



Macrophages, Chronic Inflammation, and Insulin Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
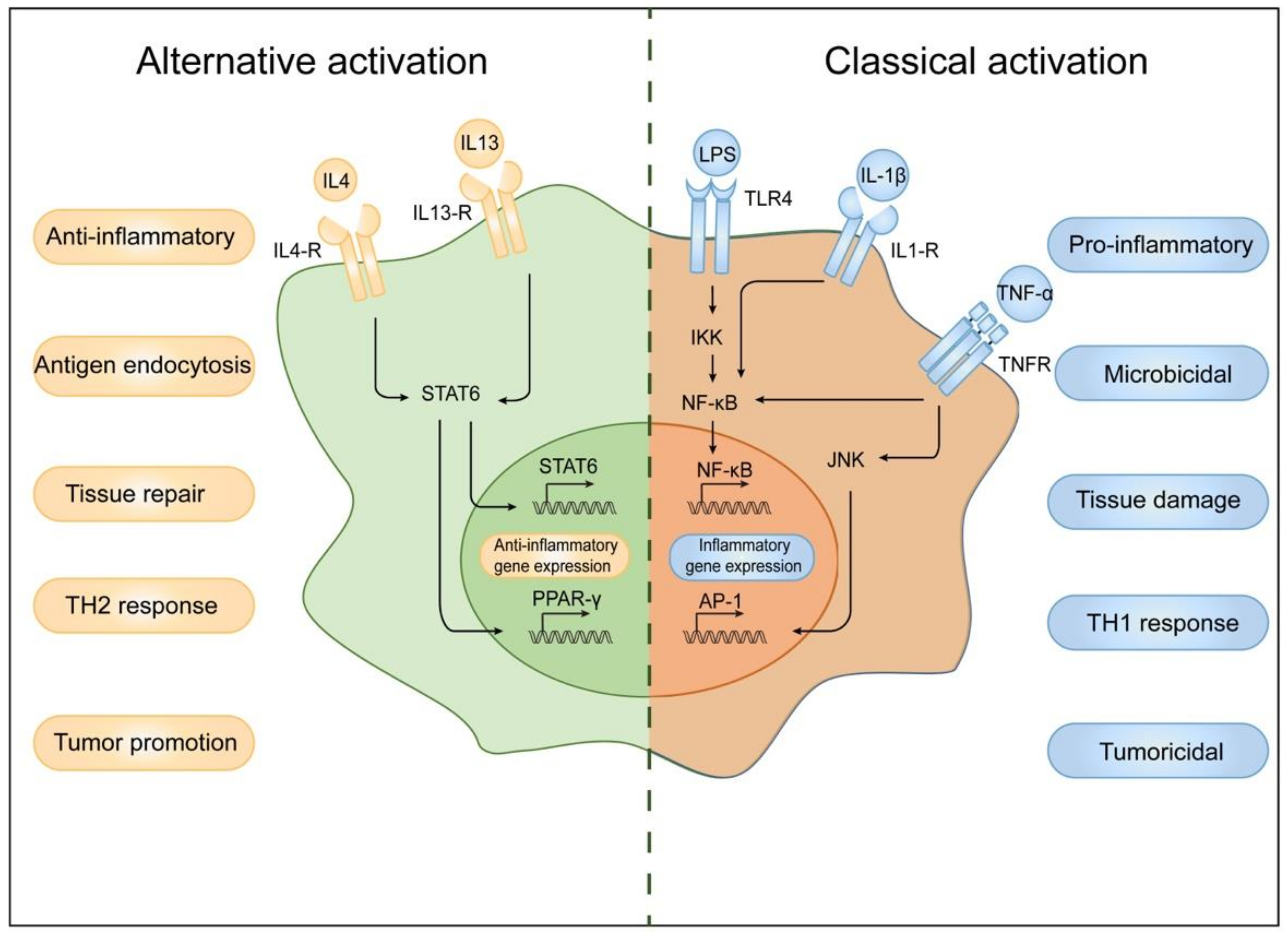
2. Classification of Macrophages
3. Macrophage- and Obesity-Induced Insulin Resistance
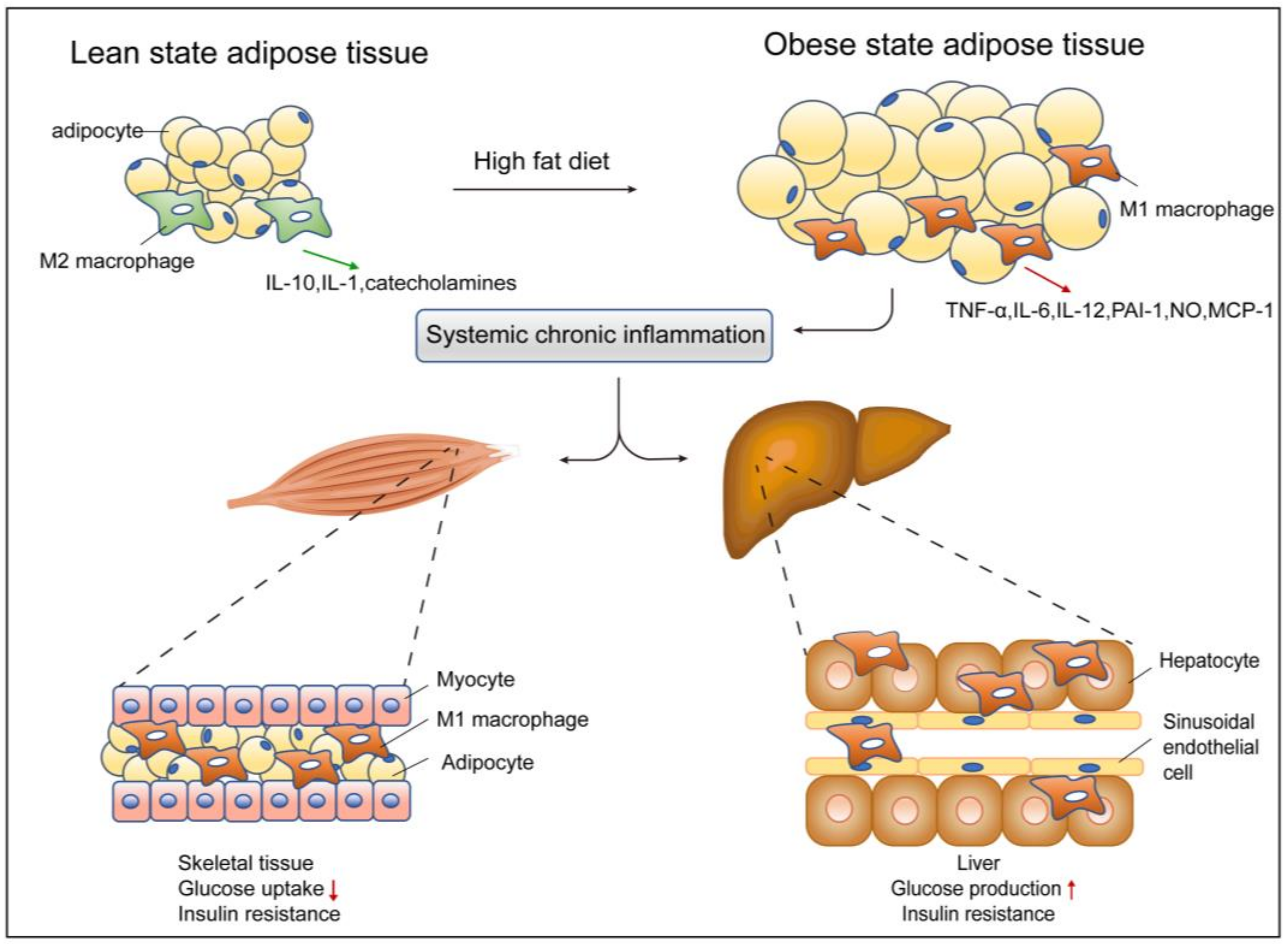
3.1. Adipose Tissue and ATMs
3.2. Liver and Kupffer Cells
3.3. Muscle and Macrophages
3.4. Pancreas and Macrophages
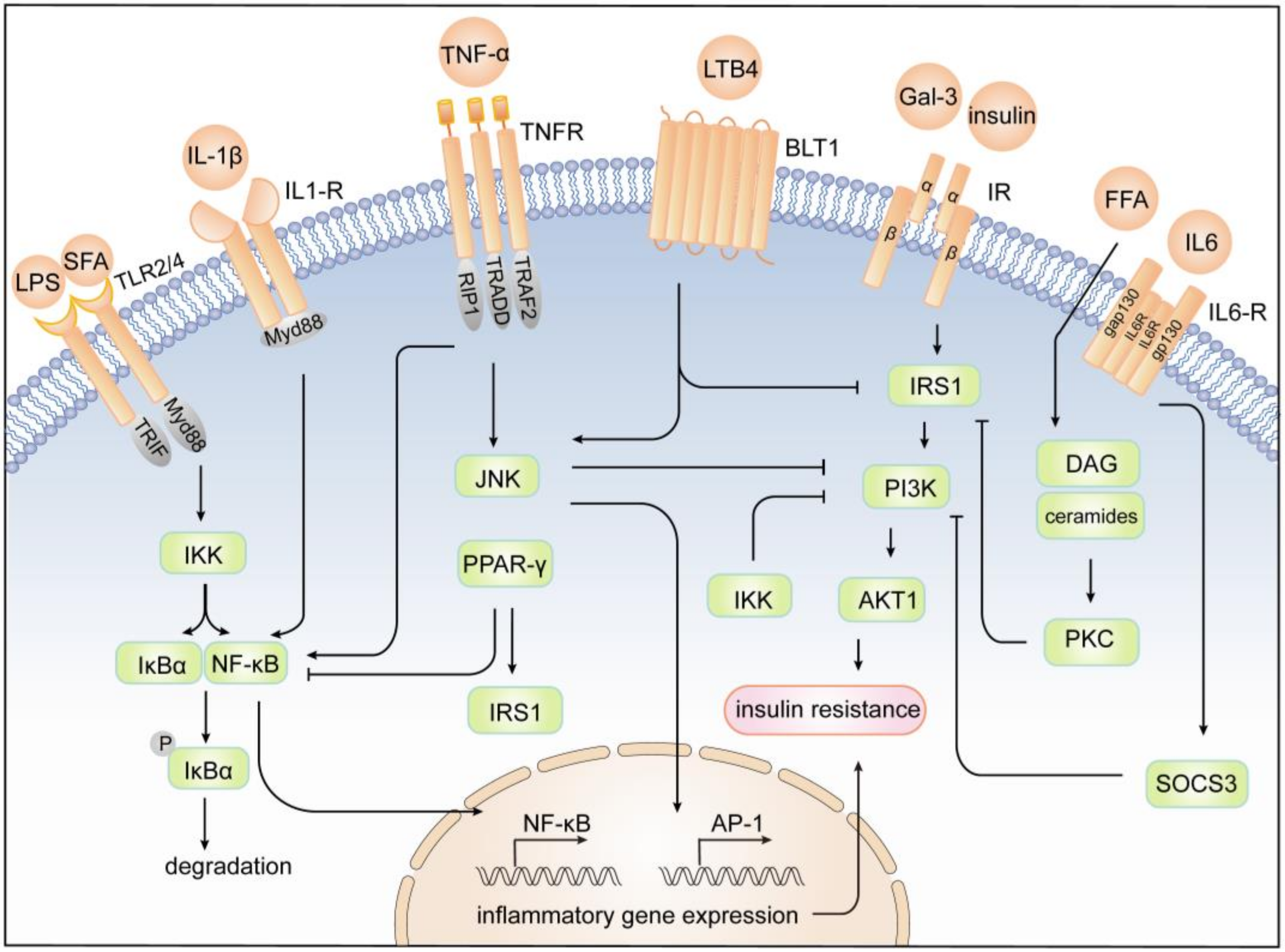
4. Mechanism of Inflammation and Insulin Resistance
4.1. JNK Pathway
4.2. IKK/NF-κB Pathway
4.3. JAK/STAT Pathway
4.4. Other Pathways
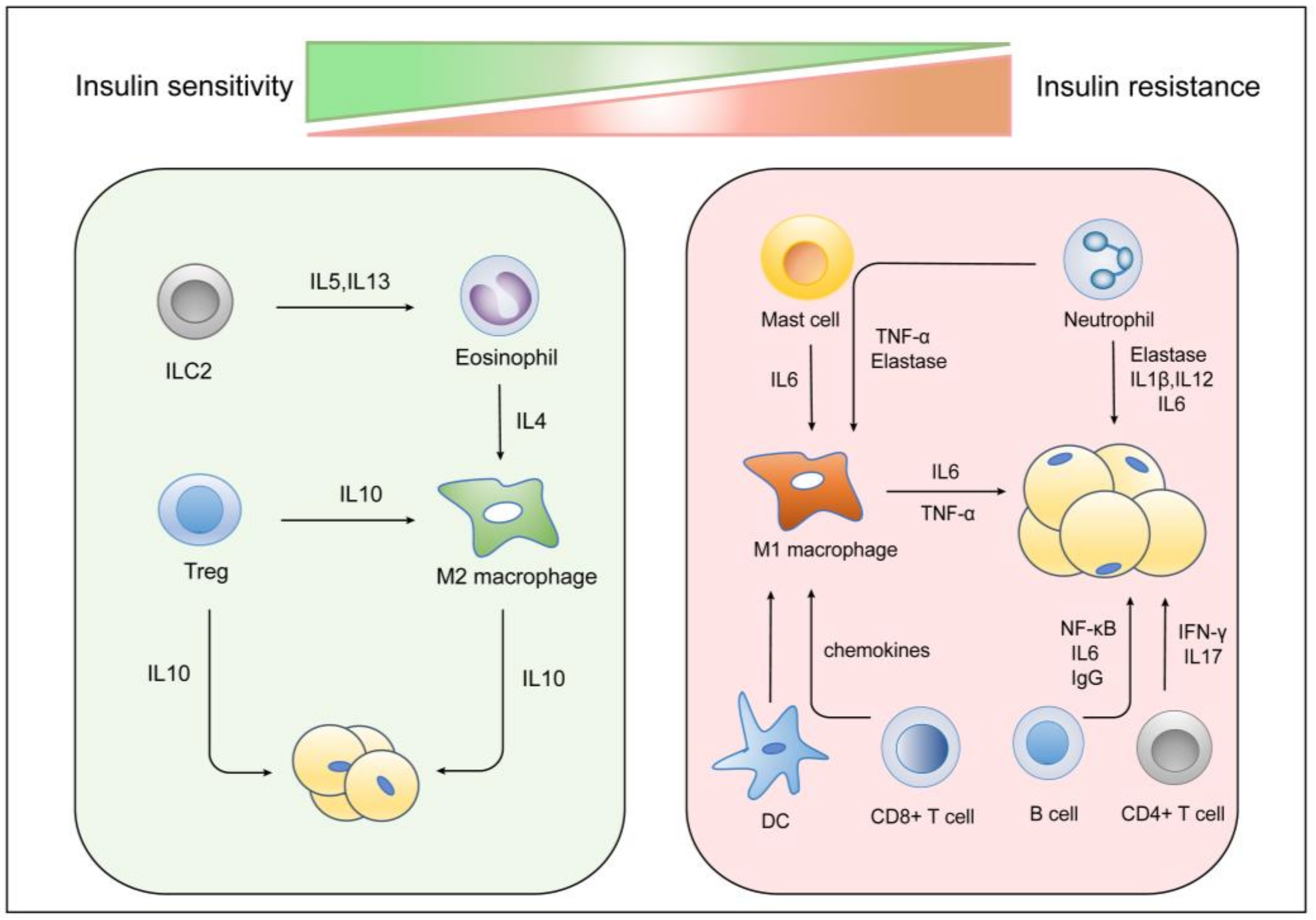
5. Other Immune Cells and Insulin Resistance
6. Macrophage-Related T2D Therapeutic Targets
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef]
- Bhurosy, T.; Jeewon, R. Overweight and obesity epidemic in developing countries: A problem with diet, physical activity, or socioeconomic status? Sci. World J. 2014, 2014, 964236. [Google Scholar] [CrossRef]
- Pulgaron, E.R.; Delamater, A.M. Obesity and type 2 diabetes in children: Epidemiology and treatment. Curr. Diabetes Rep. 2014, 14, 508. [Google Scholar] [CrossRef] [PubMed]
- Zubrzycki, A.; Cierpka-Kmiec, K.; Kmiec, Z.; Wronska, A. The role of low-calorie diets and intermittent fasting in the treatment of obesity and type-2 diabetes. J. Physiol. Pharmacol. 2018, 69, 663–683. [Google Scholar]
- Singh, G.M.; Danaei, G.; Farzadfar, F.; Stevens, G.A.; Woodward, M.; Wormser, D.; Kaptoge, S.; Whitlock, G.; Qiao, Q.; Lewington, S.; et al. The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: A pooled analysis. PLoS ONE 2013, 8, e65174. [Google Scholar] [CrossRef] [PubMed]
- Patell, R.; Dosi, R.; Joshi, H.; Sheth, S.; Shah, P.; Jasdanwala, S. Non-Alcoholic Fatty Liver Disease (NAFLD) in Obesity. J. Clin. Diagn. Res. 2014, 8, 62–66. [Google Scholar] [PubMed]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Lu, F.B.; Hu, E.D.; Xu, L.M.; Chen, L.; Wu, J.L.; Li, H.; Chen, D.Z.; Chen, Y.P. The relationship between obesity and the severity of non-alcoholic fatty liver disease: Systematic review and meta-analysis. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Teoh, H.; Verma, S. Obesity and atherosclerosis: Mechanistic insights. Can. J. Cardiol. 2015, 31, 177–183. [Google Scholar] [CrossRef]
- Reardon, C.A.; Lingaraju, A.; Schoenfelt, K.Q.; Zhou, G.; Cui, C.; Jacobs-El, H.; Babenko, I.; Hoofnagle, A.; Czyz, D.; Shuman, H.; et al. Obesity and Insulin Resistance Promote Atherosclerosis through an IFNgamma-Regulated Macrophage Protein Network. Cell Rep. 2018, 23, 3021–3030. [Google Scholar] [CrossRef]
- El-Khani, U.; Ahmed, A.; Hakky, S.; Nehme, J.; Cousins, J.; Chahal, H.; Purkayastha, S. The impact of obesity surgery on musculoskeletal disease. Obes. Surg. 2014, 24, 2175–2192. [Google Scholar] [CrossRef]
- Salaun, H.; Thariat, J.; Vignot, M.; Merrouche, Y.; Vignot, S. Obesity and cancer. Bull. Cancer 2017, 104, 30–41. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity-Cancer Link-A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef]
- Barazzoni, R.; Gortan Cappellari, G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight Disord. 2018, 23, 149–157. [Google Scholar] [CrossRef]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef]
- Rafaqat, S.; Rafaqat, S.; Rafaqat, S. Pathophysiological aspects of insulin resistance in Atrial Fibrillation: Novel therapeutic approaches. Int. J. Arrhythm. 2022, 23, 6. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. North Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Dufour, S.; Petersen, K.F.; LeBon, V.; Enoksson, S.; Ma, Y.Z.; Savoye, M.; Rothman, D.L.; Shulman, G.I.; Caprio, S. Assessment of skeletal muscle triglyceride content by (1)H nuclear magnetic resonance spectroscopy in lean and obese adolescents: Relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes 2002, 51, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Bernroider, E. Hepatic glucose metabolism in humans—Its role in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 365–383. [Google Scholar] [CrossRef]
