
Platelets and the Cybernetic Regulation of Ischemic Inflammatory Responses through PNC Formation Regulated by Extracellular Nucleotide Metabolism and Signaling

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethic Statement
2.2. Isolation of Human Platelets
2.3. ADP Release from Human Platelets
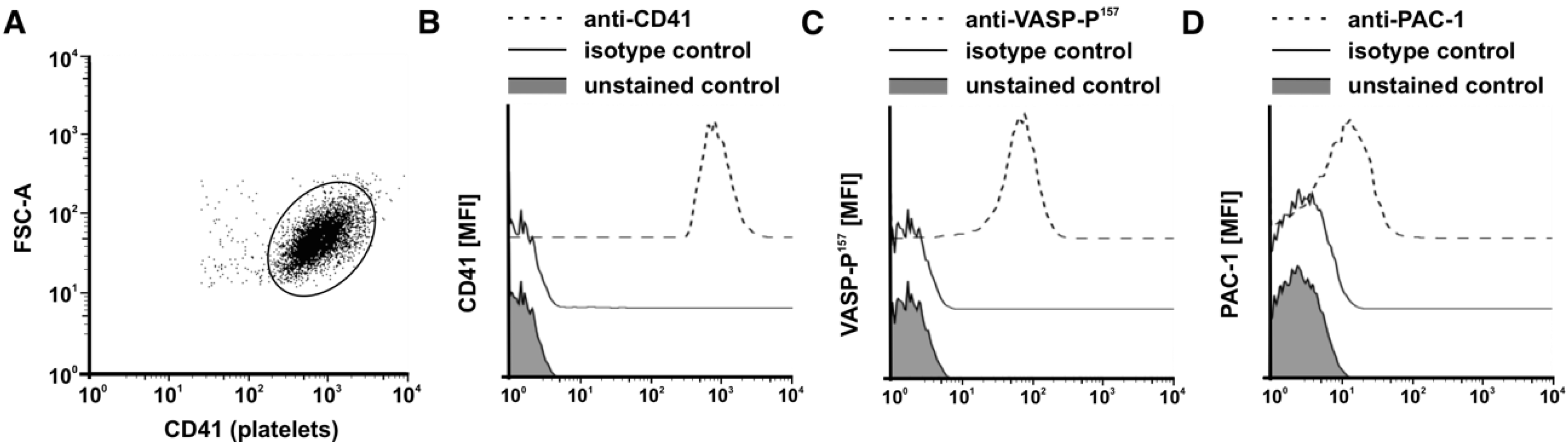
2.4. Flow Cytometry of Human PRP
2.5. Flow Cytometry of Murine Whole Blood Samples
2.6. Hepatic and Myocardial Ischemia Reperfusion Model in Mice
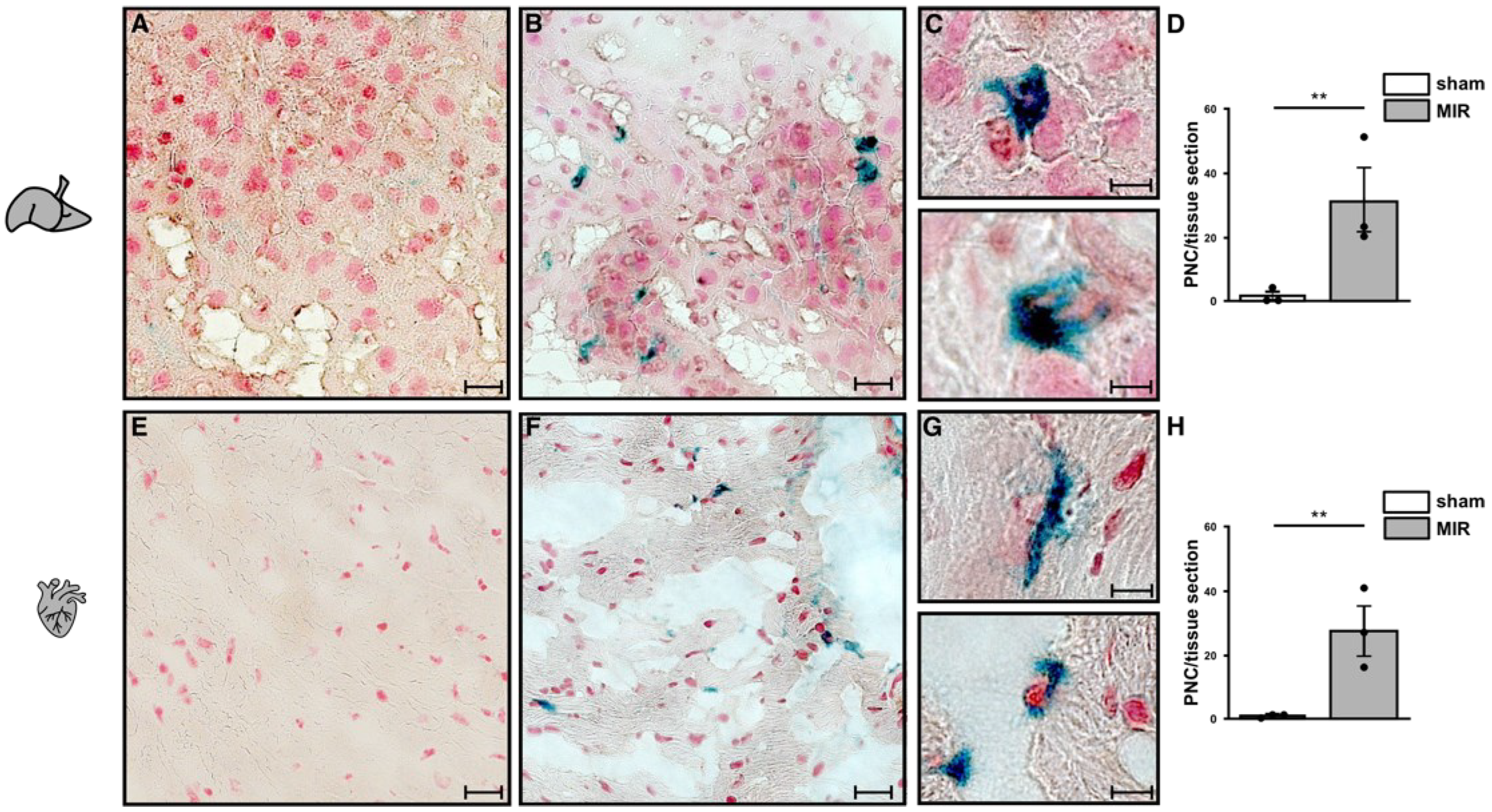
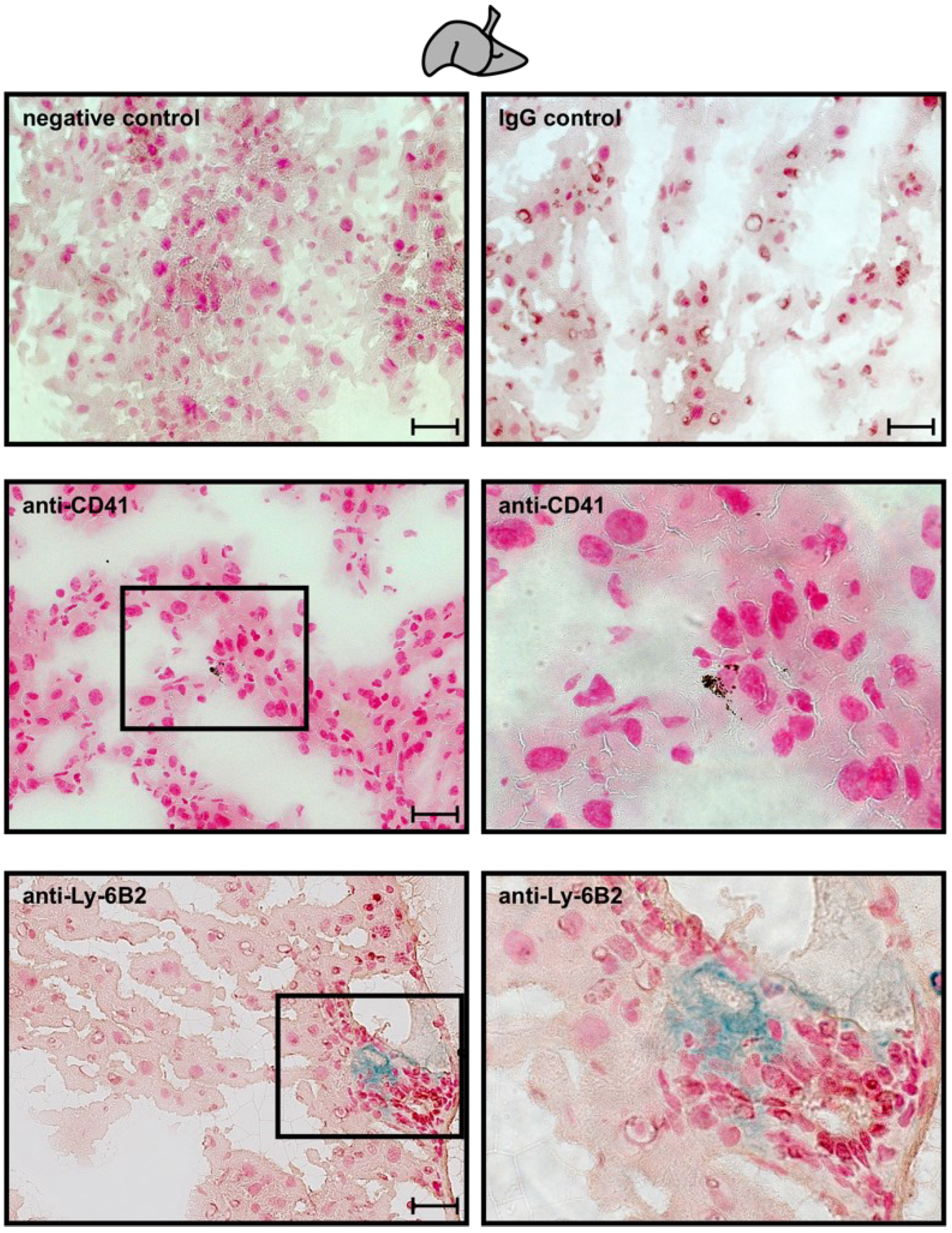
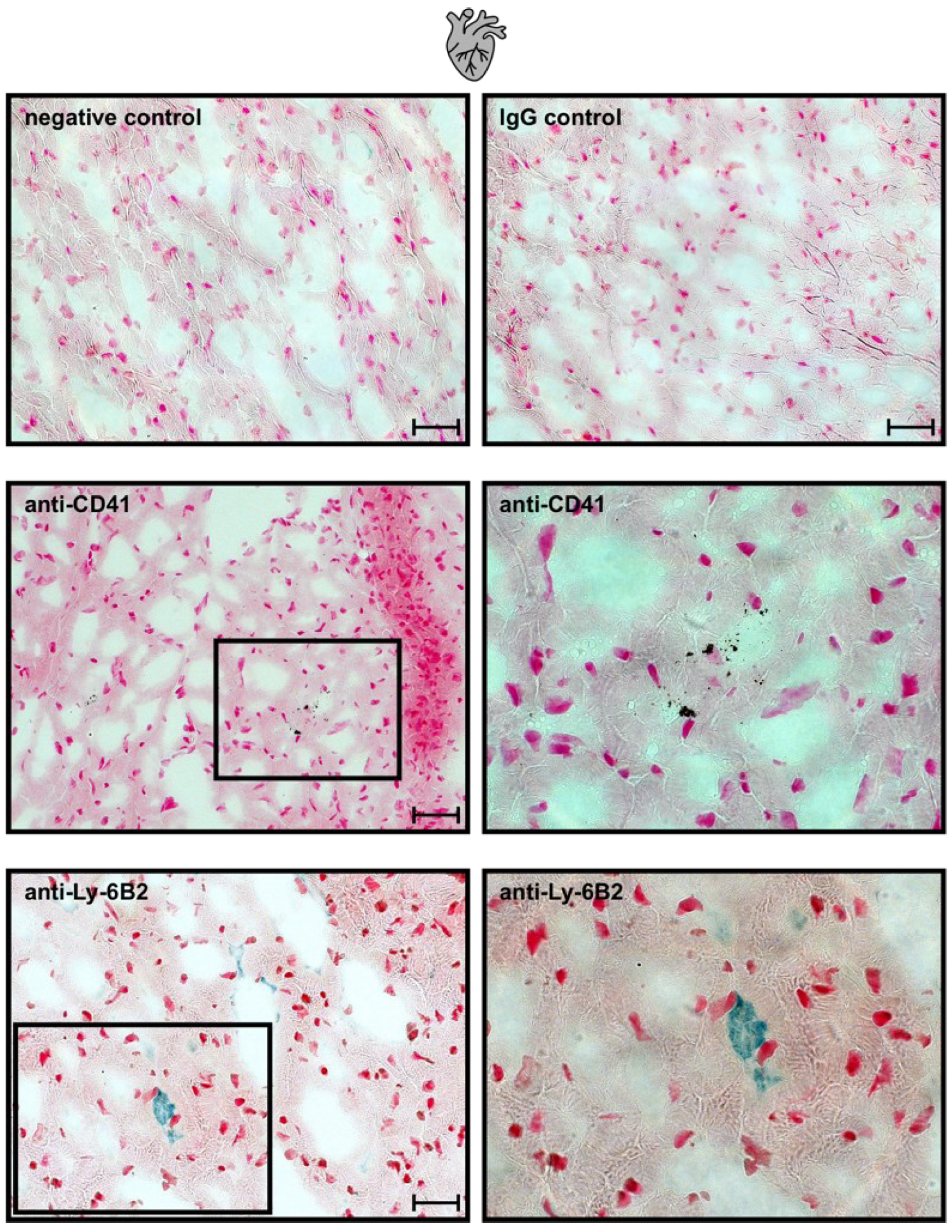
2.7. Neutrophils and Platelet Staining in Murine Hepatic and Myocardial Tissues
2.8. Detection of Adenosine Receptors (ADORAs) in Human Platelets by Immunoblot Analysis
2.9. Data Analysis
3. Results
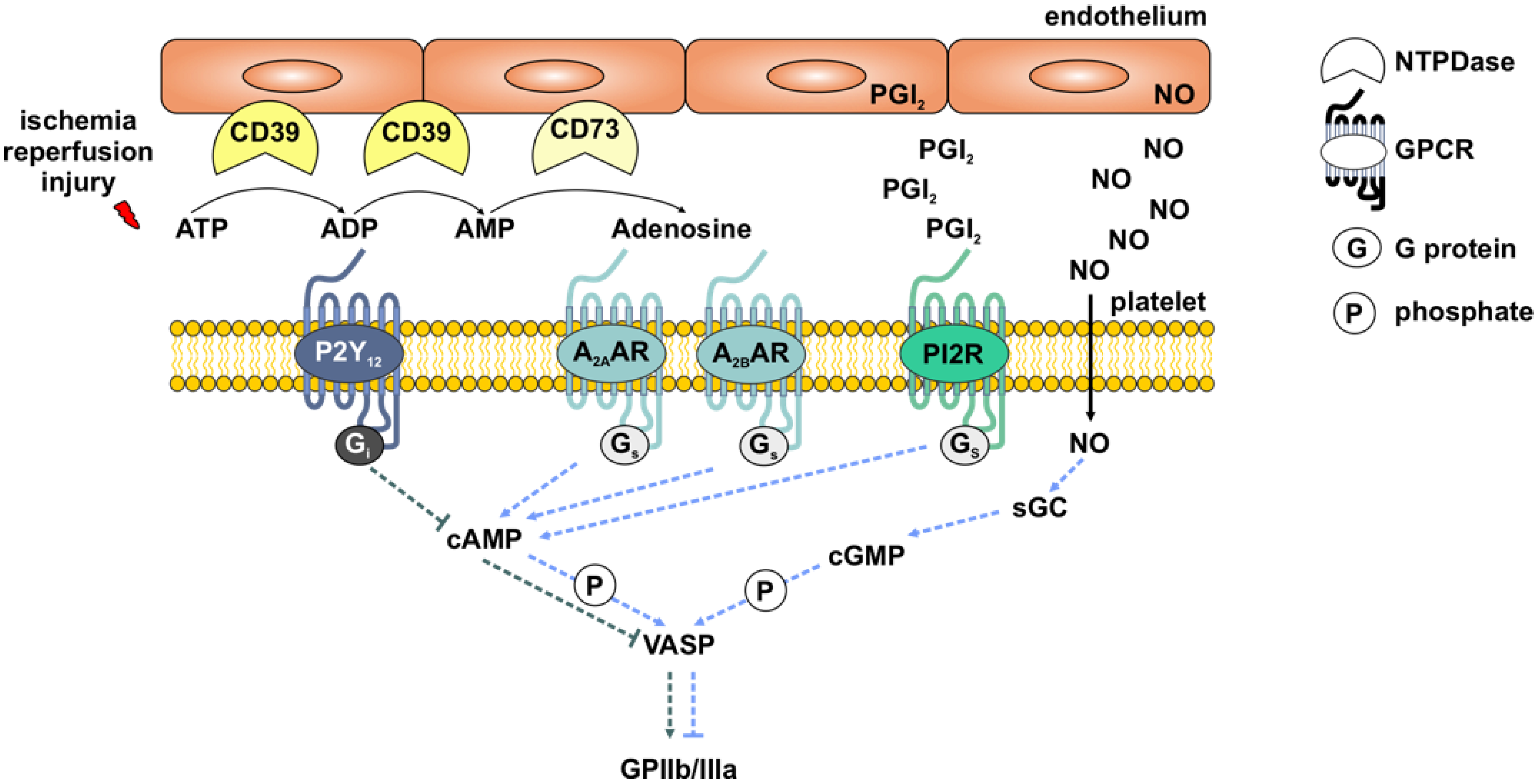
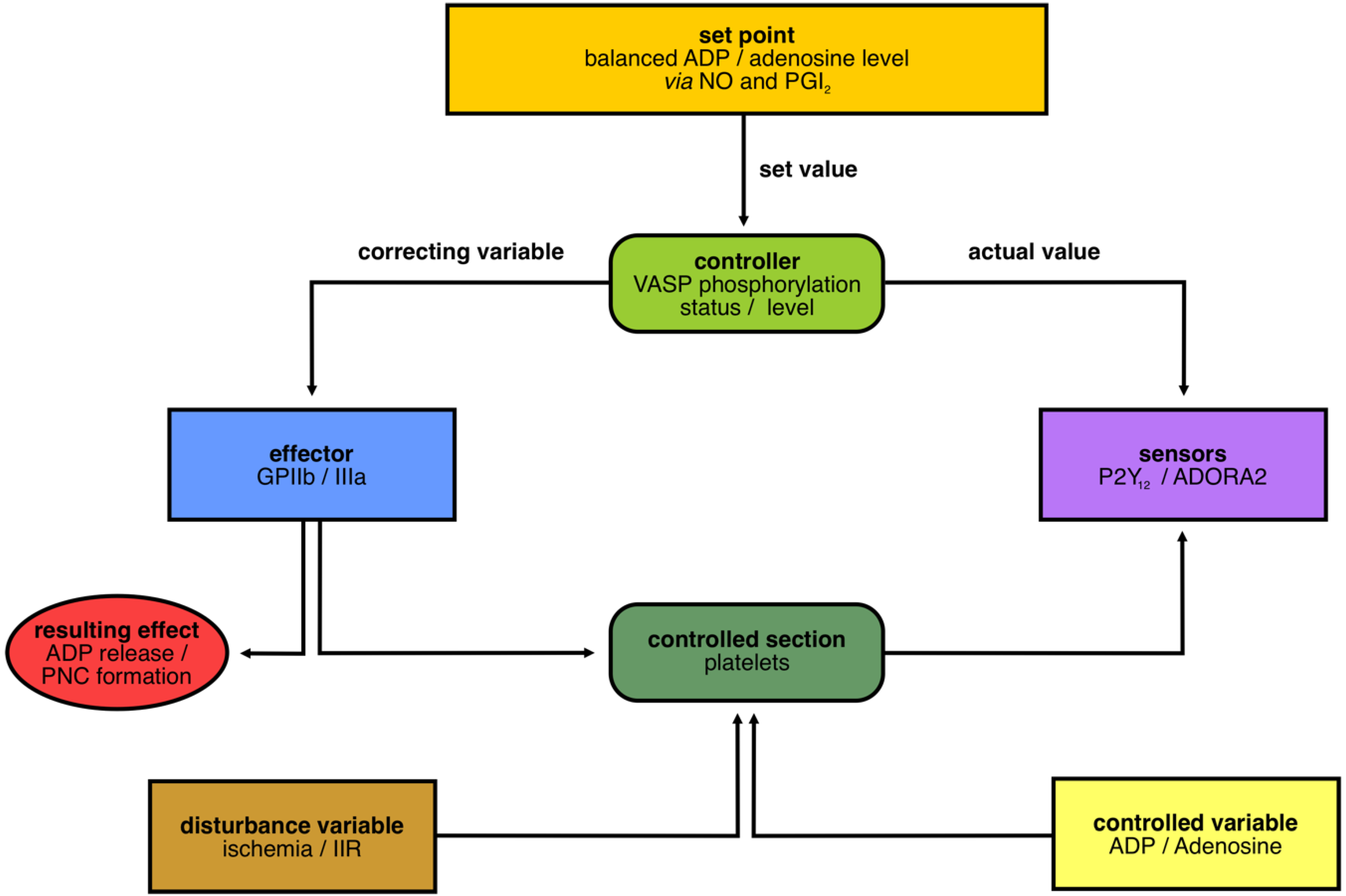
3.1. Layout of the Cybernetic System/Control Circuit “Platelets in IIR”
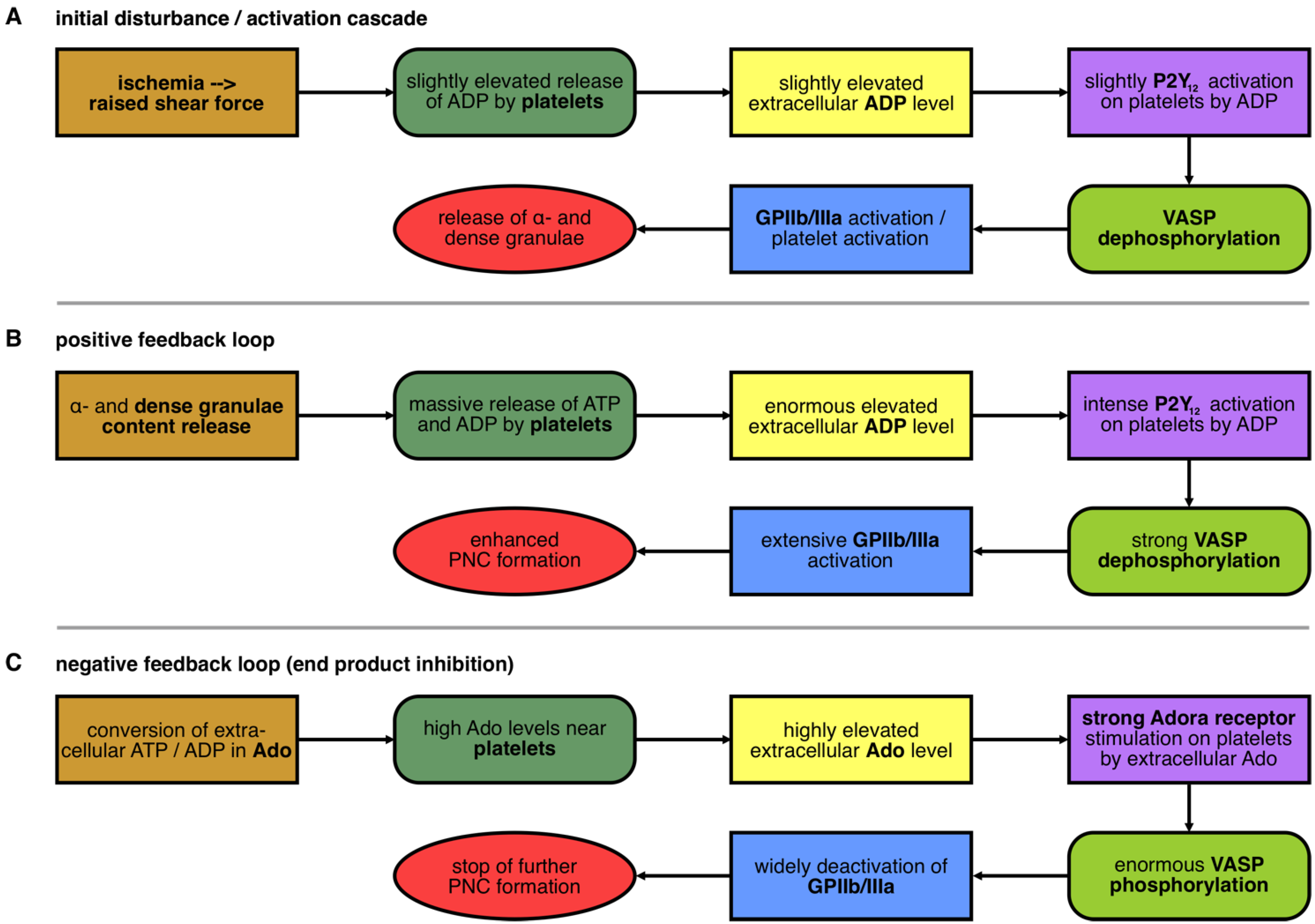
3.2. Initial Disturbance and the Positive Feedback Loop in the Nucleotide-Controlled Cybernetic System
3.3. The Negative Feedback Loop in the Nucleotide-Controlled Cybernetic System
3.4. Analysis of Set Point Components for Homeostasis
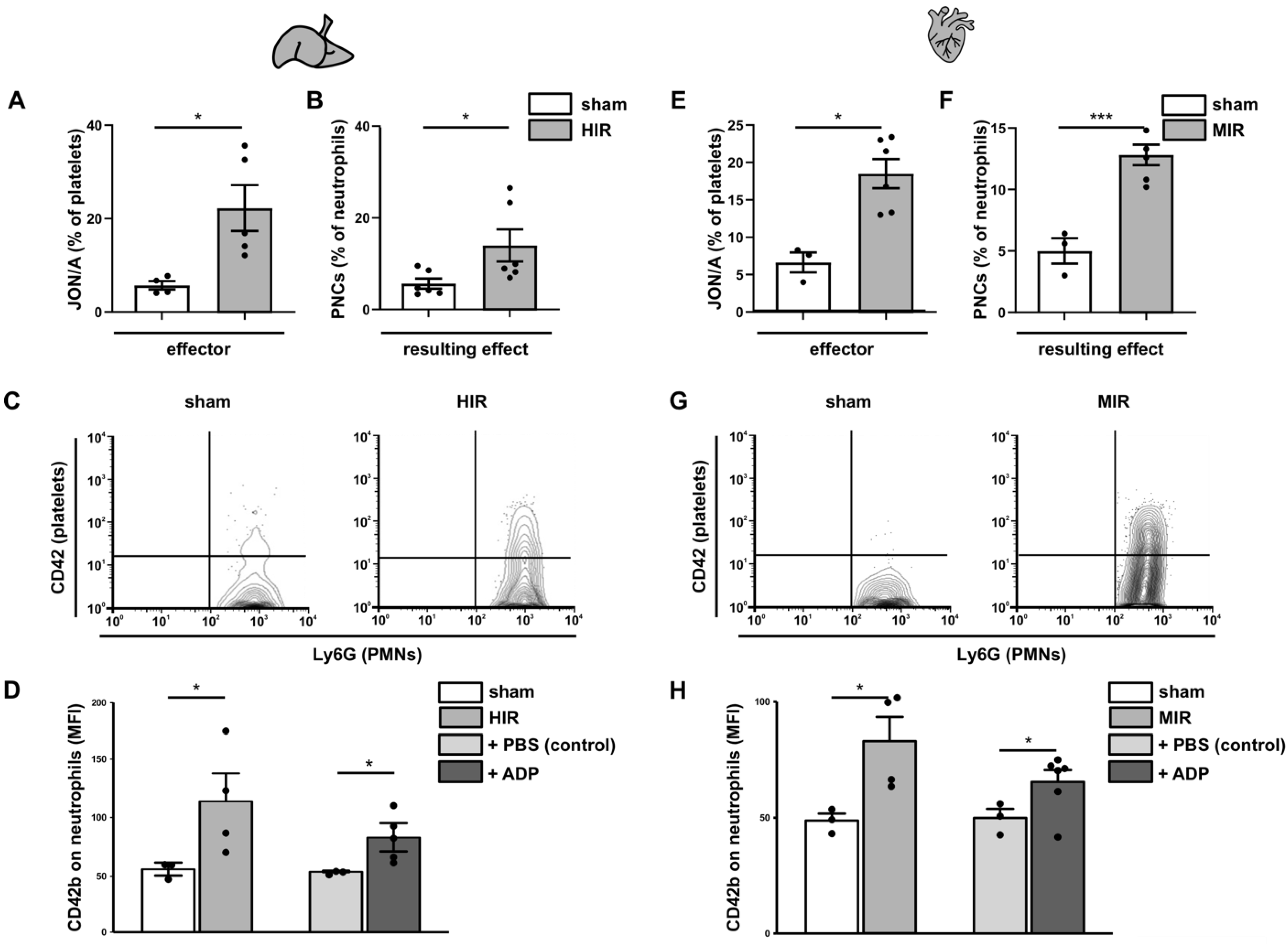
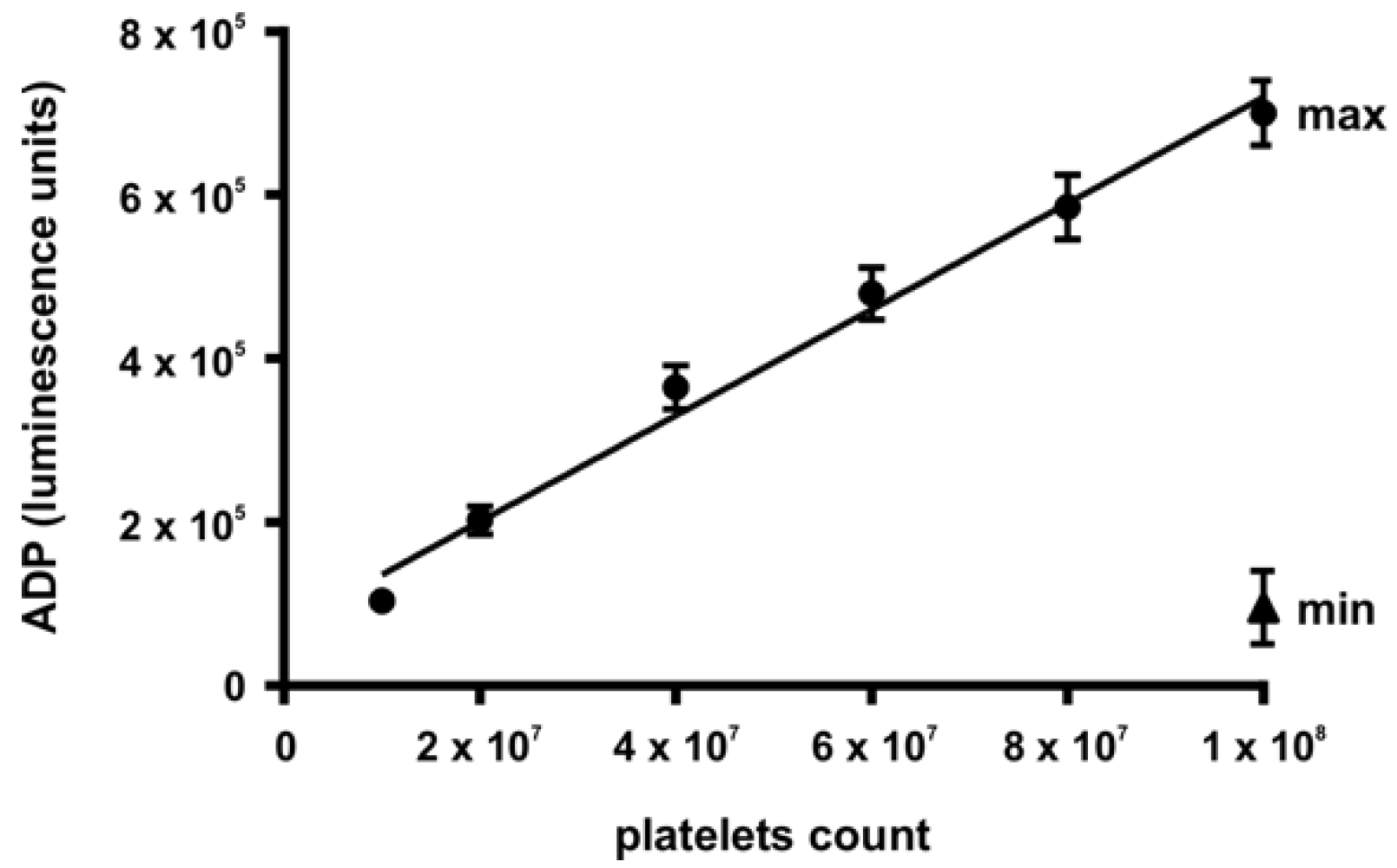
3.5. Analysis of the Initial Disturbance Induced by Ischemia
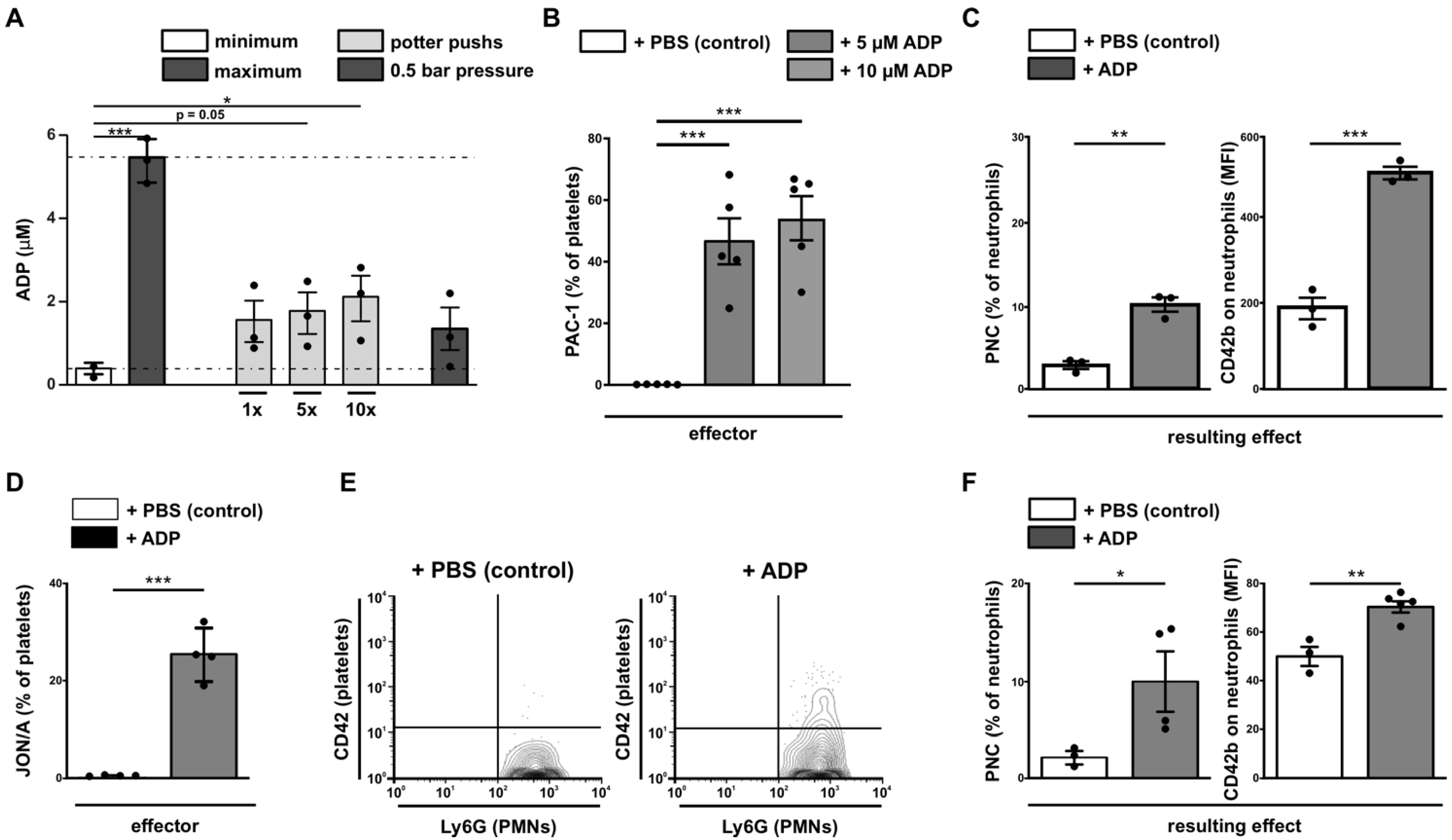
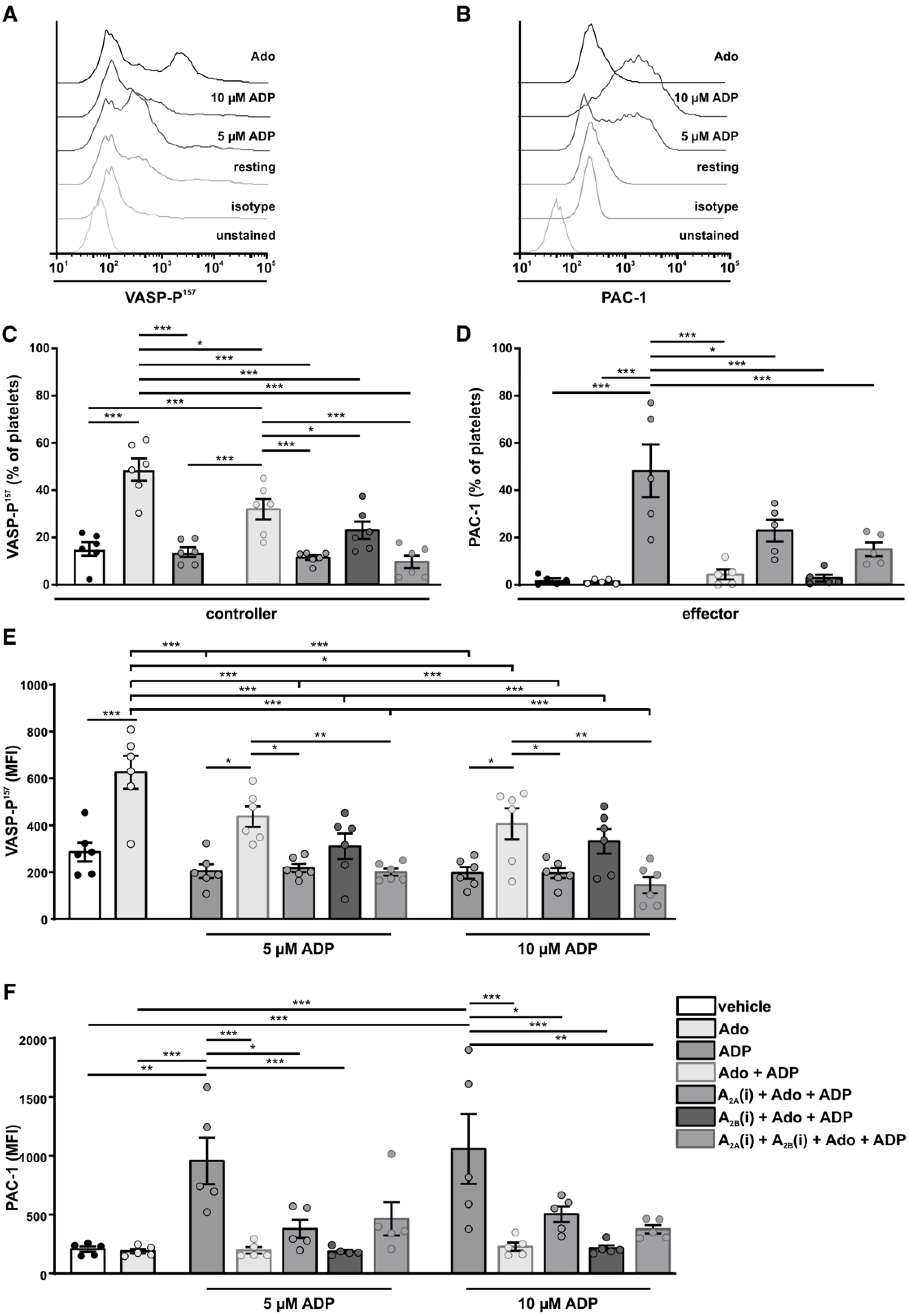
3.6. Analysis of the Positive Feedback Loop
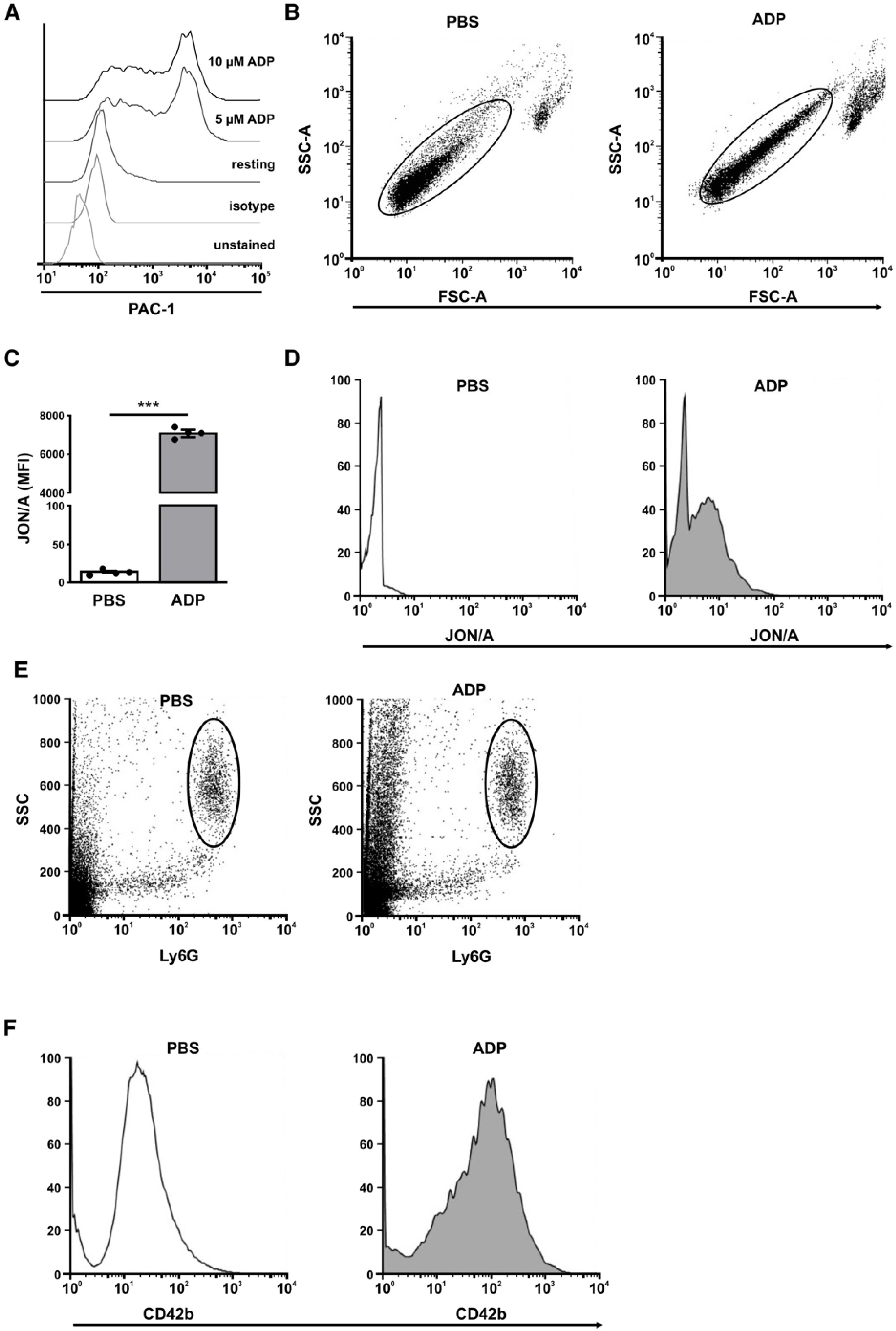
3.7. Ischemia-Induced Changes in Platelet Activation Results in PNC Formation
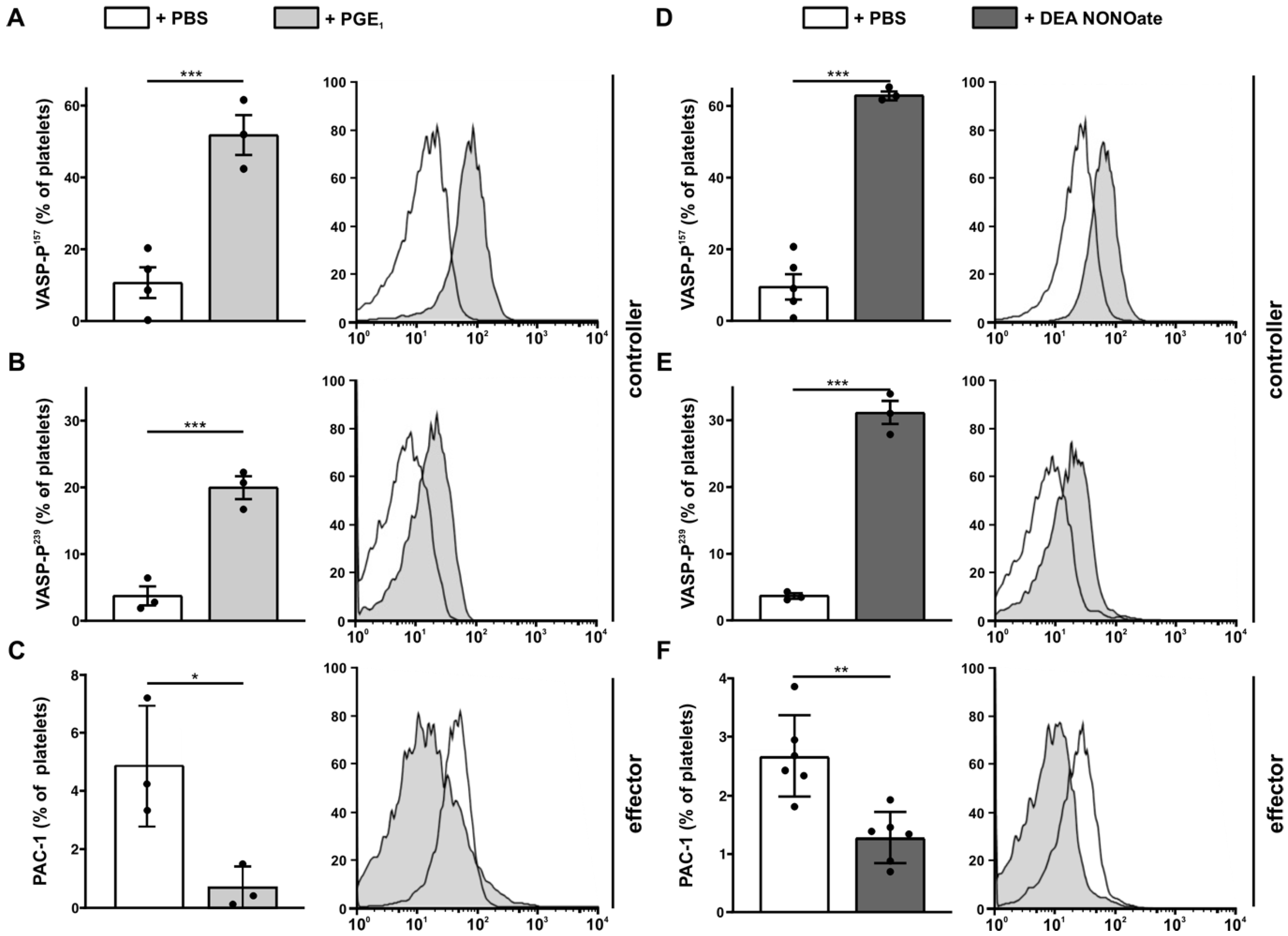
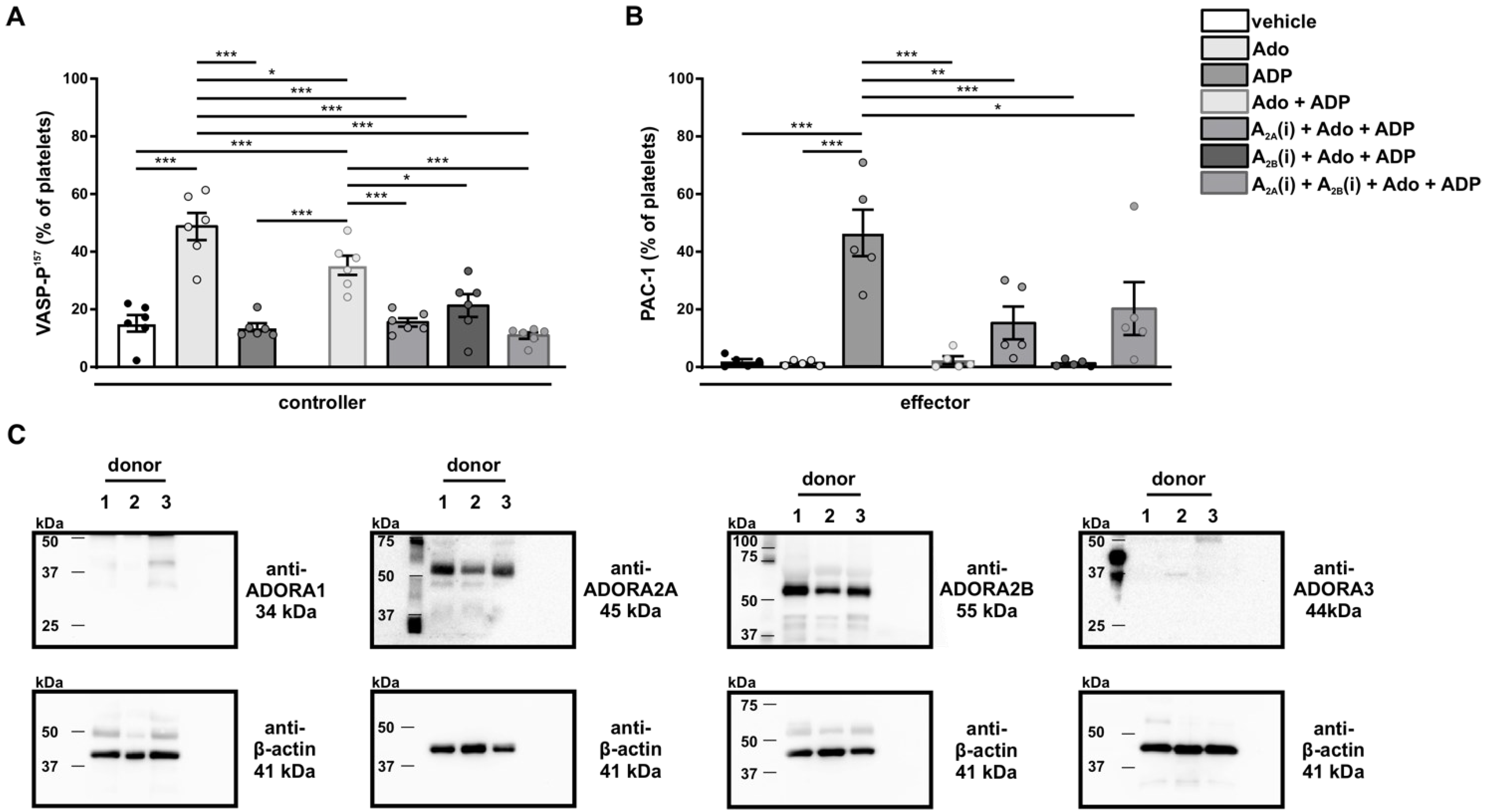
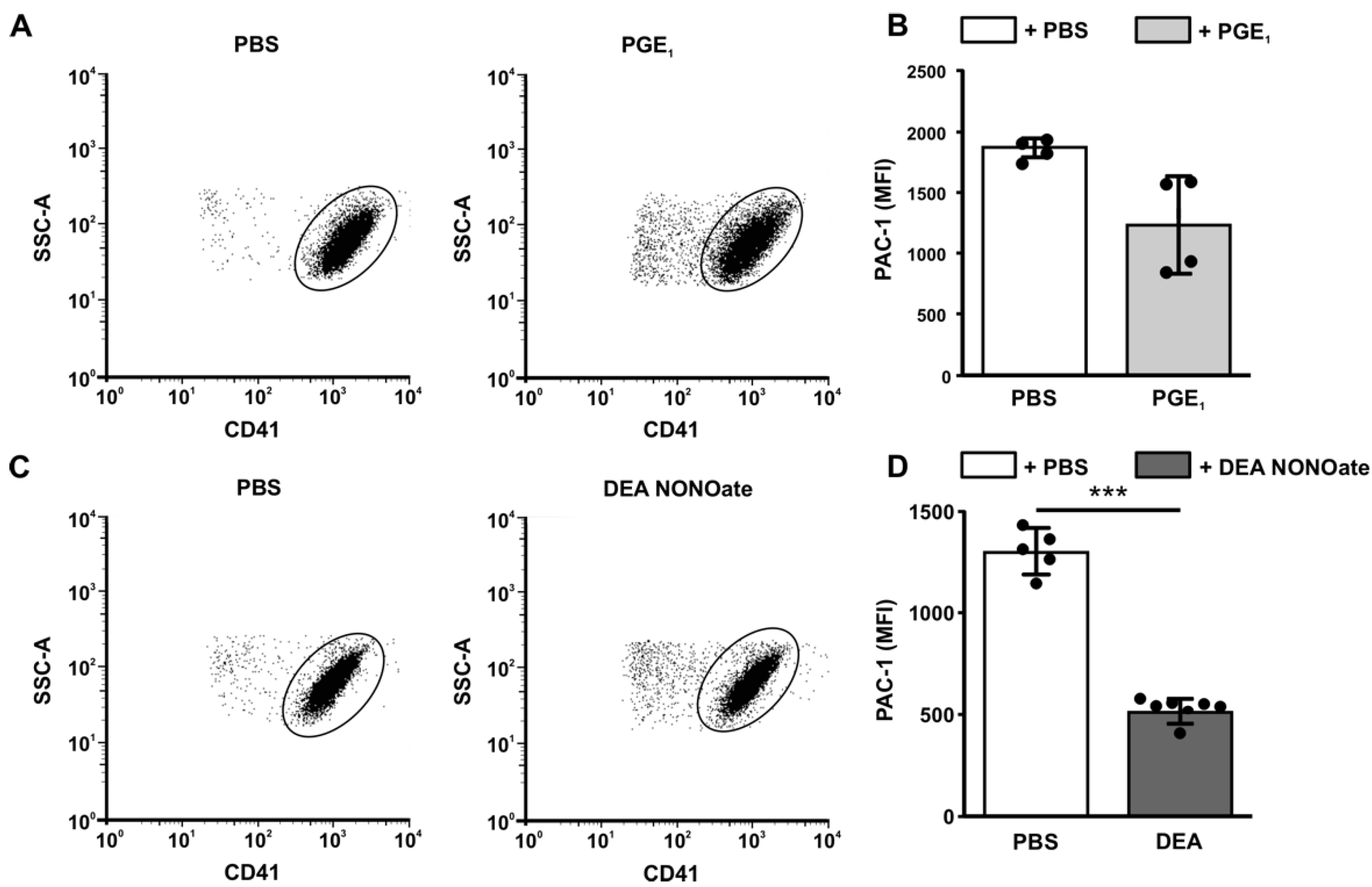
3.8. Analysis of the Negative Feedback Loop
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A







References
- Wiener, N. Cybernetics. Sci. Am. 1948, 179, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Punchard, N.A.; Whelan, C.J.; Adcock, I. The Journal of Inflammation. J. Inflamm. 2004, 1, 1. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Granja, T.; Magunia, H.; Schüssel, P.; Fischer, C.; Prüfer, T.; Schibilsky, D.; Serna-Higuita, L.; Wendel, H.P.; Schlensak, C.; Häberle, H.; et al. Left ventricular assist device implantation causes platelet dysfunction and proinflammatory platelet-neutrophil interaction. Platelets 2020, 33, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Magunia, H.; Schüssel, P.; Fischer, C.; Prüfer, T.; Schibilsky, D.; Serna-Higuita, L.; Wendel, H.P.; Schlensak, C.; Häberle, H.; et al. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Amison, R.; Page, C.; Pitchford, S. Pharmacological modulation of the inflammatory actions of platelets. Handb. Exp. Pharmacol. 2012, 210, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Taub, D.D. Chemokine-leukocyte interactions. The voodoo that they do so well. Cytokine Growth Factor Rev. 1996, 7, 355–376. [Google Scholar] [CrossRef]
