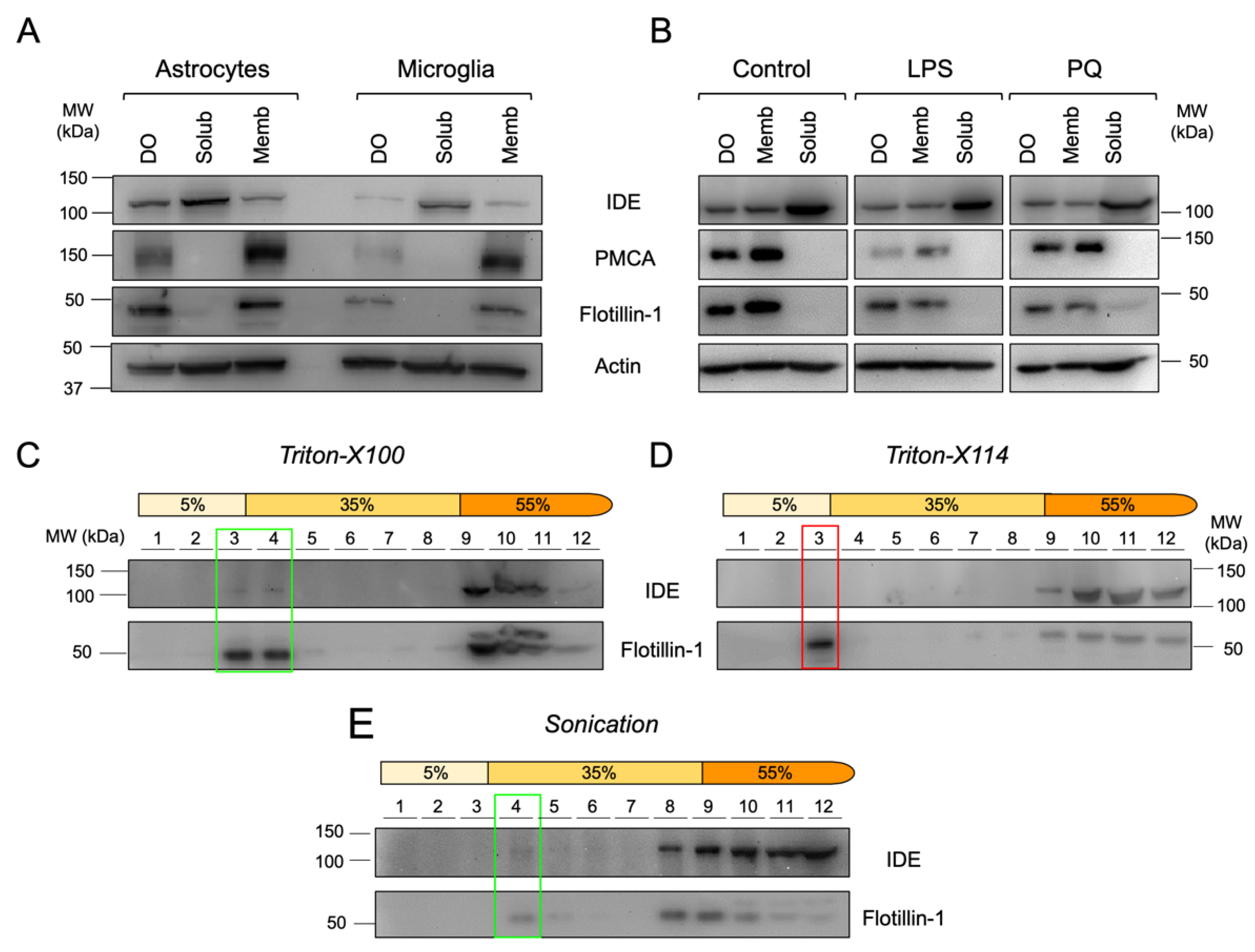
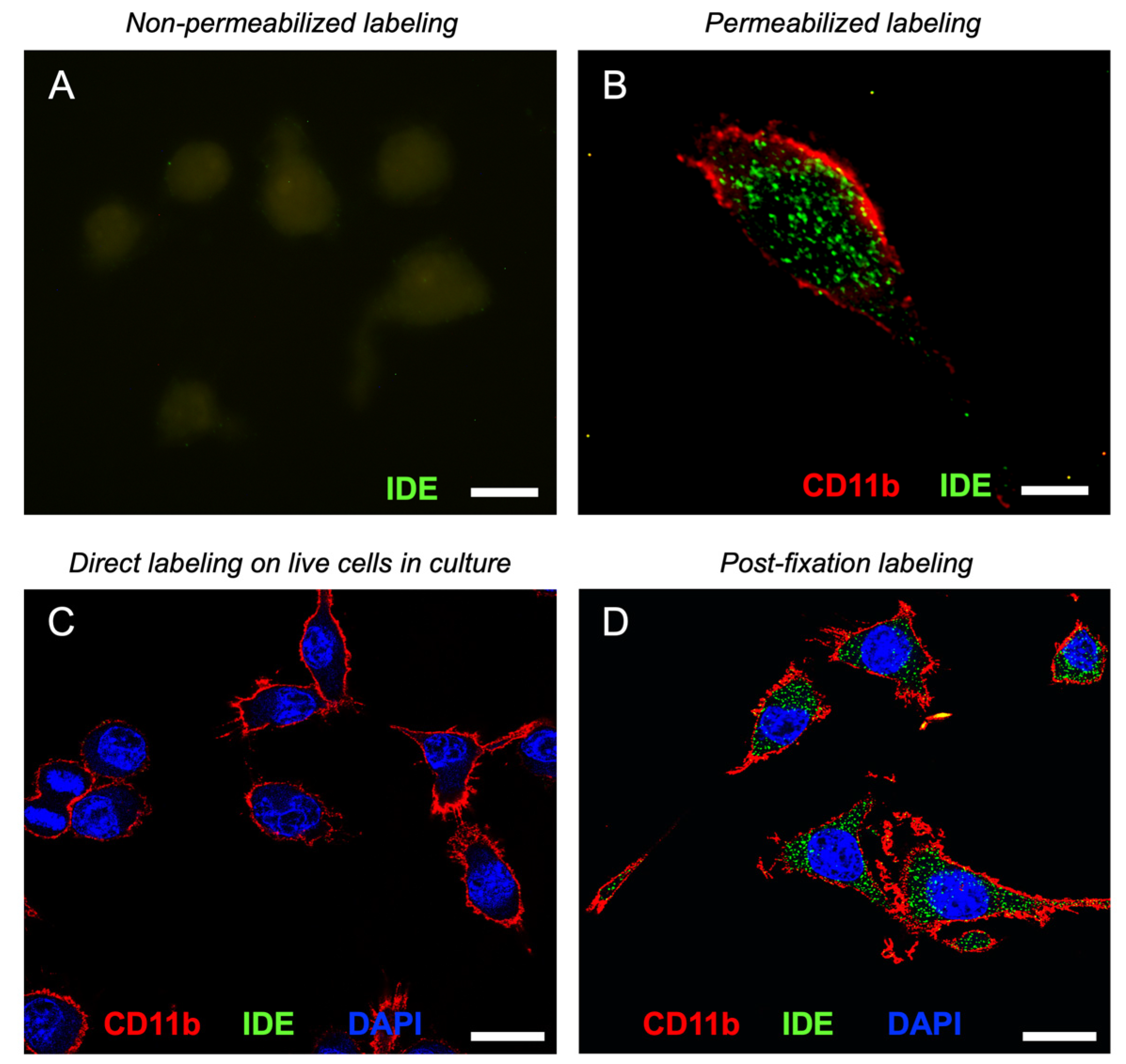
3.5. Microglial Cells Display IDE in Cytosol, Membranes and Lipid Rafts
3.6. IDE Associates to Membranes by Their Cytosolic Side
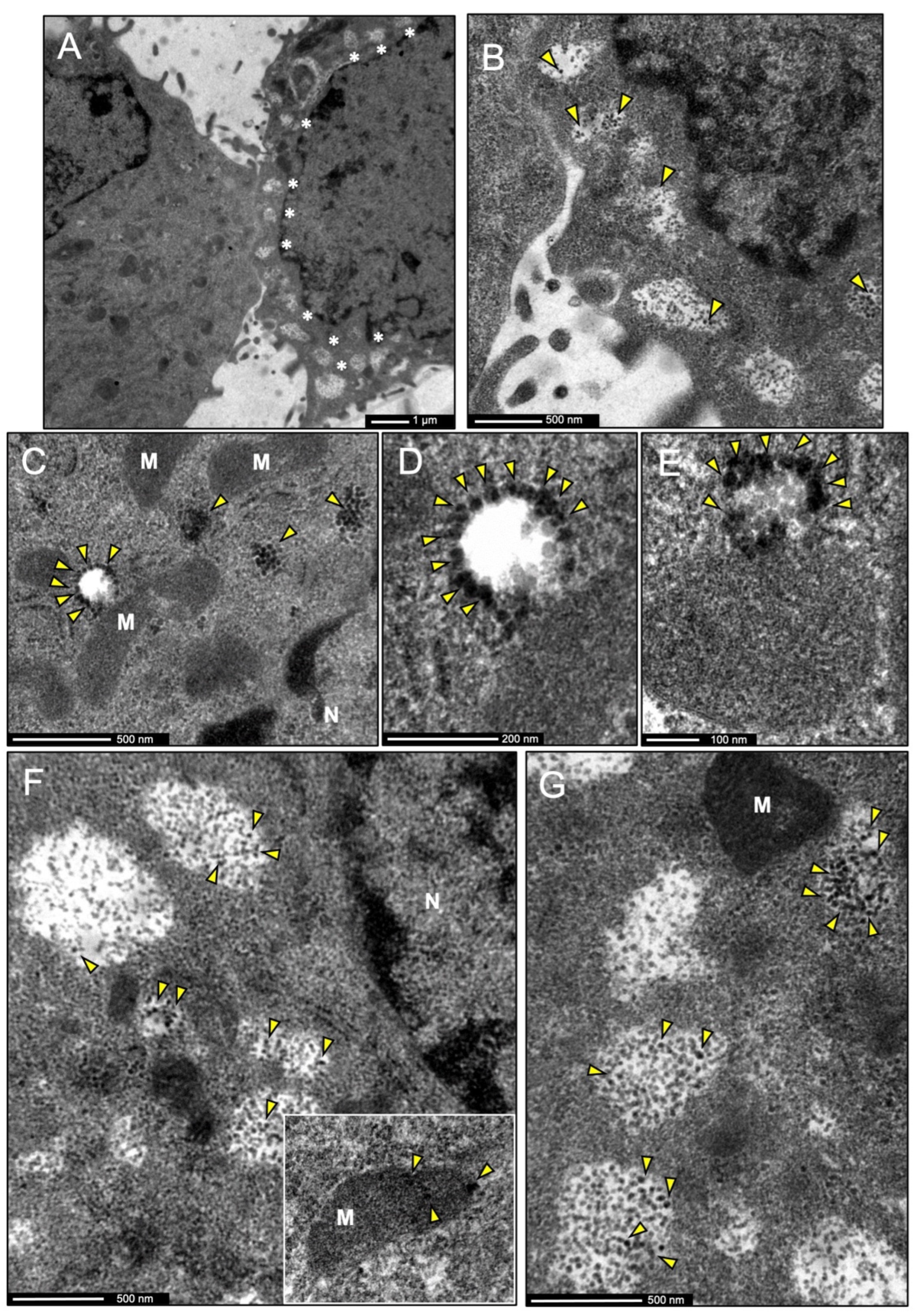
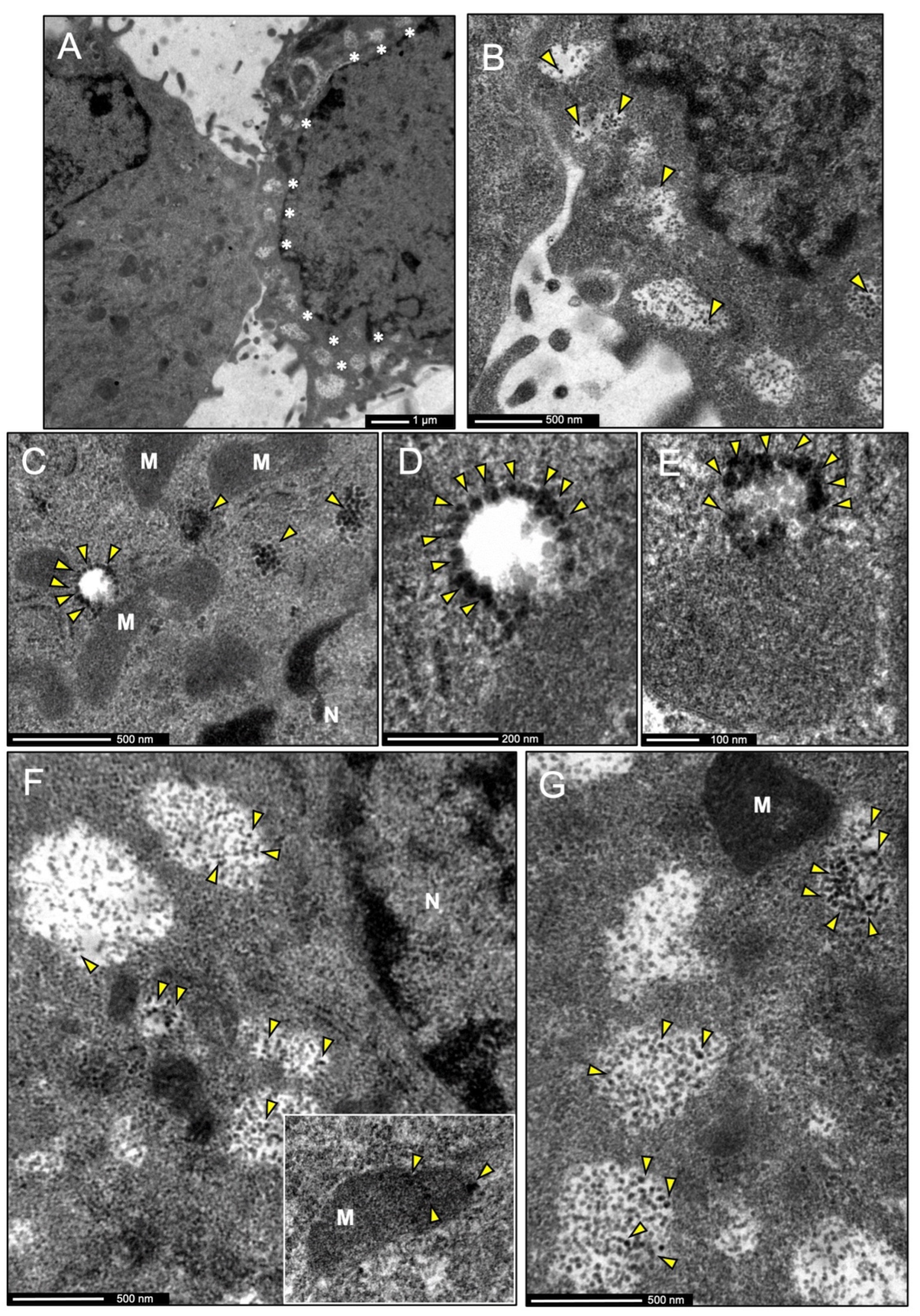
3.7. IDE Is Found in Multivesicular Bodies and Their Microvesicles in Secretory Microglia
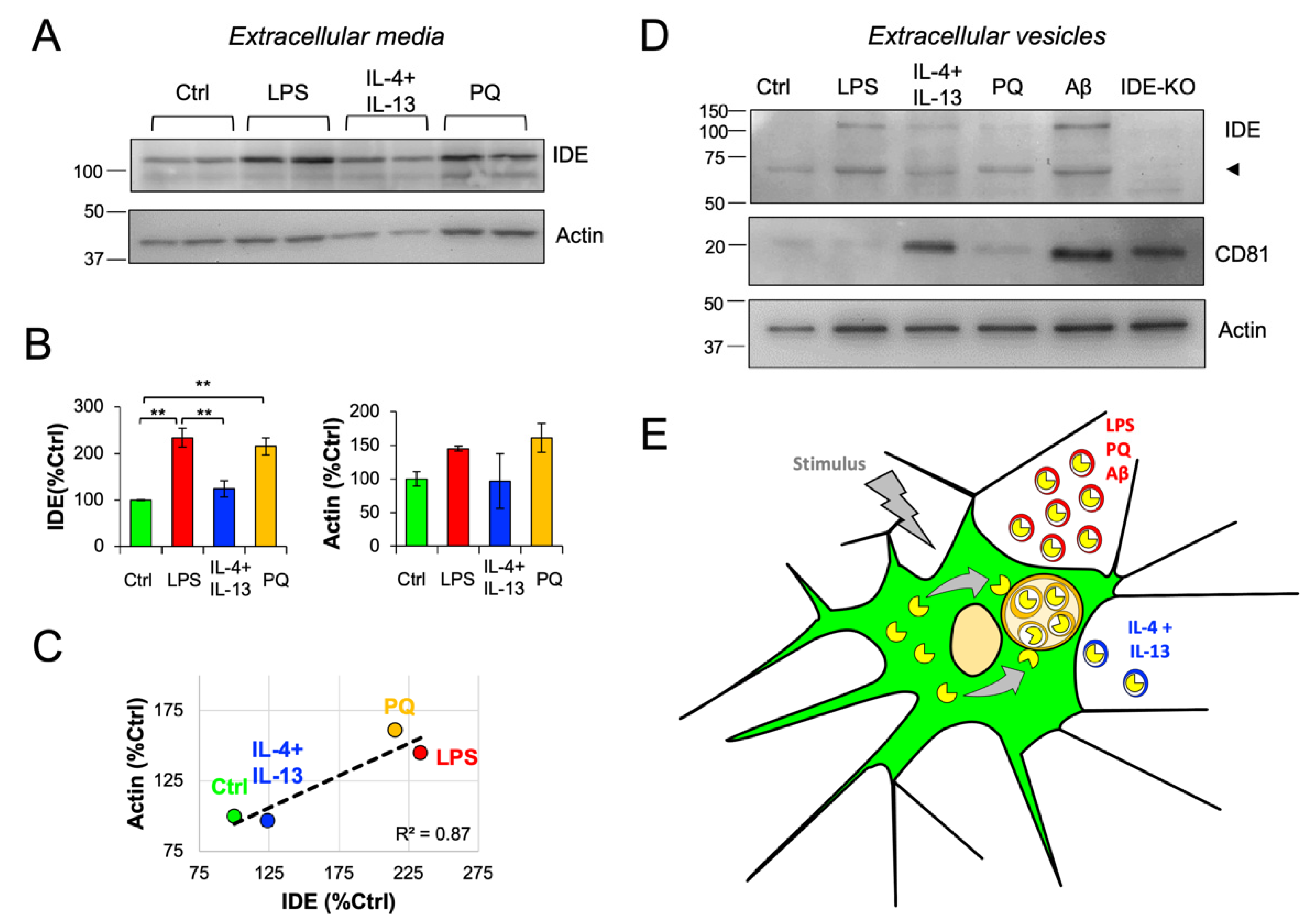
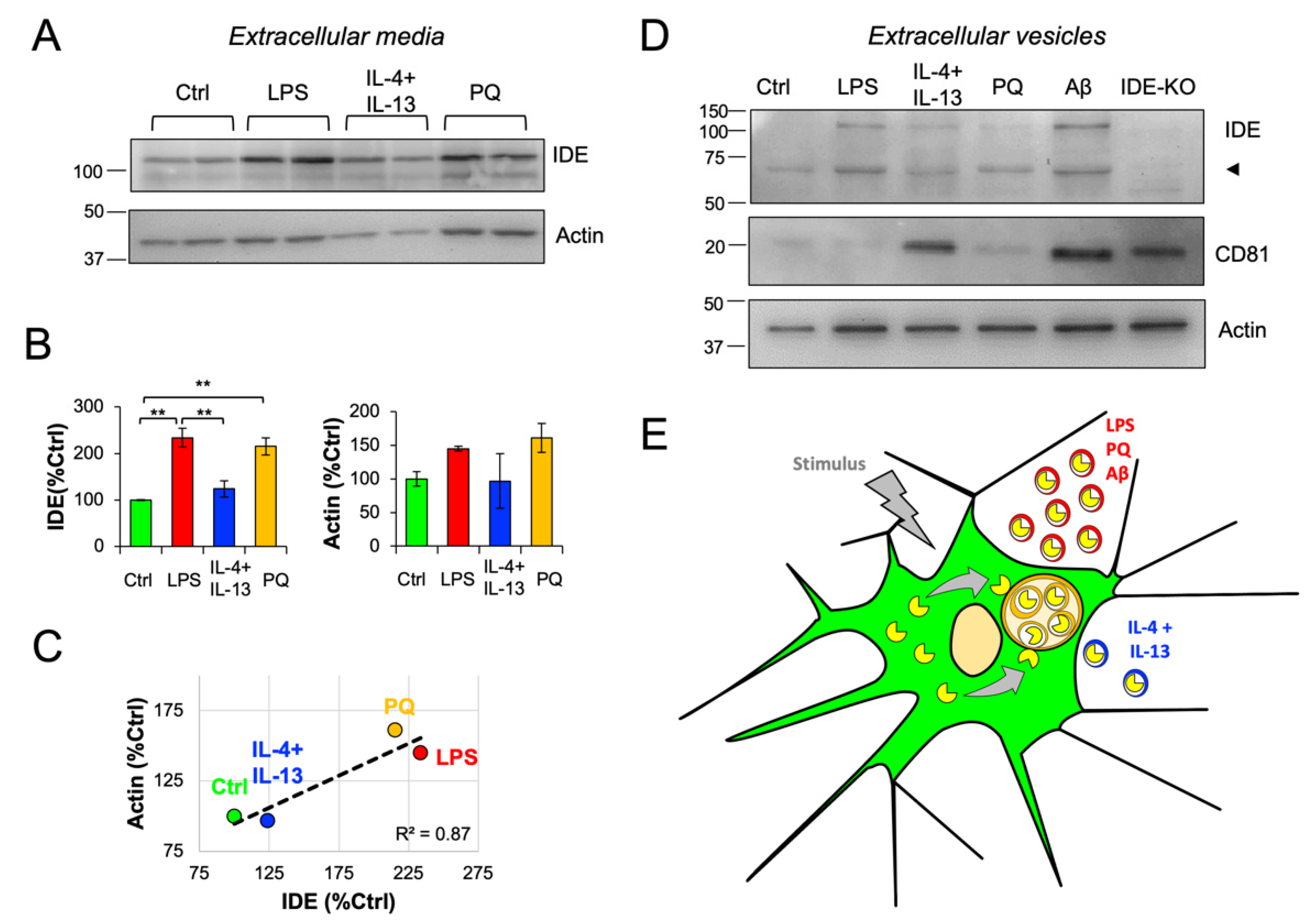
3.8. IDE Exportation in Extracellular Vesicles Is Regulated by the Activation State of Microglial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broh-Kahn, R.; Mirsky, I. The Inactivation of Insulin by Tissue Extracts; the Effect of Fasting on the Insulinase Content of Rat Liver. Arch. Biochem. 1949, 20, 10–14. [Google Scholar] [PubMed]
- Hooper, N.M. Families of Zinc metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Morton, F.R.; Kok, C.Y.; Kong, J.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2007, 36, D320–D325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, N.D.; Barrett, A.J. Introduction: Metallopeptidases and Their Clans. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 325–370. ISBN 978-0-12-382219-2. [Google Scholar]
- Kuo, W.L.; Montag, A.G.; Rosner, M.R. Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 1993, 132, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Folstein, M. Insulin, insulin-degrading enzyme and amyloid-β peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Shii, K.; Yokono, K.; Baba, S.; Roth, R.A. Purification and characterization of insulin-degrading enzyme from human erythrocytes. Diabetes 1986, 35, 675–683. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurochkin, I.V.; Goto, S. Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Shii, K.; Yokono, K.; Yonezawa, K.; Sato, S.; Watanabe, K.; Baba, S. Cellular localization of insulin-degrading enzyme in rat liver using monoclonal antibodies specific for this enzyme. Biochem. Biophys. Res. Commun. 1988, 155, 914–922. [Google Scholar] [CrossRef]
- Duckworth, W.C. Insulin Degradation: Mechanisms, products, and significance. Endocr. Rev. 1988, 9, 319–345. [Google Scholar] [CrossRef]
- Leissring, M.A.; Farris, W.; Wu, X.; Christodoulou, D.C.; Haigis, M.C.; Guarente, L.; Selkoe, D.J. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem. J. 2004, 383, 439–446. [Google Scholar] [CrossRef]
- Hamel, F.G.; Mahoney, M.J.; Duckworth, W.C. Degradation of intraendosomal insulin by insulin-degrading enzyme without acidification. Diabetes 1991, 40, 436–443. [Google Scholar] [CrossRef]
- Seabright, P.J.; Smith, G.D. The characterization of endosomal insulin degradation intermediates and their sequence of production. Biochem. J. 1996, 320, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Song, E.S.; Jang, H.; Guo, H.-F.; Juliano, M.A.; Juliano, L.; Morris, A.J.; Galperin, E.; Rodgers, D.W.; Hersh, L.B. Inositol phosphates and phosphoinositides activate insulin-degrading enzyme, while phosphoinositides also mediate binding to endosomes. Proc. Natl. Acad. Sci. USA 2017, 114, E2826–E2835. [Google Scholar] [CrossRef] [Green Version]