- Araujo, E.P.; De Souza, C.T.; Ueno, M.; Cintra, D.E.; Bertolo, M.B.; Carvalheira, J.B.; Saad, M.J.; Velloso, L.A. Infliximab restores glucose homeostasis in an animal model of diet-induced obesity and diabetes. Endocrinology 2007, 148, 5991–5997. [Google Scholar] [CrossRef]
- Wu, M.; Wang, X.; Duan, Q.; Lu, T. Arachidonic acid can significantly prevent early insulin resistance induced by a high-fat diet. Ann. Nutr. Metab. 2007, 51, 270–276. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Banerji, M.A.; Bray, G.A.; Buchanan, T.A.; Clement, S.; Henry, R.R.; Kitabchi, A.E.; Mudaliar, S.; Musi, N.; Ratner, R.; et al. Determinants of glucose tolerance in impaired glucose tolerance at baseline in the Actos Now for Prevention of Diabetes (ACT NOW) study. Diabetologia 2010, 53, 435–445. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Asghar, A.; Sheikh, N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell. Immunol. 2017, 315, 18–26. [Google Scholar] [CrossRef]
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73. [Google Scholar] [CrossRef]
- Unanue, E.R. Antigen-presenting function of the macrophage. Annu. Rev. Immunol. 1984, 2, 395–428. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Vogel, D.Y.; Glim, J.E.; Stavenuiter, A.W.; Breur, M.; Heijnen, P.; Amor, S.; Dijkstra, C.D.; Beelen, R.H. Human macrophage polarization in vitro: Maturation and activation methods compared. Immunobiology 2014, 219, 695–703. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef]
- Shaul, M.E.; Bennett, G.; Strissel, K.J.; Greenberg, A.S.; Obin, M.S. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet--induced obesity in mice. Diabetes 2010, 59, 1171–1181. [Google Scholar] [CrossRef]
- Verreck, F.A.W.; de Boer, T.; Langenberg, D.M.L.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Mosser, D.M. The many faces of macrophage activation. J. Leukoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Subramanian, M.; Tabas, I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur. J. Immunol. 2011, 41, 2515–2518. [Google Scholar] [CrossRef]
- Avila-Ponce de León, U.; Vázquez-Jiménez, A.; Matadamas-Guzman, M.; Pelayo, R.; Resendis-Antonio, O. Transcriptional and microenvironmental landscape of macrophage transition in cancer: A boolean analysis. Front Immunol. 2021, 12, 642842. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Galvan-Pena, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar]
- Jaitin, D.A.; Adlung, L.; Thaiss, C.A.; Weiner, A.; Li, B.; Descamps, H.; Lundgren, P.; Bleriot, C.; Liu, Z.; Deczkowska, A.; et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 2019, 178, 686–698.e14. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Rao, V.H.; Shao, Z.; Agrawal, D.K. Dendritic Cells Expressing Triggering Receptor Expressed on Myeloid Cells-1 Correlate with Plaque Stability in Symptomatic and Asymptomatic Patients with Carotid Stenosis. PLoS ONE 2016, 11, e0154802. [Google Scholar] [CrossRef]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 Nuclear Factor- B Overexpression in Tumor-Associated Macrophages Inhibits M1 Inflammatory Responses and Antitumor Resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Escribese, M.M.; Casas, M.; Corbi, A.L. Influence of low oxygen tensions on macrophage polarization. Immunobiology 2012, 217, 1233–1240. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Raes, G.; Van den Bergh, R.; De Baetselier, P.; Ghassabeh, G.H.; Scotton, C.; Locati, M.; Mantovani, A.; Sozzani, S. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J. Immunol. 2005, 174, 6561. [Google Scholar] [CrossRef]
- Beattie, L.; Sawtell, A.; Mann, J.; Frame, T.C.M.; Teal, B.; de Labastida Rivera, F.; Brown, N.; Walwyn-Brown, K.; Moore, J.; MacDonald, S.; et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J. Hepatol. 2016, 65, 758–768. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Charo, I.F. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab. 2007, 6, 96–98. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Patsouris, D.; Li, P.-P.; Thapar, D.; Chapman, J.; Olefsky, J.M.; Neels, J.G. Ablation of CD11c-Positive Cells Normalizes Insulin Sensitivity in Obese Insulin Resistant Animals. Cell Metab. 2008, 8, 301–309. [Google Scholar] [CrossRef]