- Gopalakrishnan, M.; Saurabh, S. Is red blood cell a mediator of remote ischaemic preconditioning? Med. Hypotheses 2014, 83, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.F.; Granger, D.N. Role of blood cells in ischaemia-reperfusion induced endothelial barrier failure. Cardiovasc. Res. 2010, 87, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Gorsuch, W.B.; Chrysanthou, E.; Schwaeble, W.; Stahl, G.L. The complement system in ischemia-reperfusion injuries. Immunobiology 2012, 217, 1026–1033. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Mendolicchio, G.L. Adhesion mechanisms in platelet function. Circ. Res. 2007, 100, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Jesty, J.; Beltrami, E. Positive feedbacks of coagulation: Their role in threshold regulation. Arter. Thromb. Vasc. Biol. 2005, 25, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Kanthi, Y.; Hyman, M.C.; Liao, H.; Baek, A.E.; Visovatti, S.H.; Sutton, N.; Goonewardena, S.N.; Neral, M.K.; Jo, H.; Pinsky, D.J. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J. Clin. Investig. 2015, 125, 3027–3036. [Google Scholar] [CrossRef]
- Rossaint, J.; Zarbock, A. Platelets in leucocyte recruitment and function. Cardiovasc. Res. 2015, 107, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Herter, J.M.; Rossaint, J.; Zarbock, A. Platelets in inflammation and immunity. J. Thromb. Haemost. 2014, 12, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Mezger, M.; Nording, H.; Sauter, R.; Graf, T.; Heim, C.; Von Bubnoff, N.; Ensminger, S.M.; Langer, H.F. Platelets and Immune Responses During Thromboinflammation. Front. Immunol. 2019, 10, 1731. [Google Scholar] [CrossRef]
- Bazzoni, G.; Dejana, E.; del Maschio, A. Platelet-neutrophil interactions. Possible relevance in the pathogenesis of thrombosis and inflammation. Haematologica 1991, 76, 491–499. [Google Scholar]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A. Neutrophil-platelet interactions in inflammation. Receptor 1994, 4, 3–7. [Google Scholar]
- Peters, M.J.; Dixon, G.; Kotowicz, K.T.; Hatch, D.J.; Heyderman, R.S.; Klein, N.J. Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br. J. Haematol. 1999, 106, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Kohler, D.; Straub, A.; Weissmüller, T.; Faigle, M.; Bender, S.; Lehmann, R.; Wendel, H.-P.; Kurz, J.; Walter, U.; Zacharowski, K.; et al. Phosphorylation of vasodilator-stimulated phosphoprotein prevents platelet-neutrophil complex formation and dampens myocardial ischemia-reperfusion injury. Circulation 2011, 123, 2579–2590. [Google Scholar] [CrossRef] [PubMed]
- Kohler, D.; Birk, P.; König, K.; Straub, A.; Eldh, T.; Morote-García, J.C.; Rosenberger, P. Phosphorylation of vasodilator-stimulated phosphoprotein (VASP) dampens hepatic ischemia-reperfusion injury. PLoS ONE 2011, 6, e29494. [Google Scholar] [CrossRef] [PubMed]