- Authier, F.; Bergeron, J.J.; Ou, W.J.; Rachubinski, R.A.; Posner, B.I.; Walton, P.A. Degradation of the cleaved leader peptide of thiolase by a peroxisomal proteinase. Proc. Natl. Acad. Sci. USA 1995, 92, 3859–3863. [Google Scholar] [CrossRef] [Green Version]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lütjohann, D.; Wunderlich, P.; et al. Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulloj, A.; Leal, M.C.; Xu, H.; Castaño, E.M.; Morelli, L. Insulin-degrading enzyme sorting in exosomes: A secretory pathway for a key brain amyloid-β degrading protease. J. Alzheimers Dis. 2010, 19, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, W.C. Insulin degradation by liver cell membranes. Endocrinology 1979, 104, 1758–1764. [Google Scholar] [CrossRef] [PubMed]
- Yokono, K.; Imamura, Y.; Sakai, H.; Baba, S. Insulin-degrading activity of plasma membranes from rat skeletal muscle: Its Isolation, characterization, and biologic significance. Diabetes 1979, 28, 810–817. [Google Scholar] [CrossRef]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Bulloj, A.; Leal, M.C.; Surace, E.I.; Zhang, X.; Xu, H.; Ledesma, M.D.; Castaño, E.M.; Morelli, L. Detergent resistant membrane-associated IDE in brain tissue and cultured cells: Relevance to Aβ and insulin degradation. Mol. Neurodegener. 