- Subramanian, V.; Ferrante, A.W., Jr. Obesity, inflammation, and macrophages. Nestle Nutr. Workshop Ser. Pediatr. Program. 2009, 63, 151–159. [Google Scholar]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Hassnain Waqas, S.F.; Noble, A.; Hoang, A.C.; Ampem, G.; Popp, M.; Strauss, S.; Guille, M.; Röszer, T. Adipose tissue macrophages develop from bone marrow-independent progenitors in Xenopus laevis and mouse. J. Leukoc. Biol. 2017, 102, 845–855. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Cho, C.H.; Koh, Y.J.; Han, J.; Sung, H.K.; Jong Lee, H.; Morisada, T.; Schwendener, R.A.; Brekken, R.A.; Kang, G.; Oike, Y.; et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ. Res. 2007, 100, e47–e57. [Google Scholar] [CrossRef]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Hill, D.A.; Lim, H.W.; Kim, Y.H.; Ho, W.Y.; Foong, Y.H.; Nelson, V.L.; Nguyen, H.; Chegireddy, K.; Kim, J.; Habertheuer, A.; et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 2018, 115, E5096–E5105. [Google Scholar] [CrossRef]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef]
- Fujisaka, S. The role of adipose tissue M1/M2 macrophages in type 2 diabetes mellitus. Diabetol. Int. 2021, 12, 74–79. [Google Scholar] [CrossRef]
- Wu, D.; Molofsky, A.B.; Liang, H.E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W., Jr.; et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef]
- Liang, W.; Qi, Y.; Yi, H.; Mao, C.; Meng, Q.; Wang, H.; Zheng, C. The Roles of Adipose Tissue Macrophages in Human Disease. Front. Immunol. 2022, 13, 908749. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Qiu, Y.; Cui, X.; Goh, Y.P.; Mwangi, J.; David, T.; Mukundan, L.; Brombacher, F.; Locksley, R.M.; Chawla, A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011, 480, 104–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, P.; Cui, R.; Zhang, M.; Li, H.; Qian, C.; Sheng, C.; Qu, S.; Bu, L. Eosinophils Reduce Chronic Inflammation in Adipose Tissue by Secreting Th2 Cytokines and Promoting M2 Macrophages Polarization. Int. J. Endocrinol. 2015, 2015, 565760. [Google Scholar] [CrossRef]
- Fischer, K.; Ruiz, H.H.; Jhun, K.; Finan, B.; Oberlin, D.J.; van der Heide, V.; Kalinovich, A.V.; Petrovic, N.; Wolf, Y.; Clemmensen, C.; et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 2017, 23, 623–630. [Google Scholar] [CrossRef]
- Camell, C.D.; Sander, J.; Spadaro, O.; Lee, A.; Nguyen, K.Y.; Wing, A.; Goldberg, E.L.; Youm, Y.H.; Brown, C.W.; Elsworth, J.; et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017, 550, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Macrophages dispose of catecholamines in adipose tissue. Nat. Med. 2017, 23, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Gornicka, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Kashyap, S.; Schauer, P.R.; Feldstein, A.E. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 2010, 285, 3428–3438. [Google Scholar] [CrossRef]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef]
- Poret, J.M.; Souza-Smith, F.; Marcell, S.J.; Gaudet, D.A.; Tzeng, T.H.; Braymer, H.D.; Harrison-Bernard, L.M.; Primeaux, S.D. High fat diet consumption differentially affects adipose tissue inflammation and adipocyte size in obesity-prone and obesity-resistant rats. Int. J. Obes. 2018, 42, 535–541. [Google Scholar] [CrossRef]
- Ramkhelawon, B.; Hennessy, E.J.; Ménager, M.; Ray, T.D.; Sheedy, F.J.; Hutchison, S.; Wanschel, A.; Oldebeken, S.; Geoffrion, M.; Spiro, W.; et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 2014, 20, 377–384. [Google Scholar] [CrossRef]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef]