- Kohler, D.; Bibli, S.-I.; Klammer, L.P.; Roth, J.M.; Lehmann, R.; Fleming, I.; Granja, T.F.; Straub, A.; Benz, P.M.; Rosenberger, P. Phosphorylation of vasodilator-stimulated phosphoprotein contributes to myocardial ischemic preconditioning. Basic Res. Cardiol. 2018, 113, 11. [Google Scholar] [CrossRef]
- Kohler, D.; Granja, T.; Volz, J.; Koeppen, M.; Langer, H.F.; Hansmann, G.; Legchenko, E.; Geisler, T.; Bakchoul, T.; Eggstein, C.; et al. Red blood cell-derived semaphorin 7A promotes thrombo-inflammation in myocardial ischemia-reperfusion injury through platelet GPIb. Nat. Commun. 2020, 11, 1315. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef]
- Gachet, C. Regulation of platelet functions by P2 receptors. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 277–300. [Google Scholar] [CrossRef]
- Devanathan, V.; Hagedorn, I.; Köhler, D.; Pexa, K.; Cherpokova, D.; Kraft, P.; Singh, M.; Rosenberger, P.; Stoll, G.; Birnbaumer, L.; et al. Platelet Gi protein Gαi2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 6491–6496. [Google Scholar] [CrossRef]
- Evangelista, V.; Manarini, S.; Dell’Elba, G.; Martelli, N.; Napoleone, E.; Di Santo, A.; Lorenzet, P.S.R. Clopidogrel inhibits platelet-leukocyte adhesion and platelet-dependent leukocyte activation. Thromb. Haemost. 2005, 94, 568–577. [Google Scholar]
- Burnstock, G. Blood cells: An historical account of the roles of purinergic signalling. Purinergic Signal. 2015, 11, 411–434. [Google Scholar] [CrossRef] [PubMed]
- Koziak, K.; Sévigny, J.; Robson, S.C.; Siegel, J.B.; Kaczmarek, E. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb. Haemost. 1999, 82, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, B.; Dwyer, K.; Enjyoji, K.; Robson, S.C. Ecto-nucleotidases of the CD39/NTPDase family modulate platelet activation and thrombus formation: Potential as therapeutic targets. Blood Cells Mol. Dis. 2006, 36, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Sundberg, C.; Braun, N.; Guckelberger, O.; Csizmadia, E.; Qawi, I.; Imai, M.; Zimmermann, H.; Robson, S.C. Differential catalytic properties and vascular topography of murine nucleoside triphosphate diphosphohydrolase 1 (NTPDase1) and NTPDase2 have implications for thromboregulation. Blood 2002, 99, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Kohler, D.; Eckle, T.; Faigle, M.; Grenz, A.; Mittelbronn, M.; Laucher, S.; Hart, M.L.; Robson, S.C.; Müller, C.E.; Eltzschig, H.K. CD39/Ectonucleoside Triphosphate Diphosphohydrolase 1 Provides Myocardial Protection During Cardiac Ischemia/Reperfusion Injury. Circulation 2007, 16, 1784–1794. [Google Scholar] [CrossRef]
- Eckle, T.; Köhler, D.; Lehmann, R.; El Kasmi, K.C.; Eltzschig, H.K. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation 2008, 118, 166–175. [Google Scholar] [CrossRef]