2008, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Yokono, K.; Roth, R.A.; Baba, S. Identification of insulin-degrading enzyme on the surface of cultured human lymphocytes, rat hepatoma cells, and primary cultures of rat hepatocytes. Endocrinology 1982, 111, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, I.D.; Williams, J.A.; Bailey, A.C.; Wong, K.Y.; Iwamoto, Y.; Yokono, K.; Baba, S.; Roth, R.A. Degradation of insulin by isolated mouse pancreatic acini: Evidence for cell surface protease activity. Diabetes 1984, 33, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Glebov, K.; Löchner, M.; Jabs, R.; Lau, T.; Merkel, O.; Schloss, P.; Steinhäuser, C.; Walter, J. Serotonin stimulates secretion of exosomes from microglia cells: Serotonin stimulates microglial exosome release. Glia 2015, 63, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 7, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hatje, K.; Keller, O.; Hammesfahr, B.; Pillmann, H.; Waack, S.; Kollmar, M. Cross-species protein sequence and gene structure prediction with fine-tuned Webscipio 2.0 and scipio. BMC Res. Notes 2011, 4, 265. [Google Scholar] [CrossRef] [Green Version]
- Hammesfahr, B.; Odronitz, F.; Mühlhausen, S.; Waack, S.; Kollmar, M. GenePainter: A Fast tool for aligning gene structures of eukaryotic protein families, visualizing the alignments and mapping gene structures onto protein structures. BMC Bioinform. 2013, 14, 77. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Holm, L. Using Dali for protein structure comparison. In Structural Bioinformatics; Gáspári, Z., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2112, pp. 29–42. ISBN 978-1-07-160269-0. [Google Scholar]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hay, S.O.; Kang, D.; McBride, M.; Li, L.; Zhao, J.; Leissring, M.A. Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. PLoS ONE 2011, 6, e20818. [Google Scholar] [CrossRef] [Green Version]
- Saura, J.; Tusell, J.M.; Serratosa, J. High-yield isolation of murine microglia by mild trypsinization. Glia 2003, 44, 183–189. [Google Scholar] [CrossRef]
- Núñez, L.; Calvo-Rodríguez, M.; Caballero, E.; García-Durillo, M.; Villalobos, C. Neurotoxic Ca2+ Signaling induced by amyloid–β oligomers in aged hippocampal neurons in vitro. In Amyloid Proteins; Sigurdsson, E.M., Calero, M., Gasset, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1779, pp. 341–354. ISBN 978-1-4939-7815-1. [Google Scholar]
- Grassi, S.; Giussani, P.; Mauri, L.; Prioni, S.; Prinetti, A. Isolation and analysis of lipid rafts from neural cells and tissues. In Lipid Rafts; Bieberich, E., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2187, pp. 1–25. ISBN 978-1-07-160813-5. [Google Scholar]
- Fernandez-Diaz, C.M.; Merino, B.; Lopez-Acosta, J.F.; Cidad, P. Pancreatic beta-cell-specific deletion of insulin-degrading enzyme leads to dysregulated insulin secretion and beta-cell functional immaturity. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E805–E819. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, S.; Geisert, E.E.; Gispen, W.H.; Bär, P.R.; Joosten, E.A.J. Up-regulation of CD81 (target of the antiproliferative antibody; TAPA) by reactive microglia and astrocytes after spinal cord injury in the rat. J. Comp. Neurol. 2000, 428, 266–277. [Google Scholar] [CrossRef]