- Gao, B. Basic liver immunology. Cell. Mol. Immunol. 2016, 13, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, H.; Wang, J.; Burhan, D.; Shang, R.; Wang, P.; Zhou, Y.; Li, R.; Liang, B.; Evert, K.; et al. mTORC2 signaling is necessary for timely liver regeneration after partial hepatectomy. Am. J. Pathol. 2020, 190, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.L.J.; Xu, Y.; Cao, H. Role of macrophages in experimental liver injury and repair in mice. Exp. Ther. Med. 2019, 17, 3835–3847. [Google Scholar] [CrossRef]
- Jager, J.; Aparicio-Vergara, M.; Aouadi, M. Liver innate immune cells and insulin resistance: The multiple facets of Kupffer cells. J. Intern. Med. 2016, 280, 209–220. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef]
- Ying, W.; Mahata, S.; Bandyopadhyay, G.K.; Zhou, Z.; Wollam, J.; Vu, J.; Mayoral, R.; Chi, N.W.; Webster, N.; Corti, A.; et al. Catestatin Inhibits Obesity-Induced Macrophage Infiltration and Inflammation in the Liver and Suppresses Hepatic Glucose Production, Leading to Improved Insulin Sensitivity. Diabetes 2018, 67, 841–848. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Meshkani, R.; Adeli, K. Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin. Biochem. 2009, 42, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H. Fat in the liver and insulin resistance. Ann. Med. 2005, 37, 347–356. [Google Scholar] [CrossRef]
- Lanthier, N.; Molendi-Coste, O.; Horsmans, Y.; van Rooijen, N.; Cani, P.D.; Leclercq, I.A. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G107–G116. [Google Scholar] [CrossRef]
- Neyrinck, A.M.; Cani, P.D.; Dewulf, E.M.; De Backer, F.; Bindels, L.B.; Delzenne, N.M. Critical role of Kupffer cells in the management of diet-induced diabetes and obesity. Biochem. Biophys. Res. Commun. 2009, 385, 351–356. [Google Scholar] [CrossRef]
- Lopez, B.G.; Tsai, M.S.; Baratta, J.L.; Longmuir, K.J.; Robertson, R.T. Characterization of Kupffer cells in livers of developing mice. Comp. Hepatol. 2011, 10, 2. [Google Scholar] [CrossRef]
- Neyrinck, A.M.; Gomez, C.; Delzenne, N.M. Precision-cut liver slices in culture as a tool to assess the physiological involvement of Kupffer cells in hepatic metabolism. Comp. Hepatol. 2004, 3 (Suppl. 1), S45. [Google Scholar] [CrossRef][Green Version]
- Wunderlich, F.T.; Strohle, P.; Konner, A.C.; Gruber, S.; Tovar, S.; Bronneke, H.S.; Juntti-Berggren, L.; Li, L.S.; van Rooijen, N.; Libert, C.; et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010, 12, 237–249. [Google Scholar] [CrossRef]
- Van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflug. Arch. 2017, 469, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Haines, M.S.; Dichtel, L.E.; Santoso, K.; Torriani, M.; Miller, K.K.; Bredella, M.A. Association between muscle mass and insulin sensitivity independent of detrimental adipose depots in young adults with overweight/obesity. Int. J. Obes. 2020, 44, 1851–1858. [Google Scholar] [CrossRef]
- Son, J.W.; Lee, S.S.; Kim, S.R.; Yoo, S.J.; Cha, B.Y.; Son, H.Y.; Cho, N.H. Low muscle mass and risk of type 2 diabetes in middle-aged and older adults: Findings from the KoGES. Diabetologia 2017, 60, 865–872. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Bilan, P.J.; Fink, L.N.; Klip, A. Cross-talk between skeletal muscle and immune cells: Muscle-derived mediators and metabolic implications. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E453–E465. [Google Scholar] [CrossRef]
- Khan, I.M.; Perrard, X.Y.; Brunner, G.; Lui, H.; Sparks, L.M.; Smith, S.R.; Wang, X.; Shi, Z.Z.; Lewis, D.E.; Wu, H.; et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int. J. Obes. 2015, 39, 1607–1618. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Nolen, G.T.; Phanavanh, B.; Starks, T.; Gurley, C.; Simpson, P.; McGehee, R.E., Jr.; Kern, P.A.; et al. Muscle inflammatory response and insulin resistance: Synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1300–E1310. [Google Scholar] [CrossRef]