- Morote-Garcia, J.C.; Köhler, D.; Roth, J.M.; Mirakaj, V.; Eldh, T.; Eltzschig, H.K.; Rosenberger, P. Repression of the equilibrative nucleoside transporters dampens inflammatory lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 296–305. [Google Scholar] [CrossRef]
- Kohler, D.; Streißenberger, A.; Morote-García, J.C.; Granja, T.; Schneider, M.; Straub, A.; Boison, D.; Rosenberger, P. Inhibition of Adenosine Kinase Attenuates Acute Lung Injury. Crit. Care Med. 2016, 44, e181–e189. [Google Scholar] [CrossRef]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Iyu, D.; Glenn, J.R.; White, A.E.; Fox, S.C.; Heptinstall, S. Adenosine derived from ADP can contribute to inhibition of platelet aggregation in the presence of a P2Y12 antagonist. Arter. Thromb. Vasc. Biol. 2011, 31, 416–422. [Google Scholar] [CrossRef]
- Yang, D.; Chen, H.; Koupenova, M.; Carroll, S.H.; Eliades, A.; Freedman, J.E.; Toselli, P.; Ravid, K. A new role for the A2b adenosine receptor in regulating platelet function. J. Thromb. Haemost. 2010, 8, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Granja, T.F.; Köhler, D.; Schad, J.; Franz, C.B.D.O.; Konrad, F.; Hoch-Gutbrod, M.; Streißenberger, A.; Rosenberger, P.; Straub, A. Adenosine Receptor Adora2b Plays a Mechanistic Role in the Protective Effect of the Volatile Anesthetic Sevoflurane during Liver Ischemia/Reperfusion. Anesthesiology 2016, 125, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C.; Hechler, B. Platelet Purinergic Receptors in Thrombosis and Inflammation. Hamostaseologie 2020, 40, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Rebowski, G.; Lee, S.H.; Dominguez, R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. Embo. J. 2007, 26, 4597–4606. [Google Scholar] [CrossRef]
- Bachmann, C.; Fischer, L.; Walter, U.; Reinhard, M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. JBC 1999, 274, 23549–23557. [Google Scholar] [CrossRef]
- Halbrugge, M.; Friedrich, C.; Eigenthaler, M.; Schanzenbächer, P.; Walter, U. Stoichiometric and reversible phosphorylation of a 46-kDa protein in human platelets in response to cGMP- and cAMP-elevating vasodilators. J. Biol. Chem. 1990, 265, 3088–3093. [Google Scholar] [CrossRef]
- Walter, U.; Gambaryan, S. cGMP and cGMP-dependent protein kinase in platelets and blood cells. Handb. Exp. Pharmacol. 2009, 191, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Frere, C.; Cuisset, T.; Quilici, J.; Camoin, L.; Carvajal, J.; Morange, P.E.; Lambert, M.; Juhan-Vague, I.; Bonnet, J.-L.; Alessi, M.-C. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb. Haemost. 2007, 98, 838–843. [Google Scholar] [CrossRef]
- Morel, O.; Ohlmann, P.; Jesel, L.; Desprez, D.; Grunebaum, L.; Bareiss, P.; Morel, O. Impaired platelet responsiveness to clopidogrel identified by flow cytometric vasodilator-stimulated phosphoprotein (VASP) phosphorylation in patients with subacute stent thrombosis. Thromb. Haemost. 2007, 98, 896–899. [Google Scholar]
