
A Novel Missense Mutation in ERCC8 Co-Segregates with Cerebellar Ataxia in a Consanguineous Pakistani Family

 , , , ,
, , , ,
Abstract
1. Introduction
2. Material and Methods
2.1. Participants and Procedure
2.2. Genetic Analysis
2.3. In Silico Modelling of Mutant Protein
2.4. Biochemical Analysis of Mutant ERCC8
3. Results
3.1. Clinical Presentations
3.2. Magnetic Resonance Imaging (MRI)
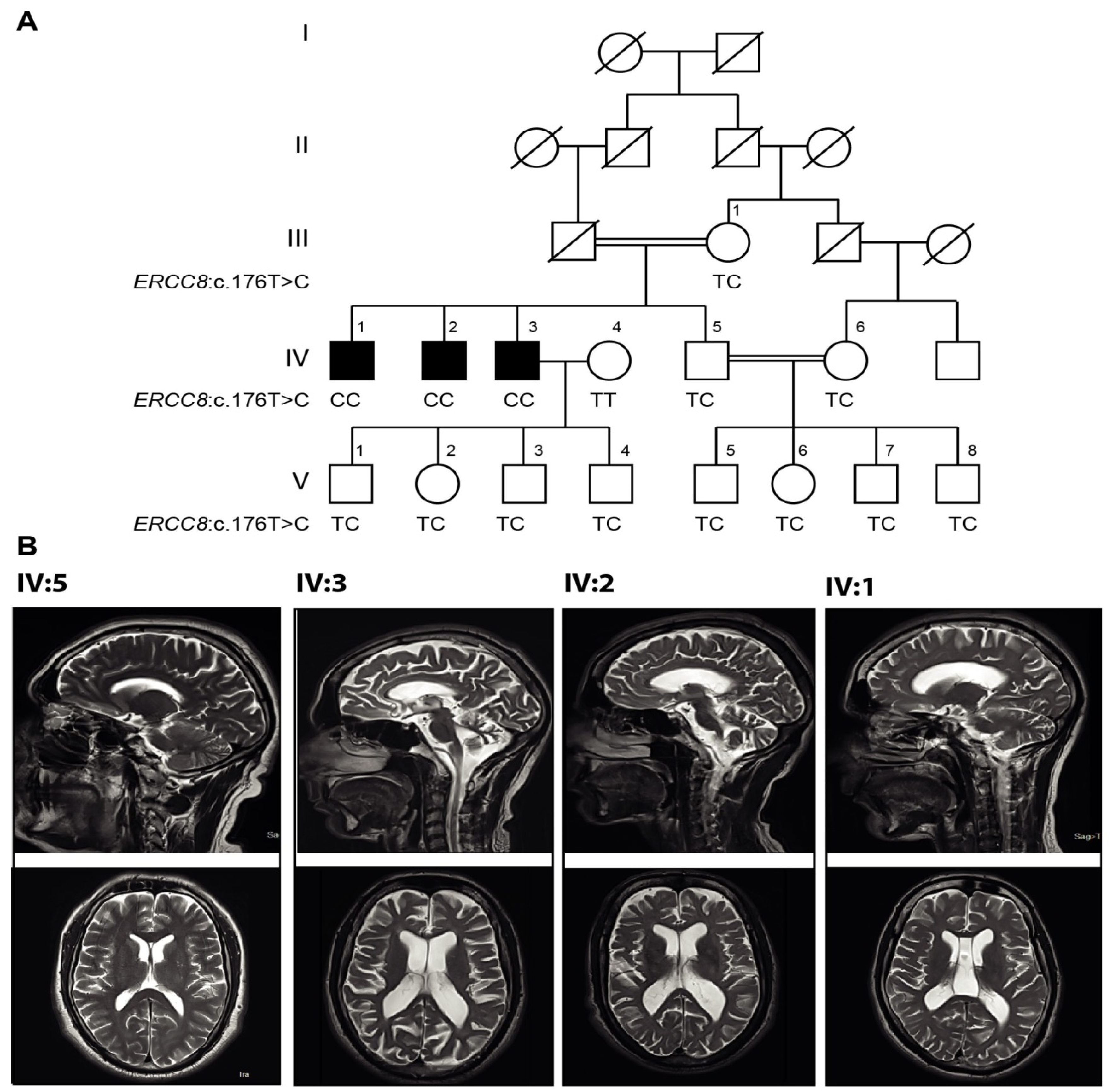
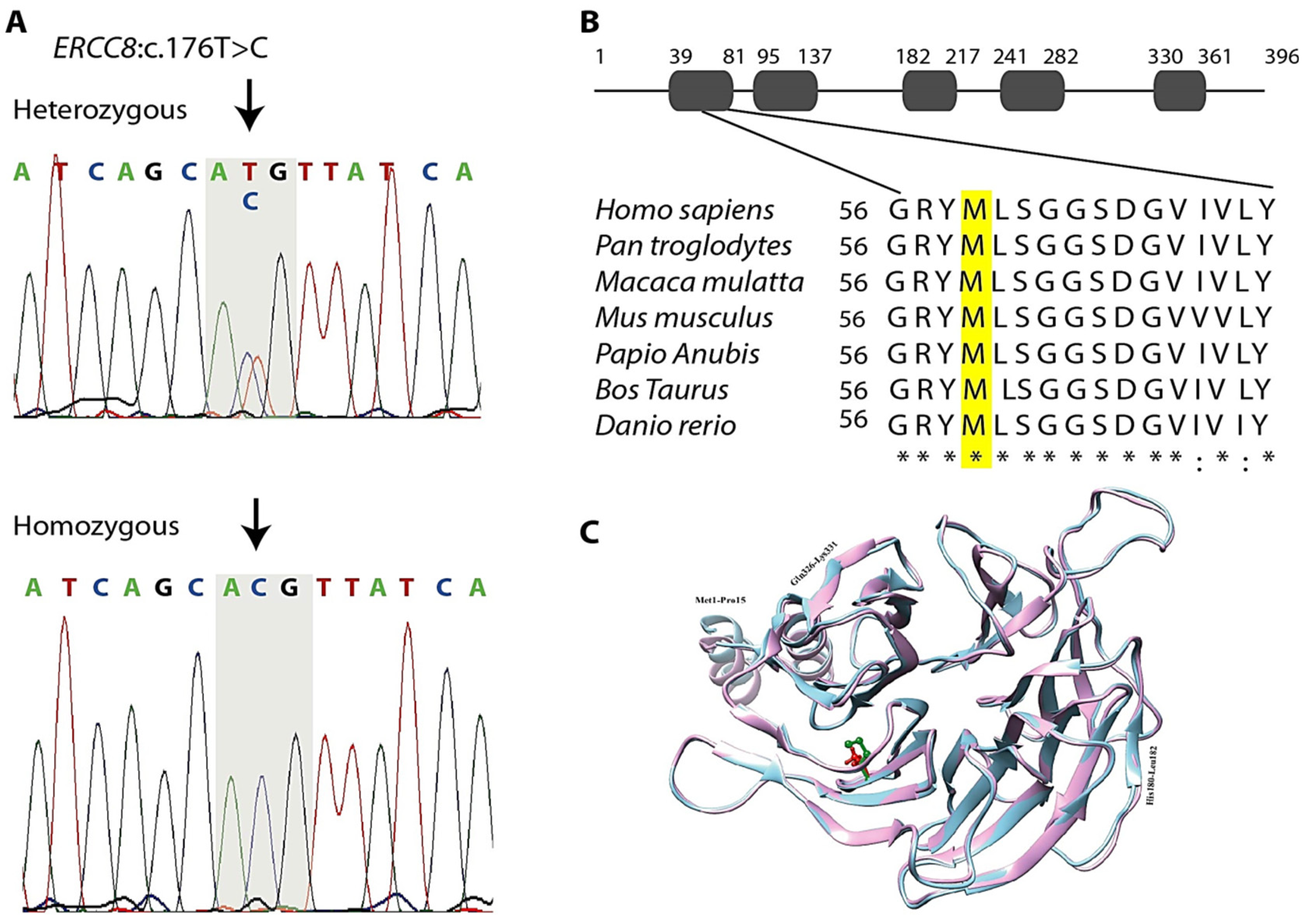
3.3. Genetic Findings
3.4. In Silico Analysis
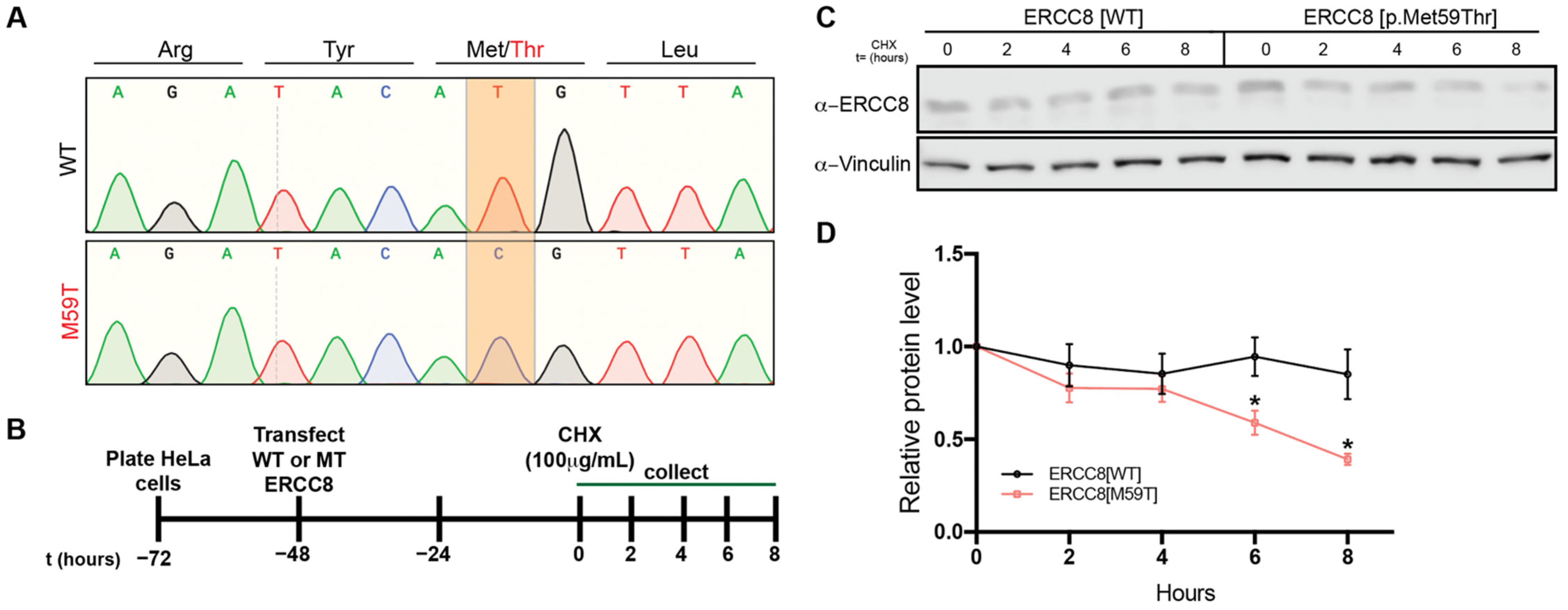
3.5. M59 Mutation Destablizes ERCC8 Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anheim, M.; Tranchant, C.; Koenig, M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012, 366, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Németh, A.H. Recessive ataxias. Handb. Clin. Neurol. 2018, 155, 73–89. [Google Scholar] [PubMed]
- Harding, A. Classification of the hereditary ataxias and paraplegias. Lancet 1983, 321, 1151–1155. [Google Scholar] [CrossRef]
- Bird, T.D. Hereditary Ataxia Overview. In Gene Reviews; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Rossi, M.; Anheim, M.; Durr, A.; Klein, C.; Koenig, M.; Synofzik, M.; Marras, C.; van de Warrenburg, B.P.; International Parkinson and Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders. The genetic nomenclature of recessive cerebellar ataxias. Mov. Disord. 2018, 33, 1056–1076. [Google Scholar] [CrossRef]
- Synofzik, M.; Puccio, H.; Mochel, F.; Schöls, L. Autosomal recessive cerebellar ataxias: Paving the way toward targeted molecular therapies. Neuron 2019, 101, 560–583. [Google Scholar] [CrossRef]
- Beaudin, M.; Matilla-Dueñas, A.; Soong, B.-W.; Pedroso, J.L.; Barsottini, O.G.; Mitoma, H.; Tsuji, S.; Schmahmann, J.D.; Manto, M.; Rouleau, G.A. The classification of autosomal recessive cerebellar ataxias: A consensus statement from the society for research on the cerebellum and ataxias task force. Cerebellum 2019, 18, 1098–1125. [Google Scholar] [CrossRef]
