
Cell Cycle Regulation by Heat Shock Transcription Factors


{kind=link}
{kind=link}
Abstract
1. Introduction
- -
- The essential genes in the cell cycle were discovered through experiments using temperature-sensitive mutant yeast cells.
- -
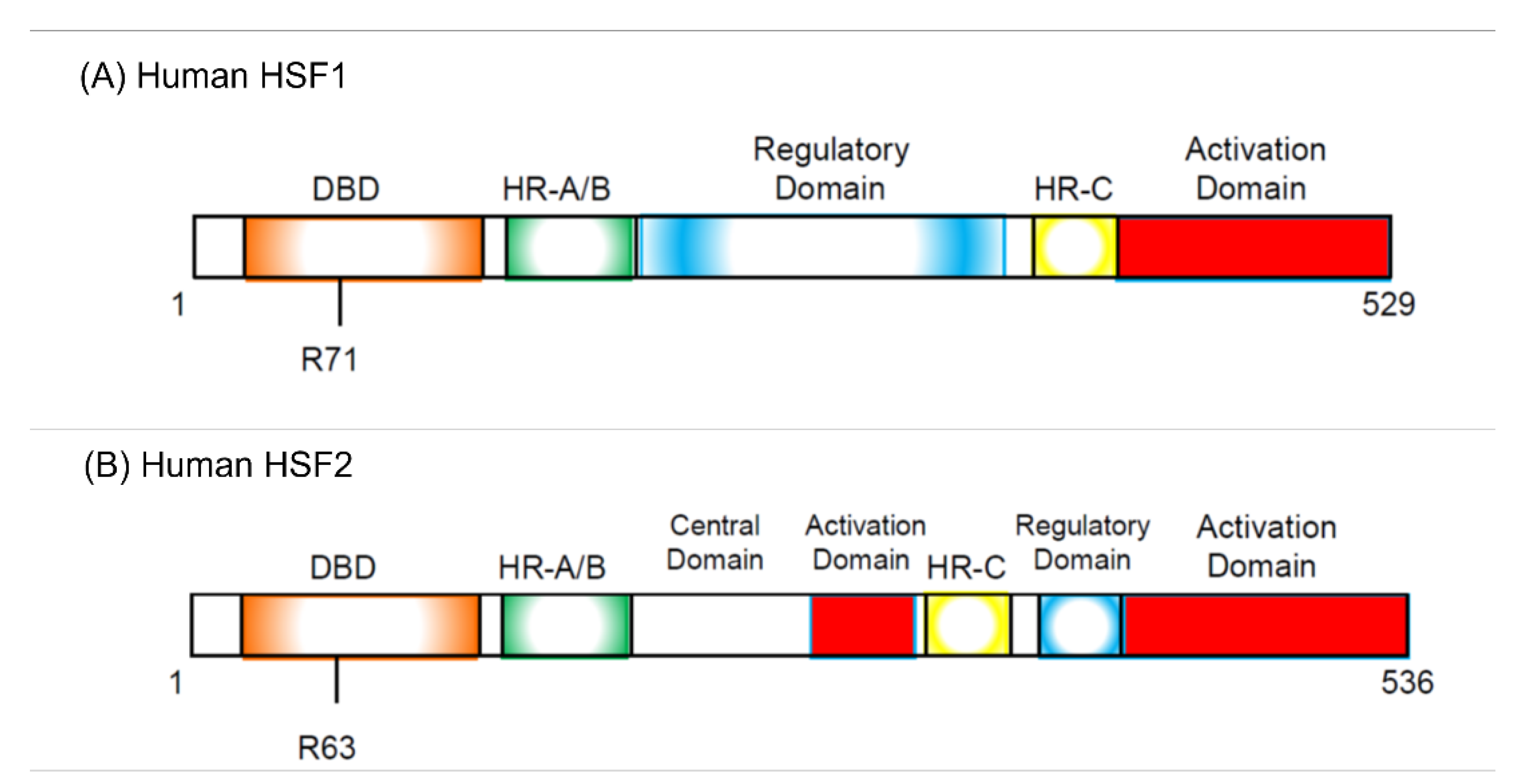
- HSFs were discovered as proteins specifically binding to HSE sequence commonly found in the HSP promoter.
2. Discovery of HSF Involvement in Cell Cycle Regulation
- -
- Mutant HSF1 affects cell cycle progression and arrest.
- -
- Higher expression of HSF1 is related to cancer and aneuploidy.
3. Bookmarking Is the Important Role of HSFs
- -
- HSF2 is the first transcription factor shown to have a gene bookmarking function and to bind to the gene promoter during mitosis.
- -
- Chromatin is not completely condensed. The structure is disturbed in some genes.