- Morel, O.; Bernhard, N.; Desprez, D.; Grunebaum, L.; Freyssinet, J.M.; Toti, F.; Bareiss, P. Residual prothrombotic status in low responder patients to clopidogrel identified by Vasodilator-Stimulated Phosphoprotein Phosphorylation (VASP) analysis? Thromb. Haemost. 2008, 99, 448–451. [Google Scholar]
- Cowan, A.Q.; Cho, D.J.; Rosenson, R.S. Importance of blood rheology in the pathophysiology of atherothrombosis. Cardiovasc. Drugs Ther. 2012, 26, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Granja, T.; Schad, J.; Schüssel, P.; Fischer, C.; Häberle, H.; Rosenberger, P.; Straub, A. Using six-colour flow cytometry to analyse the activation and interaction of platelets and leukocytes—A new assay suitable for bench and bedside conditions. Thromb. Res. 2015, 136, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.L.; Much, C.; Köhler, D.; Schittenhelm, J.; Gorzolla, I.C.; Stahl, G.L.; Eltzschig, H.K. Use of a hanging-weight system for liver ischemic preconditioning in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 294, G1431–G1440. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Grenz, A.; Köhler, D.; Redel, A.; Falk, M.; Rolauffs, B.; Osswald, H.; Kehl, F.; Eltzschig, H.K. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2533–H2540. [Google Scholar] [CrossRef]
- Kohler, D.; Devanathan, V.; de Oliveira Franz, C.B.; Eldh, T.; Novakovic, A.; Roth, J.M.; Granja, T.; Birnbaumer, L.; Rosenberger, P.; Beer-Hammer, S.; et al. Gαi2- and Gαi3-deficient mice display opposite severity of myocardial ischemia reperfusion injury. PLoS ONE 2014, 9, e98325. [Google Scholar]
- Broekman, M.J.; Eiroa, A.M.; Marcus, A.J. Inhibition of human platelet reactivity by endothelium-derived relaxing factor from human umbilical vein endothelial cells in suspension: Blockade of aggregation and secretion by an aspirin-insensitive mechanism. Blood 1991, 78, 1033–1040. [Google Scholar] [CrossRef]
- Wang, G.R.; Zhu, Y.; Halushka, P.V.; Lincoln, T.M.; Mendelsohn, M.E. Mechanism of platelet inhibition by nitric oxide: In vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 4888–4893. [Google Scholar] [CrossRef]
- Riddell, D.R.; Owen, J.S. Nitric oxide and platelet aggregation. Vitam. Horm. 1999, 57, 25–48. [Google Scholar]
- Gambaryan, S.; Subramanian, H.; Kehrer, L.; Mindukshev, I.; Sudnitsyna, J.; Reiss, C.; Rukoyatkina, N.; Friebe, A.; Sharina, I.; Martin, E.; et al. Erythrocytes do not activate purified and platelet soluble guanylate cyclases even in conditions favourable for NO synthesis. Cell Commun. Signal. 2016, 14, 16. [Google Scholar] [CrossRef]
- Shiravand, Y.; Walter, U.; Jurk, K. Fine-Tuning of Platelet Responses by Serine/Threonine Protein Kinases and Phosphatases-Just the Beginning. Hamostaseologie 2021, 41, 206–216. [Google Scholar] [CrossRef]