- Koch, S.; Garcia Gonzalez, O.; Assfalg, R.; Schelling, A.; Schäfer, P.; Scharffetter-Kochanek, K.; Iben, S. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle 2014, 13, 2029–2037. [Google Scholar] [CrossRef]
- Henning, K.A.; Li, L.; Iyer, N.; McDaniel, L.D.; Reagan, M.S.; Legerski, R.; Schultz, R.A.; Stefanini, M.; Lehmann, A.R.; Mayne, L.V. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 1995, 82, 555–564. [Google Scholar] [CrossRef]
- Calmels, N.; Botta, E.; Jia, N.; Fawcett, H.; Nardo, T.; Nakazawa, Y.; Lanzafame, M.; Moriwaki, S.; Sugita, K.; Kubota, M. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 2018, 55, 329–343. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, L.; Chen, F.; Yao, Z.; Li, M. Two novel mutations in the ERCC8 gene in a patient with ultraviolet-sensitive syndrome. Acta Derm. Venereol. 2019, 99, 117–118. [Google Scholar] [CrossRef]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med. Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Planas-Ballvé, A.; Morales-Briceño, H.; Crespo-Cuevas, A.M.; Fung, V.S. Movement Disorders and Spinal Cord Atrophy in Cockayne Syndrome with Prolonged Survival. Mov. Disord. Clin. Pract. 2018, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Chikhaoui, A.; Kraoua, I.; Calmels, N.; Bouchoucha, S.; Obringer, C.; Zayoud, K.; Montagne, B.; M’rad, R.; Abdelhak, S.; Laugel, V. Heterogeneous clinical features in Cockayne syndrome patients and siblings carrying the same CSA mutations. Orphanet J. Rare. Dis. 2022, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Dai, L.; Zhou, Z.; Hu, J.; Bai, Y.; Guo, H. Homozygosity mapping and whole exome sequencing reveal a novel ERCC8 mutation in a Chinese consanguineous family with unique cerebellar ataxia. Clin. Chim. Acta 2019, 494, 64–70. [Google Scholar] [CrossRef]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 393. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. In Structural Genomics; Springer: Amsterdam, The Netherlands, 2021; pp. 239–255. [Google Scholar]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Dong, T.; Tejwani, L.; Jung, Y.; Kokubu, H.; Luttik, K.; Driessen, T.M.; Lim, J. Microglia regulate brain progranulin levels through the endocytosis/lysosomal pathway. JCI Insight 2021, 6, e136147. [Google Scholar] [CrossRef]
- Algahtani, H.; Shirah, B.; Almatrafi, S.; Al-Qahtani, M.H.; Abdulkareem, A.A.; Naseer, M.I. A novel variant in CWF19L1 gene in a family with late-onset autosomal recessive cerebellar ataxia 17. Neurol. Res. 2021, 43, 141–147. [Google Scholar] [CrossRef]
- Fousteri, M.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008, 18, 73–84. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing. Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. UV-sensitive syndrome. Mutat. Res. 2005, 577, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Yousefipour, F.; Mozhdehipanah, H.; Mahjoubi, F. Identification of two novel homozygous nonsense mutations in TRAPPC9 in two unrelated consanguineous families with intellectual Disability from Iran. Mol. Genet. Genomic. Med. 2021, 9, e1610. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Scrima, A.; Böhm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Saijo, M. The role of Cockayne syndrome group A (CSA) protein in transcription-coupled nucleotide excision repair. Mech. Ageing. Dev. 2013, 134, 196–201. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244.e24. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient ID (Sex) | IV:1 (Male) | IV:2 (Male) | IV:3 (Male) |
|---|---|---|---|
| ERCC8 mutation | c.176T>C homozygous | c.176T>C homozygous | c.176T>C homozygous |
| Parental Consanguinity | + | + | + |
| Age of onset (years) | 10 | 12 | 15 |