4. Change in HSF1 Expression Level Induces Cell Cycle Arrest
4.1. Decreased HSF1 Expression Suppresses Cancer Cell Proliferation
4.2. Overexpression of HSF1 Also Causes Suppression of Cancer Cell Proliferation
- -
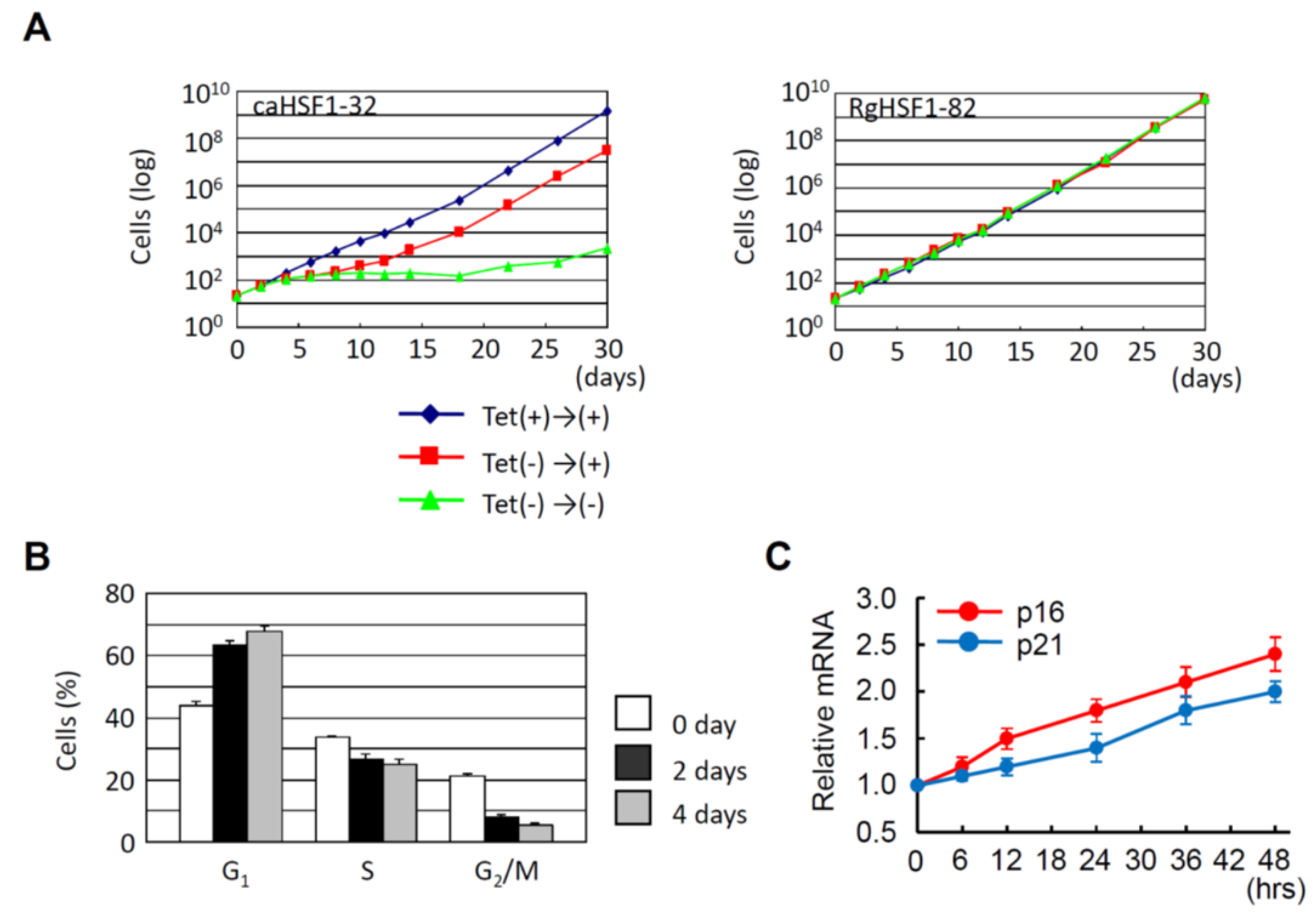
- Reduced HSF1 expression (induced by knockdown) inhibits the proliferation of cancer cells but not normal cells.
- -
- Constitutive active HSF1 expression also inhibits the proliferation of cancer cells probably through the induction of G1 cell cycle arrest.
5. Degradation of HSFs during Cell Cycle
5.1. Degradation of Cyclins and CDKs, Subunits of Cyclin-CDK Complex
5.2. HSFs Are Degraded by APC/C Complex
- -
- Both HSF1 and HSF2 are degraded by the APC/C complex with the same mechanism as cyclins.
6. HSFs Are Important for Meiosis
- -
- HSF1 and HSF2 are involved in meiosis as well as mitosis.
- -
- Spermatogenesis is affected by both HSF1 and HSF2. It is not clear whether oogenesis is also affected by both HSFs.