- Hönigschmid, P.; Bykova, N.; Schneider, R.; Ivankov, D.; Frishman, D. Evolutionary interplay between symbiotic relationships and patterns of signal peptide gain and loss. Genome Biol. Evol. 2018, 10, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A. Pitrilysin. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1412–1414. ISBN 978-0-12-382219-2. [Google Scholar]
- Mzhavia, N.; Devi, L.A. Eupitrilysin. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1457–1459. ISBN 978-0-12-382219-2. [Google Scholar]
- Kutejová, E.; Kučera, T.; Matušková, A.; Janata, J. Mitochondrial processing peptidase. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1435–1442. ISBN 978-0-12-382219-2. [Google Scholar]
- Lamppa, G.; Zhong, R. Chloroplast stromal processing peptidase. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1442–1447. ISBN 978-0-12-382219-2. [Google Scholar]
- Glaser, E.; Kmiec, B.; Teixeira, P.F. Mitochondrial and chloroplastic targeting peptides peptidase, PreP. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1426–1430. ISBN 978-0-12-382219-2. [Google Scholar]
- Fu, H.; Liu, B.; Li, L.; Lemere, C.A. Microglia do not take up soluble amyloid-beta peptides, but partially degrade them by secreting insulin-degrading enzyme. Neuroscience 2020, 443, 30–43. [Google Scholar] [CrossRef]