| Age at examination | 30 | 37 | 43 |
| Symptoms at onset | Involuntary movement of hands | Involuntary hands and head movement | Involuntary hands movement |
| Marital status | Single | Single | Married |
| Microcephaly | − | − | − |
| Kyphosis | − | − | − |
| Ankylosis | − | − | − |
| Optic atrophy | − | − | − |
| Growth retardation | − | − | − |
| Facial features | Normal | Normal | Normal |
| Sun burn | + | + | + |
| Cognition | Normal | Normal | Normal |
| Impaired coordination of legs & arms | + | + | + |
| Difficulty in writing | + | + | + |
| Difficulty in handling objects | + | + | + |
| Dysarthria | + | + | + |
| Dysphagia | + | + | + |
| Nystagmus | − | − | − |
| Diplopia | + | + | + |
| Visual loss | − | − | − |
| Grip | Normal | Normal | Normal |
| Reflexes | Intact | Upper limb depressed, normal knee absent at ankle | Upper limb and ankle jerk depressed, knee jerk positive |
| Plantar response | Down-going | Withdrawal, nonspecific | Down-going |
| Finger nose test | + | + | + |
| Heel shin test | + | + | + |
| Romberg test | − | − | − |
| Gait ataxia | + | + | + |
| Skin pigmentation | Normal | Normal | Hyper |
| Fallen wound sign | − | − | + |
| Cranial nerve | Normal | Normal | Normal |
| Pin prick | Normal | Normal | Normal |
| others | Divergent squint | Head titubation | None |
| Cerebellar atrophy | very mild | + | + |
| Cerebral atrophy | − | + | + |
| Gene | Chr | Pos | Ref Allele | Alt Allele | Mutation Type | Aa Change | Allele Frequency | Meta Svm | Cadd Score | %Genic Intolerance Score Based on Exac | Omim/ Diseases | ACMG Classification | Segregation Status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERCC8 | 5 | 60217980 | A | G | Missense | p.M59T | 0.000008018 | D | 20.4 | 53.24 | 609412/Cockayne syndrome type-A, UV-sensitive syndrome 2 | Likely pathogenic (II) | Segregated |