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hegner, R.W.; Russell, C.P. Differential Mitoses in the Germ-Cell Cycle of Dineutes Nigrior. Proc. Natl. Acad. Sci. USA 1916, 2, 356–360. [Google Scholar] [CrossRef][Green Version]
- Brooks, F.G. THE Germ Cell Cycle of the Digenetic Trematodes. Science 1928, 68, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.W.; Leduc, E. Stimulation of mitosis in mouse liver. Anat. Rec. 1946, 96, 534. [Google Scholar]
- Koller, P.C. Abnormal mitosis in tumours. Br. J. Cancer 1947, 1, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Elftman, H. The Sertoli cell cycle in the mouse. Anat. Rec. 1950, 106, 381–392. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.J. The metabolism of cell division. Br. J. Exp. Pathol. 1950, 31, 390–396. [Google Scholar] [PubMed]
- O’Connor, R.J. The effect on cell division of inhibiting aerobic glycolysis. Br. J. Exp. Pathol. 1950, 31, 449–453. [Google Scholar]
- Roberts, K.B.; Florey, H.W.; Joklik, W.K. The influence of Cortisone on cell division. Q. J. Exp. Physiol. Cogn. Med. Sci. 1952, 37, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.J. Cytoplasmic changes during cell division with reference to mitochondria and the golgi substance. Ann. N. Y. Acad. Sci. 1951, 51, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Elliot, E. Formation of new cell walls in cell division. Nature 1951, 168, 1089. [Google Scholar] [CrossRef]
- Bisset, K.A. Spurious mitotic spindles and fusion tubes in bacteria. Nature 1952, 169, 247. [Google Scholar] [CrossRef]
- DeLamater, E.D. Spurious mitotic spindles and fusion tubes in bacteria. Nature 1952, 169, 248. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, A.B. Formation of new cell walls in cell division. Nature 1952, 170, 329. [Google Scholar] [CrossRef] [PubMed]
- King, C.G. Diffuse centromere, and other cytological observations on two desmids. Nature 1953, 171, 181. [Google Scholar] [CrossRef] [PubMed]
- DeLamater, E.D. The mitotic mechanism in bacteria. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 99–100. [Google Scholar] [CrossRef]
- Boss, J. The spindle and the mechanism of chromosome separation as seen in the living cell. J. Physiol. 1953, 119, 34–36. [Google Scholar]
- Stevens, C.E.; Daoust, R.; Leblond, C.P. The desoxyribonucleic acid of interphase and dividing nuclei. Can. J. Med. Sci. 1953, 31, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, R. Production of three-group metaphases in the bone-marrow of the golden hamster. Nature 1953, 171, 1029–1030. [Google Scholar] [CrossRef]
- Fautrez, J.; Fautrez-Firlefyn, N. Deoxyribonucleic acid content of the cell nucleus and mitosis. Nature 1953, 172, 119–120. [Google Scholar] [CrossRef]
- Ogur, M.; Minckler, S.; McClary, D.O. Desoxyribonucleic acid and the budding cycle in the yeasts. J. Bacteriol. 1953, 66, 642–645. [Google Scholar] [CrossRef]
- Dawson, I.M.; Stern, H. Structure in the bacterial cell-wall during cell division. Biochim. Biophys. Acta 1954, 13, 31–40. [Google Scholar] [CrossRef]
- Hamperl, H. Three group metaphases and carcinoma in situ of the cervix uteri. Acta Unio Int. Contra Cancrum 1954, 10, 128–131. [Google Scholar]
- Hsu, T.C. Cytological studies on HeLa, a strain of human cervical carcinoma, I. Observations on mitosis and chromosomes. Tex. Rep. Biol. Med. 1954, 12, 833–846. [Google Scholar] [PubMed]
- Roles, H. Mitosis and deoxyribonucleic acid content of the nucleus. Nature 1954, 173, 1039–1040. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.J.; Culotti, J.G.; Nash, R.S.; Pringle, J.R. Forty-five years of cell-cycle genetics. Mol. Biol. Cell 2015, 26, 4307–4312. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Culotti, J.; Reid, B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. USA 1970, 66, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Mortimer, R.K.; Culotti, J.; Culotti, M. Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics 1973, 74, 267–286. [Google Scholar] [CrossRef]
- Hereford, L.M.; Hartwell, L.H. Sequential gene function in the initiation of Saccharomyces cerevisiae DNA synthesis. J. Mol. Biol. 1974, 84, 445–461. [Google Scholar] [CrossRef]
- Nasmyth, K.A.; Reed, S.I. Isolation of genes by complementation in yeast: Molecular cloning of a cell-cycle gene. Proc. Natl. Acad. Sci. USA 1980, 77, 2119–2123. [Google Scholar] [CrossRef]