- Patsouris, D.; Cao, J.J.; Vial, G.; Bravard, A.; Lefai, E.; Durand, A.; Durand, C.; Chauvin, M.A.; Laugerette, F.; Debard, C.; et al. Insulin resistance is associated with MCP1-mediated macrophage accumulation in skeletal muscle in mice and humans. PLoS ONE 2014, 9, e110653. [Google Scholar] [CrossRef]
- Hong, E.G.; Ko, H.J.; Cho, Y.R.; Kim, H.J.; Ma, Z.; Yu, T.Y.; Friedline, R.H.; Kurt-Jones, E.; Finberg, R.; Fischer, M.A.; et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 2009, 58, 2525–2535. [Google Scholar] [CrossRef]
- Rachek, L.I. Free Fatty Acids and Skeletal Muscle Insulin Resistance. Glucose Homeostatis and the Pathogenesis of Diabetes Mellitus. Prog. Mol. Biol. Transl. Sci. 2014, 121, 267–292. [Google Scholar]
- Boren, J.; Taskinen, M.R.; Olofsson, S.O.; Levin, M. Ectopic lipid storage and insulin resistance: A harmful relationship. J. Intern. Med. 2013, 274, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Banaei-Bouchareb, L.; Gouon-Evans, V.; Samara-Boustani, D.; Castellotti, M.C.; Czernichow, P.; Pollard, J.W.; Polak, M. Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J. Leukoc. Biol. 2004, 76, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Schumann, D.M.; Sauter, N.; Ellingsgaard, H.; Bosco, D.; Baertschiger, R.; Iwakura, Y.; Oberholzer, J.; Wollheim, C.B.; Gauthier, B.R.; et al. Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes 2006, 55, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef]
- Kamata, K.; Mizukami, H.; Inaba, W.; Tsuboi, K.; Tateishi, Y.; Yoshida, T.; Yagihashi, S. Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 2014, 21, 191–201. [Google Scholar] [CrossRef]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Han, Z.; Boyle, D.L.; Chang, L.; Bennett, B.; Karin, M.; Yang, L.; Manning, A.M.; Firestein, G.S. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J. Clin. Investig. 2001, 108, 73–81. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, L.; Dong, X.; Hu, X.; Zhou, L.; Liu, Q.; Song, B.; Wu, Q.; Li, L. The c-Jun N-terminal kinase (JNK) pathway is activated in human interstitial cystitis (IC) and rat protamine sulfate induced cystitis. Sci. Rep. 2016, 6, 19670. [Google Scholar] [CrossRef]
- Chen, Y.R.; Tan, T.H. The c-Jun N-terminal kinase pathway and apoptotic signaling (review). Int. J. Oncol. 2000, 16, 651–662. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Rios, R.; Silva, H.; Carneiro, N.V.Q.; Pires, A.O.; Carneiro, T.C.B.; Costa, R.D.S.; Marques, C.R.; Machado, M.; Velozo, E.; Silva, T.; et al. Solanum paniculatum L. decreases levels of inflammatory cytokines by reducing NFKB, TBET and GATA3 gene expression in vitro. J. Ethnopharmacol. 2017, 209, 32–40. [Google Scholar] [CrossRef]
- Yang, R.; Tonnesseen, T.I. DAMPs and sterile inflammation in drug hepatotoxicity. Hepatol. Int. 2019, 13, 42–50. [Google Scholar] [CrossRef]
- Franklin, T.C.; Xu, C.; Duman, R.S. Depression and sterile inflammation: Essential role of danger associated molecular patterns. Brain Behav. Immun. 2018, 72, 2–13. [Google Scholar] [CrossRef]
- Patel, S. Danger-Associated Molecular Patterns (DAMPs): The Derivatives and Triggers of Inflammation. Curr. Allerg. Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J. Cell Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- De Taeye, B.M.; Novitskaya, T.; McGuinness, O.P.; Gleaves, L.; Medda, M.; Covington, J.W.; Vaughan, D.E. Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E713–E725. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Tang, F.; Tang, G.; Xiang, J.; Dai, Q.; Rosner, M.R.; Lin, A. The absence of NF-kappaB-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 2002, 22, 8571–8579. [Google Scholar] [CrossRef]
- Bandyopadhyay, G.K.; Yu, J.G.; Ofrecio, J.; Olefsky, J.M. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes 2005, 54, 2351–2359. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. cJun NH2-terminal kinase 1 (JNK1): Roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010, 35, 490–496. [Google Scholar] [CrossRef]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.; Corbould, A.; Dunaif, A.; White, M.F. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 181–189. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef]