| MX2 | 21 | 42749772 | C | A | Missense | p.D102E | 0.000007956 | D | 16.39 | 29.62 | 147890/Nil | Variant of Uncertain Significance | No Segregation |
| PLA2G12A | 4 | 110638815 | A | G | Missense | p.Y114H | 0.00001989 | T | 24.9 | 74.21 | 611652/Nil | Variant of Uncertain Significance | No Segregation |
| PLA2G12A | 4 | 110650941 | G | A | Missense | p.L9F | 0.00002486 | T | 19.74 | 74.21 | 611652/Nil | Variant of Uncertain Significance | No Segregation |
| GEMIN8 | X | 14027232 | C | T | Missense | p.D177N | Not available | T | 16.84 | 57.84 | 300962/Nil | Likely benign (I) | No Segregation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauhar, Z.; Tejwani, L.; Abdullah, U.; Saeed, S.; Shafique, S.; Badshah, M.; Choi, J.; Dong, W.; Nelson-Williams, C.; Lifton, R.P.; et al. A Novel Missense Mutation in ERCC8 Co-Segregates with Cerebellar Ataxia in a Consanguineous Pakistani Family. Cells 2022, 11, 3090. https://doi.org/10.3390/cells11193090
Gauhar Z, Tejwani L, Abdullah U, Saeed S, Shafique S, Badshah M, Choi J, Dong W, Nelson-Williams C, Lifton RP, et al. A Novel Missense Mutation in ERCC8 Co-Segregates with Cerebellar Ataxia in a Consanguineous Pakistani Family. Cells. 2022; 11(19):3090. https://doi.org/10.3390/cells11193090
Chicago/Turabian StyleGauhar, Zeeshan, Leon Tejwani, Uzma Abdullah, Sadia Saeed, Shagufta Shafique, Mazhar Badshah, Jungmin Choi, Weilai Dong, Carol Nelson-Williams, Richard P. Lifton, and et al. 2022. "A Novel Missense Mutation in ERCC8 Co-Segregates with Cerebellar Ataxia in a Consanguineous Pakistani Family" Cells 11, no. 19: 3090. https://doi.org/10.3390/cells11193090
APA StyleGauhar, Z., Tejwani, L., Abdullah, U., Saeed, S., Shafique, S., Badshah, M., Choi, J., Dong, W., Nelson-Williams, C., Lifton, R. P., Lim, J., & Raja, G. K. (2022). A Novel Missense Mutation in ERCC8 Co-Segregates with Cerebellar Ataxia in a Consanguineous Pakistani Family. Cells, 11(19), 3090. https://doi.org/10.3390/cells11193090