- Makhoul, S.; Walter, E.; Pagel, O.; Walter, U.; Sickmann, A.; Gambaryan, S.; Smolenski, A.; Zahedi, R.P.; Jurk, K. Effects of the NO/soluble guanylate cyclase/cGMP system on the functions of human platelets. Nitric. Oxide 2018, 76, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Viisoreanu, D.; Gear, A. Effect of physiologic shear stresses and calcium on agonist-induced platelet aggregation, secretion, and thromboxane A2 formation. Thromb. Res. 2007, 120, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.J.; Mickelson, J.; Fantone, J.C.; Gallagher, K.P.; Lucchesi, B.R. Reduction of experimental canine myocardial infarct size with prostaglandin E1: Inhibition of neutrophil migration and activation. J. Pharmacol. Exp. Ther. 1988, 244, 619–624. [Google Scholar] [PubMed]
- Carini, R.; Albano, E. Recent insights on the mechanisms of liver preconditioning. Gastroenterology 2003, 125, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Jugdutt, B.I. Nitric oxide and cardioprotection during ischemia-reperfusion. Heart Fail. Rev. 2002, 7, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef]
- Asaduzzaman, M.; Lavasani, S.; Rahman, M.; Zhang, S.; Braun, O.; Jeppsson, B.; Thorlacius, H. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit. Care Med. 2009, 37, 1389–1396. [Google Scholar] [CrossRef]
- Kornerup, K.N.; Salmon, G.P.; Pitchford, S.C.; Liu, W.L.; Page, C.P. Circulating Platelet-Neutrophil Complexes Are Important for Subsequent Neutrophil Activation and Migration. J. Appl. Physiol. 2010, 109, 758–767. [Google Scholar] [CrossRef]
- Ziegler, M.; Wang, X.; Peter, K. Platelets in cardiac ischaemia/reperfusion injury: A promising therapeutic target. Cardiovasc. Res. 2019, 115, 1178–1188. [Google Scholar] [CrossRef]
- Makhoul, S.; Trabold, K.; Gambaryan, S.; Tenzer, S.; Pillitteri, D.; Walter, U.; Jurk, K. cAMP- and cGMP-elevating agents inhibit GPIbalpha-mediated aggregation but not GPIbalpha-stimulated Syk activation in human platelets. Cell Commun. Signal. 2019, 17, 122. [Google Scholar] [CrossRef]
- Fuentes, F.; Alarcón, M.; Badimon, L.; Fuentes, M.; Klotz, K.N.; Vilahur, G.; Kachler, S.; Padró, T.; Palomo, I.; Fuentes, E. Guanosine exerts antiplatelet and antithrombotic properties through an adenosine-related cAMP-PKA signaling. Int. J. Cardiol. 2017, 248, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell Signal. 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Abbracchio, M.P.; Burnstock, G.; Daly, J.W.; Harden, T.K.; A Jacobson, K.; Leff, P.; Williams, M. Nomenclature and classification of purinoceptors. Pharmacol. Rev. 1994, 46, 143–156. [Google Scholar] [PubMed]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar]
- Wolska, N.; Rozalski, M. Blood Platelet Adenosine Receptors as Potential Targets for Anti-Platelet Therapy. Int. J. Mol. Sci. 2019, 20, 5475. [Google Scholar] [CrossRef]