- Nurse, P. Genetic control of cell size at cell division in yeast. Nature 1975, 256, 547–551. [Google Scholar] [CrossRef]
- Nurse, P.; Thuriaux, P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics 1980, 96, 627–637. [Google Scholar] [CrossRef]
- Beach, D.; Durkacz, B.; Nurse, P. Functionally homologous cell cycle control genes in budding and fission yeast. Nature 1982, 300, 706–709. [Google Scholar] [CrossRef]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Swenson, K.I.; Farrell, K.M.; Ruderman, J.V. The clam embryo protein cyclin A induces entry into M phase and the resumption of meiosis in Xenopus oocytes. Cell 1986, 47, 861–870. [Google Scholar] [CrossRef]
- Pines, J.; Hunt, T. Molecular cloning and characterization of the mRNA for cyclin from sea urchin eggs. EMBO J. 1987, 6, 2987–2995. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F. New puffing pattern induced by temperature shock and DNP in Drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Ritossa, F.M. Behavior of RNA and DNA synthesis at the puff level in salivary gland chromosomes of Drosopila. Exp. Cell Res. 1964, 36, 515–523. [Google Scholar] [CrossRef]
- Ritossa, F. Discovery of the heat shock response. Cell Stress Chaperones 1996, 1, 97–98. [Google Scholar] [CrossRef]
- Tissières, A.; Mitchell, H.K.; Tracy, U.M. Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J. Mol. Biol. 1974, 84, 389–398. [Google Scholar] [CrossRef]
- Schedl, P.; Artavanis-Tsakonas, S.; Steward, R.; Gehring, W.J.; Mirault, M.E.; Goldschmidt-Clermont, M.; Moran, L.; Tissières, A. Two hybrid plasmids with D. melanogaster DNA sequences complementary to mRNA coding for the major heat shock protein. Cell 1978, 14, 921–929. [Google Scholar] [CrossRef]
- Wu, C. Activating protein factor binds in vitro to upstream control sequences in heat shock gene chromatin. Nature 1984, 311, 81–84. [Google Scholar] [CrossRef]
- Wu, C. An exonuclease protection assay reveals heat-shock element and TATA box DNA-binding proteins in crude nuclear extracts. Nature 1985, 317, 84–87. [Google Scholar] [CrossRef]
- Kingston, R.E.; Schuetz, T.J.; Larin, Z. Heat-inducible human factor that binds to a human hsp70 promoter. Mol. Cell. Biol. 1987, 7, 1530–1534. [Google Scholar]
- Sorger, P.K.; Pelham, H.R. Purification and characterization of a heat-shock element binding protein from yeast. EMBO J. 1987, 6, 3035–3041. [Google Scholar] [CrossRef]
- Sorger, P.K.; Lewis, M.J.; Pelham, H.R. Heat shock factor is regulated differently in yeast and HeLa cells. Nature 1987, 329, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Wiederrecht, G.; Shuey, D.J.; Kibbe, W.A.; Parker, C.S. The Saccharomyces and Drosophila heat shock transcription factors are identical in size and DNA binding properties. Cell 1987, 48, 507–515. [Google Scholar] [CrossRef]
- Rabindran, S.K.; Giorgi, G.; Clos, J.; Wu, C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc. Natl. Acad. Sci. USA 1991, 88, 6906–6910. [Google Scholar] [CrossRef]
- Schuetz, T.J.; Gallo, G.J.; Sheldon, L.; Tempst, P.; Kingston, R.E. Isolation of a cDNA for HSF2: Evidence for two heat shock factor genes in humans. Proc. Natl. Acad. Sci. USA 1991, 88, 6911–6915. [Google Scholar] [CrossRef]
- Sarge, K.D.; Zimarino, V.; Holm, K.; Wu, C.; Morimoto, R.I. Cloning and characterization of two mouse heat shock factors with distinct inducible and constitutive DNA-binding ability. Genes. Dev. 1991, 5, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Sistonen, L.; Sarge, K.D.; Phillips, B.; Abravaya, K.; Morimoto, R.I. Activation of heat shock factor 2 during hemin-induced differentiation of human erythroleukemia cells. Mol. Cell. Biol. 1992, 12, 4104–4111. [Google Scholar] [PubMed]
- Smith, B.J.; Yaffe, M.P. A mutation in the yeast heat-shock factor gene causes temperature-sensitive defects in both mitochondrial protein import and the cell cycle. Mol. Cell. Biol. 1991, 11, 2647–2655. [Google Scholar] [PubMed]
- Morano, K.A.; Santoro, N.; Koch, K.A.; Thiele, D.J. A trans-activation domain in yeast heat shock transcription factor is essential for cell cycle progression during stress. Mol. Cell. Biol. 1999, 19, 402–411. [Google Scholar] [CrossRef]
- Luft, J.C.; Benjamin, I.J.; Mestril, R.; Dix, D.J. Heat shock factor 1-mediated thermotolerance prevents cell death and results in G2/M cell cycle arrest. Cell Stress Chaperones 2001, 6, 326–336. [Google Scholar] [CrossRef]