- Tanti, J.F.; Gremeaux, T.; van Obberghen, E.; Le Marchand-Brustel, Y. Serine/threonine phosphorylation of insulin receptor substrate 1 modulates insulin receptor signaling. J. Biol. Chem. 1994, 269, 6051–6057. [Google Scholar] [CrossRef]
- Li, P.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Maris, M.; Ofrecio, J.M.; et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef]
- Higham, A.; Cadden, P.; Southworth, T.; Rossall, M.; Kolsum, U.; Lea, S.; Knowles, R.; Singh, D. Leukotriene B4 levels in sputum from asthma patients. ERJ Open Res. 2016, 2, 00088–2015. [Google Scholar] [CrossRef]
- Brandt, S.L.; Serezani, C.H. Too much of a good thing: How modulating LTB4 actions restore host defense in homeostasis or disease. Semin. Immunol. 2017, 33, 37–43. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.H.; Bazuine, M.; Lumeng, C.N.; Geletka, L.M.; Mowers, J.; White, N.M.; Ma, J.T.; Zhou, J.; Qi, N.; Westcott, D.; et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009, 138, 961–975. [Google Scholar] [CrossRef]
- Gao, D.; Madi, M.; Ding, C.; Fok, M.; Steele, T.; Ford, C.; Hunter, L.; Bing, C. Interleukin-1beta mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E289–E304. [Google Scholar] [CrossRef]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.; Joosten, L.A.; Tack, C.J.; Netea, M.G. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef]
- Peiro, C.; Lorenzo, O.; Carraro, R.; Sanchez-Ferrer, C.F. IL-1beta Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front. Pharmacol. 2017, 8, 363. [Google Scholar] [CrossRef]
- Boni-Schnetzler, M.; Donath, M.Y. How biologics targeting the IL-1 system are being considered for the treatment of type 2 diabetes. Br. J. Clin. Pharmacol. 2013, 76, 263–268. [Google Scholar] [CrossRef]
- Chawla, A.; Barak, Y.; Nagy, L.; Liao, D.; Tontonoz, P.; Evans, R.M. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 2001, 7, 48–52. [Google Scholar] [CrossRef]
- Olefsky, J.; Saltiel, A. PPARγ and the Treatment of Insulin Resistance. Trends Endocrinol. Metab. 2000, 11, 362–368. [Google Scholar] [CrossRef]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Henkel, J.; Neuschafer-Rube, F.; Pathe-Neuschafer-Rube, A.; Puschel, G.P. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology 2009, 50, 781–790. [Google Scholar] [CrossRef]
- Gross, B.; Staels, B. PPAR agonists: Multimodal drugs for the treatment of type-2 diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 687–710. [Google Scholar] [CrossRef]
- Sugii, S.; Olson, P.; Sears, D.D.; Saberi, M.; Atkins, A.R.; Barish, G.D.; Hong, S.H.; Castro, G.L.; Yin, Y.Q.; Nelson, M.C.; et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. USA 2009, 106, 22504–22509. [Google Scholar] [CrossRef]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPARgamma and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef]
- Feng, X.; Weng, D.; Zhou, F.; Owen, Y.D.; Qin, H.; Zhao, J.; Wen, Y.; Huang, Y.; Chen, J.; Fu, H.; et al. Activation of PPARgamma by a Natural Flavonoid Modulator, Apigenin Ameliorates Obesity-Related Inflammation Via Regulation of Macrophage Polarization. eBioMedicine 2016, 9, 61–76. [Google Scholar] [CrossRef]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef]
- Nonogaki, K.; Fuller, G.M.; Fuentes, N.L.; Moser, A.H.; Staprans, I.; Grunfeld, C.; Feingold, K.R. Interleukin-6 stimulates hepatic triglyceride secretion in rats. Endocrinology 1995, 136, 2143–2149. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334 Pt 2, 297–314. [Google Scholar] [CrossRef]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef]
- Wallenius, V.; Wallenius, K.; Ahren, B.; Rudling, M.; Carlsten, H.; Dickson, S.L.; Ohlsson, C.; Jansson, J.O. Interleukin-6-deficient mice develop mature-onset obesity. Nat. Med. 2002, 8, 75–79. [Google Scholar] [CrossRef]