- Horn, M.; Bertling, A.; Brodde, M.F.; Müller, A.; Roth, J.; VAN Aken, H.; Jurk, K.; Heilmann, C.; Peters, G.; Kehrel, B.E. Human neutrophil alpha-defensins induce formation of fibrinogen and thrombospondin-1 amyloid-like structures and activate platelets via glycoprotein IIb/IIIa. J. Thromb. Haemost. 2012, 10, 647–661. [Google Scholar] [CrossRef]
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111. [Google Scholar] [CrossRef]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar]
- Rahman, I.; MacNee, W. Role of transcription factors in inflammatory lung diseases. Thorax 1998, 53, 601–612. [Google Scholar] [CrossRef]
- Liverani, E.; Rico, M.C.; Tsygankov, A.Y.; Kilpatrick, L.E.; Kunapuli, S.P. P2Y12 Receptor Modulates Sepsis-Induced Inflammation. Arter. Thromb. Vasc. Biol. 2016, 36, 961–971. [Google Scholar] [CrossRef]
- Yan, S.L.; Russell, J.; Harris, N.R.; Senchenkova, E.Y.; Yildirim, A.; Granger, D.N. Platelet abnormalities during colonic inflammation. Inflamm. Bowel Dis. 2013, 19, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Irving, P.M.; Macey, M.G.; Feakins, R.M.; Knowles, C.H.; Frye, J.N.; Liyanage, S.H.; Dorudi, S.; Williams, N.S.; Rampton, D.S. Platelet-leucocyte aggregates form in the mesenteric vasculature in patients with ulcerative colitis. Eur. J. Gastroenterol. Hepatol. 2008, 20, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front. Immunol. 2019, 10, 1260. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef]
- Li, J.; Kim, K.; Barazia, A.; Tseng, A.; Cho, J. Platelet-neutrophil interactions under thromboinflammatory conditions. Cell. Mol. Life Sci. 2015, 72, 2627–2643. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef]
- Evangelista, V.; Manarini, S.; Sideri, R.; Rotondo, S.; Martelli, N.; Piccoli, A.; Totani, L.; Piccardoni, P.; Vestweber, D.; De Gaetano, G.; et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: Role of PSGL-1 as a signaling molecule. Blood 1999, 93, 876–885. [Google Scholar] [CrossRef]
- Theoret, J.F.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. P-selectin antagonism with recombinant p-selectin glycoprotein ligand-1 (rPSGL-Ig) inhibits circulating activated platelet binding to neutrophils induced by damaged arterial surfaces. J. Pharmacol. Exp. Ther. 2001, 298, 658–664. [Google Scholar]
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511. [Google Scholar] [CrossRef]
- Vickers, J.D. Binding of polymerizing fibrin to integrin alpha(IIb)beta(3) on chymotrypsin-treated rabbit platelets decreases phosphatidylinositol 4,5-bisphosphate and increases cytoskeletal actin. Platelets 1999, 10, 228–237. [Google Scholar] [CrossRef]
- Vickers, J.D.; Kinlough-Rathbone, R.L.; Packham, M.A.; Mustard, J.F. Changes in Phosphoinositides in Rabbit Platelets during Clot Formation. Comparison of Platelets Stimulated by ADP or by Thrombin in the Presence of Polymerising Fibrin. Platelets 1990, 1, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L.; Nicolini, F.A.; Donnelly, W.H.; Nichols, W.W. Platelet-leukocyte-endothelial interactions in coronary artery disease. Am. J. Cardiol. 1992, 69, 8B–13B. [Google Scholar] [CrossRef]