- Li, Q.; Martinez, J.D. Loss of HSF1 results in defective radiation-induced G(2) arrest and DNA repair. Radiat. Res. 2011, 176, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.L.; Chen, C.; Xie, Y.; Zhong, R.; Wang, Y.Q.; Stevenson, M.A.; Calderwood, S.K. Activation of heat shock transcription factor 1 to a DNA binding form during the G(1)phase of the cell cycle. Cell Stress Chaperones 1999, 4, 36–45. [Google Scholar] [CrossRef]
- He, L.; Fox, M.H. Activation of heat-shock transcription factor 1 in heated Chinese hamster ovary cells is dependent on the cell cycle and is inhibited by sodium vanadate. Radiat. Res. 1999, 151, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Venturi, C.B.; Erkine, A.M.; Gross, D.S. Cell cycle-dependent binding of yeast heat shock factor to nucleosomes. Mol. Cell. Biol. 2000, 20, 6435–6448. [Google Scholar] [CrossRef]
- Hoang, A.T.; Huang, J.; Rudra-Ganguly, N.; Zheng, J.; Powell, W.C.; Rabindran, S.K.; Wu, C.; Roy-Burman, P. A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am. J. Pathol. 2000, 156, 857–864. [Google Scholar] [CrossRef]
- Wang, Y.; Theriault, J.R.; He, H.; Gong, J.; Calderwood, S.K. Expression of a dominant negative heat shock factor-1 construct inhibits aneuploidy in prostate carcinoma cells. J. Biol. Chem. 2004, 279, 32651–32659. [Google Scholar] [CrossRef]
- Morano, K.A.; Thiele, D.J. Heat shock factor function and regulation in response to cellular stress, growth, and differentiation signals. Gene. Expr. 1999, 7, 271–282. [Google Scholar]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef]
- Pirkkala, L.; Nykänen, P.; Sistonen, L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001, 15, 1118–1131. [Google Scholar] [CrossRef]
- Xing, H.; Wilkerson, D.C.; Mayhew, C.N.; Lubert, E.J.; Skaggs, H.S.; Goodson, M.L.; Hong, Y.; Park-Sarge, O.K.; Sarge, K.D. Mechanism of hsp70i gene bookmarking. Science 2005, 307, 421–423. [Google Scholar] [CrossRef]
- Goodson, M.L.; Hong, Y.; Rogers, R.; Matunis, M.J.; Park-Sarge, O.K.; Sarge, K.D. Sumo-1 modification regulates the DNA binding activity of heat shock transcription factor 2, a promyelocytic leukemia nuclear body associated transcription factor. J. Biol. Chem. 2001, 276, 18513–18518. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, E.F.; Sanford, S.; Levens, D. Marking of active genes on mitotic chromosomes. Nature 1997, 388, 895–899. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Workman, J.L. Bookmarking genes for activation in condensed mitotic chromosomes. Bioessays 1998, 20, 275–279. [Google Scholar] [CrossRef]
- Follmer, N.E.; Francis, N.J. Speed reading for genes: Bookmarks set the pace. Dev. Cell 2011, 21, 807–808. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weintraub, H. Assembly of an active chromatin structure during replication. Nucleic Acids Res. 1979, 7, 781–792. [Google Scholar] [CrossRef]
- Struhl, G. A gene product required for correct initiation of segmental determination in Drosophila. Nature 1981, 293, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Groudine, M.; Weintraub, H. Propagation of globin DNAase I-hypersensitive sites in absence of factors required for induction: A possible mechanism for determination. Cell 1982, 30, 131–139. [Google Scholar] [CrossRef]
- Kadauke, S.; Udugama, M.I.; Pawlicki, J.M.; Achtman, J.C.; Jain, D.P.; Cheng, Y.; Hardison, R.C.; Blobel, G.A. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 2012, 150, 725–737. [Google Scholar] [CrossRef]
- Kadauke, S.; Blobel, G.A. Mitotic bookmarking by transcription factors. Epigenetics Chromatin 2013, 6, 6–10. [Google Scholar] [CrossRef]
- Caravaca, J.M.; Donahue, G.; Becker, J.S.; He, X.; Vinson, C.; Zaret, K.S. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013, 27, 251–260. [Google Scholar] [CrossRef]
- Lodhi, N.; Ji, Y.; Tulin, A. Mitotic bookmarking: Maintaining post-mitotic reprogramming of transcription reactivation. Curr. Mol. Biol. Rep. 2016, 2, 10–16. [Google Scholar] [CrossRef]
- Hong, Y.; Lubert, E.J.; Rodgers, D.W.; Sarge, K.D. Molecular basis of competition between HSF2 and catalytic subunit for binding to the PR65/A subunit of PP2A. Biochem. Biophys. Res. Commun. 2000, 272, 84–89. [Google Scholar] [CrossRef]