- Kurauti, M.A.; Costa-Junior, J.M.; Ferreira, S.M.; Santos, G.J.; Sponton, C.H.G.; Carneiro, E.M.; Telles, G.D.; Chacon-Mikahil, M.; Cavaglieri, C.R.; Rezende, L.F.; et al. Interleukin-6 increases the expression and activity of insulin-degrading enzyme. Sci. Rep. 2017, 7, 46750. [Google Scholar] [CrossRef]
- Chen, X.; Xun, K.; Chen, L.; Wang, Y. TNF-alpha, a potent lipid metabolism regulator. Cell Biochem. Funct. 2009, 27, 407–416. [Google Scholar] [CrossRef]
- Starnes, H.F., Jr.; Warren, R.S.; Jeevanandam, M.; Gabrilove, J.L.; Larchian, W.; Oettgen, H.F.; Brennan, M.F. Tumor necrosis factor and the acute metabolic response to tissue injury in man. J. Clin. Investig. 1988, 82, 1321–1325. [Google Scholar] [CrossRef]
- Wedell-Neergaard, A.S.; Lang Lehrskov, L.; Christensen, R.H.; Legaard, G.E.; Dorph, E.; Larsen, M.K.; Launbo, N.; Fagerlind, S.R.; Seide, S.K.; Nymand, S.; et al. Exercise-Induced Changes in Visceral Adipose Tissue Mass Are Regulated by IL-6 Signaling: A Randomized Controlled Trial. Cell Metab. 2019, 29, 844–855.e3. [Google Scholar] [CrossRef]
- Wolsk, E.; Mygind, H.; Grondahl, T.S.; Pedersen, B.K.; van Hall, G. IL-6 selectively stimulates fat metabolism in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E832–E840. [Google Scholar] [CrossRef]
- Speaker, K.J.; Fleshner, M. Interleukin-1 beta: A potential link between stress and the development of visceral obesity. BMC Physiol. 2012, 12, 8. [Google Scholar] [CrossRef]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Meng, L.; Li, L.L.; Ma, L.N.; Mao, X.M. Plasma Free Fatty Acids Metabolic Profile Among Uyghurs and Kazaks With or Without Type 2 Diabetes Based on GC-MS. Exp. Clin. Endocrinol. Diabetes 2018, 126, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Makarova, E.; Makrecka-Kuka, M.; Vilks, K.; Volska, K.; Sevostjanovs, E.; Grinberga, S.; Zarkova-Malkova, O.; Dambrova, M.; Liepinsh, E. Decreases in Circulating Concentrations of Long-Chain Acylcarnitines and Free Fatty Acids During the Glucose Tolerance Test Represent Tissue-Specific Insulin Sensitivity. Front. Endocrinol. 2019, 10, 870. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Birkenfeld, A.L.; Jurczak, M.J.; Kanda, S.; Guigni, B.A.; Jiang, D.C.; Zhang, D.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc. Natl. Acad. Sci. USA 2011, 108, 5748–5752. [Google Scholar] [CrossRef]
- Metcalfe, L.K.; Smith, G.C.; Turner, N. Defining lipid mediators of insulin resistance—Controversies and challenges. J. Mol. Endocrinol. 2018, 62, R65–R82. [Google Scholar] [CrossRef]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef]
- Kim, J.K.; Fillmore, J.J.; Sunshine, M.J.; Albrecht, B.; Higashimori, T.; Kim, D.W.; Liu, Z.X.; Soos, T.J.; Cline, G.W.; O’Brien, W.R.; et al. PKC-θ knockout mice are protected from fat-induced insulin resistance. J. Clin. Investig. 2004, 114, 823–827. [Google Scholar] [CrossRef]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef]
- Fox, T.E.; Houck, K.L.; O’Neill, S.M.; Nagarajan, M.; Stover, T.C.; Pomianowski, P.T.; Unal, O.; Yun, J.K.; Naides, S.J.; Kester, M. Ceramide recruits and activates protein kinase C ζ (PKCζ) within structured membrane microdomains. J. Biol. Chem. 2007, 282, 12450–12457. [Google Scholar] [CrossRef]
- Chavez, J.A.; Knotts, T.A.; Wang, L.P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef]
- Li, P.; Liu, S.; Lu, M.; Bandyopadhyay, G.; Oh, D.; Imamura, T.; Johnson, A.; Sears, D.; Shen, Z.; Cui, B.; et al. Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell 2016, 167, 973–984.e12. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int. J. Mol. Med. 2018, 41, 599–614. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef]
- Bonamichi, B.; Lee, J. Unusual Suspects in the Development of Obesity-Induced Inflammation and Insulin Resistance: NK cells, iNKT cells, and ILCs. Diabetes Metab. J. 2017, 41, 229–250. [Google Scholar] [CrossRef]