- Kuroda, T.; Shiohara, E.; Homma, T.; Furukawa, Y.; Chiba, S. Effects of leukocyte and platelet depletion on ischemia--reperfusion injury to dog pancreas. Gastroenterology 1994, 107, 1125–1134. [Google Scholar] [CrossRef]
- Kuroda, T.; Shiohara, E. Leukocyte and platelet depletion protects the liver from damage induced by cholestasis and ischemia-reperfusion in the dog. Scand. J. Gastroenterol. 1996, 31, 182–190. [Google Scholar] [CrossRef]
- Guo, Y.; Tukaye, D.N.; Wu, W.J.; Zhu, X.; Book, M.; Tan, W.; Jones, S.P.; Rokosh, G.; Narumiya, S.; Li, Q.; et al. The COX-2/PGI2 receptor axis plays an obligatory role in mediating the cardioprotection conferred by the late phase of ischemic preconditioning. PLoS ONE 2012, 7, e41178. [Google Scholar] [CrossRef]
- Lee, J.; Ahn, E.; Park, W.K.; Park, S. Phosphoproteome Profiling of SH-SY5y Neuroblastoma Cells Treated with Anesthetics: Sevoflurane and Isoflurane Affect the Phosphorylation of Proteins Involved in Cytoskeletal Regulation. PLoS ONE 2016, 11, e0162214. [Google Scholar] [CrossRef]
- Tsutsumi, Y.M.; Patel, H.H.; Lai, N.C.; Takahashi, T.; Head, B.P.; Roth, D.M. Isoflurane produces sustained cardiac protection after ischemia-reperfusion injury in mice. Anesthesiology 2006, 104, 495–502. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granja, T.F.; Köhler, D.; Leiss, V.; Eggstein, C.; Nürnberg, B.; Rosenberger, P.; Beer-Hammer, S. Platelets and the Cybernetic Regulation of Ischemic Inflammatory Responses through PNC Formation Regulated by Extracellular Nucleotide Metabolism and Signaling. Cells 2022, 11, 3009. https://doi.org/10.3390/cells11193009
Granja TF, Köhler D, Leiss V, Eggstein C, Nürnberg B, Rosenberger P, Beer-Hammer S. Platelets and the Cybernetic Regulation of Ischemic Inflammatory Responses through PNC Formation Regulated by Extracellular Nucleotide Metabolism and Signaling. Cells. 2022; 11(19):3009. https://doi.org/10.3390/cells11193009
Chicago/Turabian StyleGranja, Tiago F., David Köhler, Veronika Leiss, Claudia Eggstein, Bernd Nürnberg, Peter Rosenberger, and Sandra Beer-Hammer. 2022. "Platelets and the Cybernetic Regulation of Ischemic Inflammatory Responses through PNC Formation Regulated by Extracellular Nucleotide Metabolism and Signaling" Cells 11, no. 19: 3009. https://doi.org/10.3390/cells11193009
APA StyleGranja, T. F., Köhler, D., Leiss, V., Eggstein, C., Nürnberg, B., Rosenberger, P., & Beer-Hammer, S. (2022). Platelets and the Cybernetic Regulation of Ischemic Inflammatory Responses through PNC Formation Regulated by Extracellular Nucleotide Metabolism and Signaling. Cells, 11(19), 3009. https://doi.org/10.3390/cells11193009