- Lubert, E.J.; Hong, Y.; Sarge, K.D. Interaction between protein phosphatase 5 and the A subunit of protein phosphatase 2A: Evidence for a heterotrimeric form of protein phosphatase 5. J. Biol. Chem. 2001, 276, 38582–38587. [Google Scholar] [CrossRef]
- Kimura, K.; Hirano, M.; Kobayashi, R.; Hirano, T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 1998, 282, 487–490. [Google Scholar] [CrossRef]
- Kimura, K.; Cuvier, O.; Hirano, T. Chromosome condensation by a human condensin complex in Xenopus egg extracts. J. Biol. Chem. 2001, 276, 5417–5420. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Vanderford, N.L.; Sarge, K.D. The TBP-PP2A mitotic complex bookmarks genes by preventing condensin action. Nat. Cell Biol. 2008, 10, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.A.; Wilkerson, D.C.; Hong, Y.; Sarge, K.D. PRC1 associates with the hsp70i promoter and interacts with HSF2 during mitosis. Exp. Cell Res. 2008, 314, 2224–2230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, W.; Jimenez, G.; Wells, N.J.; Hope, T.J.; Wahl, G.M.; Hunter, T.; Fukunaga, R. PRC1: A human mitotic spindle-associated CDK substrate protein required for cytokinesis. Mol. Cell 1998, 2, 877–885. [Google Scholar] [CrossRef]
- Mollinari, C.; Kleman, J.P.; Jiang, W.; Schoehn, G.; Hunter, T.; Margolis, R.L. PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J. Cell Biol. 2002, 157, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, N. Set1/MLL complex is indispensable for the transcriptional ability of heat shock transcription factor 2. Biochem. Biophys. Res. Commun. 2015, 467, 805–812. [Google Scholar] [CrossRef]
- Blobel, G.A.; Kadauke, S.; Wang, E.; Lau, A.W.; Zuber, J.; Chou, M.M.; Vakoc, C.R. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell 2009, 36, 970–983. [Google Scholar] [CrossRef]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef]
- Nakamura, Y.; Fujimoto, M.; Hayashida, N.; Takii, R.; Nakai, A.; Muto, M. Silencing HSF1 by short hairpin RNA decreases cell proliferation and enhances sensitivity to hyperthermia in human melanoma cell line. J. Dermatol. Sci. 2010, 60, 187–192. [Google Scholar] [CrossRef]
- van Bergen en Henegouwen, P.M.; Linnemans, A.M. Heat shock gene expression and cytoskeletal alterations in mouse neuroblastoma cells. Exp. Cell Res. 1987, 171, 367–375. [Google Scholar] [CrossRef]
- Comolli, R.; Frigerio, M.; Alberti, P. Heat shock, protein synthesis and ribosomal protein S6 phosphorylation in vitro in Yoshida AH 130 ascites hepatoma cells. Cell Biol. Int. Rep. 1988, 12, 907–917. [Google Scholar] [CrossRef]
- Ullrich, S.J.; Moore, S.K.; Appella, E. Transcriptional and translational analysis of the murine 84- and 86-kDa heat shock proteins. J. Biol. Chem. 1989, 264, 6810–6816. [Google Scholar] [CrossRef]
- Cairo, G.; Schiaffonati, L.; Rappocciolo, E.; Tacchini, L.; Bernelli-Zazzera, A. Expression of different members of heat shock protein 70 gene family in liver and hepatomas. Hepatology 1989, 9, 740–746. [Google Scholar] [CrossRef]
- Teves, S.S.; An, L.; Hansen, A.S.; Xie, L.; Darzacq, X.; Tjian, R. A dynamic mode of mitotic bookmarking by transcription factors. Elife 2016, 5, e22280. [Google Scholar] [CrossRef]
- Karagianni, P.; Moulos, P.; Schmidt, D.; Odom, D.T.; Talianidis, I. Bookmarking by Non-pioneer Transcription Factors during Liver Development Establishes Competence for Future Gene Activation. Cell Rep. 2020, 30, 1319–1328. [Google Scholar] [CrossRef]
- Das, S.; Min, S.; Prahlad, V. Gene bookmarking by the heat shock transcription factor programs the insulin-like signaling pathway. Mol. Cell 2021, 81, 1–18. [Google Scholar] [CrossRef]
- Fujimoto, M.; Takaki, E.; Hayashi, T.; Kitaura, Y.; Tanaka, Y.; Inouye, S.; Nakai, A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 2005, 280, 34908–34916. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, N.; Fujimoto, M.; Tan, K.; Prakasam, R.; Shinkawa, T.; Li, L.; Ichikawa, H.; Takii, R.; Nakai, A. Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J. 2010, 29, 3459–3469. [Google Scholar] [CrossRef]
- Momonaka, M.; Hayashida, K.; Hayashida, N. Unexpected Inhibition of cervical carcinoma cell proliferation by expression of heat shock transcription factor 1. Biomed. Res. Clin. Pract. 2016, 1, 2–6. [Google Scholar] [CrossRef]