- De Dios, C.; Bartolessis, I.; Roca-Agujetas, V.; Barbero-Camps, E.; Mari, M.; Morales, A.; Colell, A. Oxidative inactivation of amyloid beta-degrading proteases by cholesterol-enhanced mitochondrial stress. Redox Biol. 2019, 26, 101283. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Cha, M.-Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.-S.; Park, S.A.; Mook-Jung, I. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in alzheimer disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. Insulin-degrading enzyme is not secreted from cultured cells. Sci. Rep. 2018, 8, 2335. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Kuo, W.-L.; Yousef, M.; Rosner, M.R.; Tang, W.-J. The C-terminal domain of human insulin degrading enzyme is required for dimerization and substrate recognition. Biochem. Biophys. Res. Commun. 2006, 343, 1032–1037. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Grasso, G.; Satriano, C.; Milardi, D. A neglected modulator of insulin-degrading enzyme activity and conformation: The pH. Biophys. Chem. 2015, 203–204, 33–40. [Google Scholar] [CrossRef]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Vicente Miranda, H. Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Family | Organism | Sequence ID |
|---|---|---|---|
| Pitrilysin | M16A | Escherichia coli | QIF70255 |
| Nardilysin | M16A | Homo sapiens | O43847 |
| IDE | M16A | Homo sapiens | P14735 |
| Ste23 | M16A | Saccharomyces cerevisiae | EWG94525 |
| Axl1p | M16A | Saccharomyces cerevisiae | NP_015447 |
| Plinsulysin | M16A | Solanum lycopersicum | NP_001233926 |
| SPP | M16B | Arabidopsis thaliana | Q9FIH8 |
| MPPA | M16B | Homo sapiens | Q10713 |
| MPPB | M16B | Homo sapiens | O75439 |