- Venkatakrishnan, C.D.; Dunsmore, K.; Wong, H.; Roy, S.; Sen, C.K.; Wani, A.; Zweier, J.L.; Ilangovan, G. HSP27 regulates p53 transcriptional activity in doxorubicin-treated fibroblasts and cardiac H9c2 cells: p21 upregulation and G2/M phase cell cycle arrest. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1736–H1744. [Google Scholar] [CrossRef] [PubMed]
- Arellano, M.; Moreno, S. Regulation of CDK/cyclin complexes during the cell cycle. Int. J. Biochem. Cell Biol. 1997, 29, 559–573. [Google Scholar] [CrossRef]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How proteolysis drives the cell cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Yu, H.; Kirschner, M.W. Control of mitotic transitions by the anaphase-promoting complex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1583–1590. [Google Scholar] [CrossRef]
- Kramer, E.R.; Scheuringer, N.; Podtelejnikov, A.V.; Mann, M.; Peters, J.M. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 2000, 11, 1555–1569. [Google Scholar] [CrossRef]
- Schwab, M.; Lutum, A.S.; Seufert, W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 1997, 90, 683–693. [Google Scholar] [CrossRef]
- Sigrist, S.J.; Lehner, C.F. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell 1997, 90, 671–681. [Google Scholar] [CrossRef]
- Visintin, R.; Prinz, S.; Amon, A. CDC20 and CDH1: A family of substrate-specific activators of APC-dependent proteolysis. Science 1997, 278, 460–463. [Google Scholar] [CrossRef]
- Fang, G.; Yu, H.; Kirschner, M.W. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell 1998, 2, 163–171. [Google Scholar] [CrossRef]
- Kramer, E.R.; Gieffers, C.; Hölzl, G.; Hengstschläger, M.; Peters, J.M. Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr. Biol. 1998, 8, 1207–1210. [Google Scholar] [CrossRef]
- Zachariae, W.; Schwab, M.; Nasmyth, K.; Seufert, W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 1998, 282, 1721–1724. [Google Scholar] [CrossRef]
- Jaspersen, S.L.; Charles, J.F.; Morgan, D.O. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol. 1999, 9, 227–236. [Google Scholar] [CrossRef]
- Gieffers, C.; Peters, B.H.; Kramer, E.R.; Dotti, C.G.; Peters, J.M. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 11317–11322. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, H.J.; Lee, J.S.; Jeoung, D.; Kang, C.M.; Bae, S.; Lee, S.J.; Kwon, S.H.; Kang, D.; Lee, Y.S. A novel function for HSF1-induced mitotic exit failure and genomic instability through direct interaction between HSF1 and Cdc20. Oncogene 2008, 27, 2999–3009. [Google Scholar] [CrossRef]
- King, R.W.; Peters, J.M.; Tugendreich, S.; Rolfe, M.; Hieter, P.; Kirschner, M.W. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 1995, 81, 279–288. [Google Scholar] [CrossRef]
- Yu, H.; Peters, J.M.; King, R.W.; Page, A.M.; Hieter, P.; Kirschner, M.W. Identification of a cullin homology region in a subunit of the anaphase-promoting complex. Science 1998, 279, 1219–1222. [Google Scholar] [CrossRef]
- Kazemi-Sefat, G.E.; Keramatipour, M.; Talebi, S.; Kavousi, K.; Sajed, R.; Kazemi-Sefat, N.A. Mousavizadeh, K.; The importance of CDC27 in cancer: Molecular pathology and clinical aspects. Cancer Cell Int. 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, E.H.; Lee, J.S.; Jeoung, D.; Bae, S.; Kwon, S.H.; Lee, Y.S. HSF1 as a mitotic regulator: Phosphorylation of HSF1 by Plk1 is essential for mitotic progression. Cancer Res. 2008, 68, 7550–7560. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Björk, J.K.; Elsing, A.N.; Aspelin, C.; Kallio, M.; Roos-Mattjus, P.; Sistonen, L. Anaphase-promoting complex/cyclosome participates in the acute response to protein-damaging stress. Mol. Cell. Biol. 2010, 30, 5608–5620. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Sarge, K.D. Regulation of protein phosphatase 2A activity by heat shock transcription factor 2. J. Biol. Chem. 1999, 274, 12967–12970. [Google Scholar] [CrossRef] [PubMed]
- Lee, J. Roles of cohesin and condensin in chromosome dynamics during mammalian meiosis. J. Reprod. Dev. 2013, 59, 431–436. [Google Scholar] [CrossRef]
- Cloutier, J.M.; Turner, J.M. Meiotic sex chromosome inactivation. Curr. Biol. 2010, 20, R962–R963. [Google Scholar] [CrossRef]
- Akerfelt, M.; Vihervaara, A.; Laiho, A.; Conter, A.; Christians, E.S.; Sistonen, L.; Henriksson, E. Heat shock transcription factor 1 localizes to sex chromatin during meiotic repression. J. Biol. Chem. 2010, 285, 34469–34476. [Google Scholar] [CrossRef]
- Bierkamp, C.; Luxey, M.; Metchat, A.; Audouard, C.; Dumollard, R.; Christians, E. Lack of maternal Heat Shock Factor 1 results in multiple cellular and developmental defects, including mitochondrial damage and altered redox homeostasis, and leads to reduced survival of mammalian oocytes and embryos. Dev. Biol. 2010, 339, 338–353. [Google Scholar] [CrossRef]
- Christians, E.; Davis, A.A.; Thomas, S.D.; Benjamin, I.J. Maternal effect of Hsf1 on reproductive success. Nature 2000, 407, 693–694. [Google Scholar] [CrossRef] [PubMed]
- Kallio, M.; Chang, Y.; Manuel, M.; Alastalo, T.P.; Rallu, M.; Gitton, Y.; Pirkkala, L.; Loones, M.T.; Paslaru, L.; Larney, S.; et al. Brain abnormalities, defective meiotic chromosome synapsis and female subfertility in HSF2 null mice. EMBO J. 2002, 21, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J.; Moskophidis, D.; Mivechi, N.F. Targeted disruption of the heat shock transcription factor (hsf)-2 gene results in increased embryonic lethality, neuronal defects, and reduced spermatogenesis. Genesis 2003, 36, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashida, N.; Katoh, T.; Oshima, K.; Shinkawa, T.; Prakasam, R.; Tan, K.; Inouye, S.; Takii, R.; Nakai, A. A novel mouse HSF3 has the potential to activate nonclassical heat-shock genes during heat shock. Mol. Biol. Cell 2010, 21, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Sasai, N.; Nagata, K.; Liu, X.D.; Liu, P.C.; Thiele, D.J.; Nakai, A. The mammalian HSF4 gene generates both an activator and a repressor of heat shock genes by alternative splicing. J. Biol. Chem. 1999, 274, 27845–27856. [Google Scholar] [CrossRef] [PubMed]
- Tu, N.; Hu, Y.; Mivechi, N.F. Heat shock transcription factor (Hsf)-4b recruits Brg1 during the G1 phase of the cell cycle and regulates the expression of heat shock proteins. J. Cell Biochem. 2006, 98, 1528–1542. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Logan, I.R.; McNeill, H.V.; Cook, S.; Lu, X.; Meek, D.W.; Fuller-Pace, F.V.; Lunec, J.; Robson, C.N. Heat shock factor-1 modulates p53 activity in the transcriptional response to DNA damage. Nucleic Acids Res. 2009, 37, 2962–2973. [Google Scholar] [CrossRef]
- Xu, C.; Min, J. Structure and function of WD40 domain proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Page, A.M.; Hieter, P. The anaphase-promoting complex: New subunits and regulators. Annu. Rev. Biochem. 1999, 68, 583–609. [Google Scholar] [CrossRef]
- Higa, L.A.; Zhang, H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007, 2, 5. [Google Scholar] [CrossRef][Green Version]
- Yu, H. Cdc20: A WD40 activator for a cell cycle degradation machine. Mol. Cell 2007, 27, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Brugh, V.M.; Lipshultz, L.I. Male factor infertility: Evaluation and management. Med. Clin. N. Am. 2004, 88, 367–385. [Google Scholar] [CrossRef]
- Borgers, M.; Wolter, M.; Hentrich, A.; Bergmann, M.; Stammler, A.; Konrad, L. Role of compensatory meiosis mechanisms in human spermatogenesis. Reproduction 2014, 148, 315–320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shinka, T.; Sato, Y.; Chen, G.; Naroda, T.; Kinoshita, K.; Unemi, Y.; Tsuji, K.; Toida, K.; Iwamoto, T.; Nakahori, Y. Molecular characterization of heat shock-like factor encoded on the human Y chromosome, and implications for male infertility. Biol. Reprod. 2004, 71, 297–306. [Google Scholar] [CrossRef]
- Tessari, A.; Salata, E.; Ferlin, A.; Bartoloni, L.; Slongo, M.L.; Foresta, C. Characterization of HSFY, a novel AZFb gene on the Y chromosome with a possible role in human spermatogenesis. Mol. Hum. Reprod. 2004, 10, 253–258. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokunaga, Y.; Otsuyama, K.-I.; Hayashida, N. Cell Cycle Regulation by Heat Shock Transcription Factors. Cells 2022, 11, 203. https://doi.org/10.3390/cells11020203
Tokunaga Y, Otsuyama K-I, Hayashida N. Cell Cycle Regulation by Heat Shock Transcription Factors. Cells. 2022; 11(2):203. https://doi.org/10.3390/cells11020203
Chicago/Turabian StyleTokunaga, Yasuko, Ken-Ichiro Otsuyama, and Naoki Hayashida. 2022. "Cell Cycle Regulation by Heat Shock Transcription Factors" Cells 11, no. 2: 203. https://doi.org/10.3390/cells11020203
APA StyleTokunaga, Y., Otsuyama, K.-I., & Hayashida, N. (2022). Cell Cycle Regulation by Heat Shock Transcription Factors. Cells, 11(2), 203. https://doi.org/10.3390/cells11020203