| YmxG | M16B | Rickettsia prowazekii | O05945 |
| PreP1 | M16C | Arabidopsis thaliana | Q9LJL3 |
| Eupitrilysin | M16C | Homo sapiens | Q5JRX3 |
| Falcilysin | M16C | Plasmodium falciparum | KAF4326450 |
| G1L | M44 | Vaccinia virus | P68493.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corraliza-Gómez, M.; Lillo, C.; Cózar-Castellano, I.; Arranz, E.; Sanchez, D.; Ganfornina, M.D. Evolutionary Origin of Insulin-Degrading Enzyme and Its Subcellular Localization and Secretion Mechanism: A Study in Microglial Cells. Cells 2022, 11, 227. https://doi.org/10.3390/cells11020227
Corraliza-Gómez M, Lillo C, Cózar-Castellano I, Arranz E, Sanchez D, Ganfornina MD. Evolutionary Origin of Insulin-Degrading Enzyme and Its Subcellular Localization and Secretion Mechanism: A Study in Microglial Cells. Cells. 2022; 11(2):227. https://doi.org/10.3390/cells11020227
Chicago/Turabian StyleCorraliza-Gómez, Miriam, Concepción Lillo, Irene Cózar-Castellano, Eduardo Arranz, Diego Sanchez, and Maria D. Ganfornina. 2022. "Evolutionary Origin of Insulin-Degrading Enzyme and Its Subcellular Localization and Secretion Mechanism: A Study in Microglial Cells" Cells 11, no. 2: 227. https://doi.org/10.3390/cells11020227
APA StyleCorraliza-Gómez, M., Lillo, C., Cózar-Castellano, I., Arranz, E., Sanchez, D., & Ganfornina, M. D. (2022). Evolutionary Origin of Insulin-Degrading Enzyme and Its Subcellular Localization and Secretion Mechanism: A Study in Microglial Cells. Cells, 11(2), 227. https://doi.org/10.3390/cells11020227