- Jonckheere, A.C.; Bullens, D.M.A.; Seys, S.F. Innate lymphoid cells in asthma: Pathophysiological insights from murine models to human asthma phenotypes. Curr. Opin. Allerg. Clin. Immunol. 2019, 19, 53–60. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Nussbaum, J.C.; Liang, H.E.; Van Dyken, S.J.; Cheng, L.E.; Mohapatra, A.; Chawla, A.; Locksley, R.M. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 2013, 210, 535–549. [Google Scholar] [CrossRef]
- Liu, W.; Zeng, Q.; Chen, Y.; Luo, R.Z. Role of Leptin/Osteopontin Axis in the Function of Eosinophils in Allergic Rhinitis with Obesity. Mediat. Inflamm. 2018, 2018, 9138904. [Google Scholar] [CrossRef]
- Bolus, W.R.; Kennedy, A.J.; Hasty, A.H. Obesity-induced reduction of adipose eosinophils is reversed with low-calorie dietary intervention. Physiol. Rep. 2018, 6, e13919. [Google Scholar] [CrossRef] [PubMed]
- Zelechowska, P.; Agier, J.; Kozlowska, E.; Brzezinska-Blaszczyk, E. Mast cells participate in chronic low-grade inflammation within adipose tissue. Obes. Rev. 2018, 19, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Milling, S. Adipokines and the control of mast cell functions: From obesity to inflammation? Immunology 2019, 158, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.M.; Shi, G.P. Emerging role of mast cells and macrophages in cardiovascular and metabolic diseases. Endocr. Rev. 2012, 33, 71–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clement, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef]
- Stefanovic-Racic, M.; Yang, X.; Turner, M.S.; Mantell, B.S.; Stolz, D.B.; Sumpter, T.L.; Sipula, I.J.; Dedousis, N.; Scott, D.K.; Morel, P.A.; et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes 2012, 61, 2330–2339. [Google Scholar] [CrossRef]
- Xu, X.; Su, S.; Wang, X.; Barnes, V.; De Miguel, C.; Ownby, D.; Pollock, J.; Snieder, H.; Chen, W.; Wang, X. Obesity is associated with more activated neutrophils in African American male youth. Int. J. Obes. 2015, 39, 26–32. [Google Scholar] [CrossRef]
- Medeiros, N.I.; Mattos, R.T.; Menezes, C.A.; Fares, R.C.G.; Talvani, A.; Dutra, W.O.; Rios-Santos, F.; Correa-Oliveira, R.; Gomes, J. IL-10 and TGF-beta unbalanced levels in neutrophils contribute to increase inflammatory cytokine expression in childhood obesity. Eur. J. Nutr. 2018, 57, 2421–2430. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef]
- Pan, Y.; Choi, J.H.; Shi, H.; Zhang, L.; Su, S.; Wang, X. Discovery and Validation of a Novel Neutrophil Activation Marker Associated with Obesity. Sci. Rep. 2019, 9, 3433. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications. Cells 2022, 11, 1883. [Google Scholar] [CrossRef]
- Chen, H.; Sun, L.; Feng, L.; Yin, Y.; Zhang, W. Role of Innate lymphoid Cells in Obesity and Insulin Resistance. Front. Endocrinol. 2022, 13, 855197. [Google Scholar] [CrossRef]
- McDonnell, M.E.; Ganley-Leal, L.M.; Mehta, A.; Bigornia, S.J.; Mott, M.; Rehman, Q.; Farb, M.G.; Hess, D.T.; Joseph, L.; Gokce, N.; et al. B lymphocytes in human subcutaneous adipose crown-like structures. Obesity 2012, 20, 1372–1378. [Google Scholar] [CrossRef]
- Acosta, J.R.; Douagi, I.; Andersson, D.P.; Backdahl, J.; Ryden, M.; Arner, P.; Laurencikiene, J. Increased fat cell size: A major phenotype of subcutaneous white adipose tissue in non-obese individuals with type 2 diabetes. Diabetologia 2016, 59, 560–570. [Google Scholar] [CrossRef]
- Khan, I.M.; Dai Perrard, X.Y.; Perrard, J.L.; Mansoori, A.; Smith, C.W.; Wu, H.; Ballantyne, C.M. Attenuated adipose tissue and skeletal muscle inflammation in obese mice with combined CD4+ and CD8+ T cell deficiency. Atherosclerosis 2014, 233, 419–428. [Google Scholar] [CrossRef]
- Pacifico, L.; Di Renzo, L.; Anania, C.; Osborn, J.F.; Ippoliti, F.; Schiavo, E.; Chiesa, C. Increased T-helper interferon-gamma-secreting cells in obese children. Eur. J. Endocrinol. 2006, 154, 691–697. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Hara, K.; Ueki, K.; Sugiura, S.; Yoshimura, K.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef]
- Zhai, X.; Qian, G.; Wang, Y.; Chen, X.; Lu, J.; Zhang, Y.; Huang, Q.; Wang, Q. Elevated B Cell Activation is Associated with Type 2 Diabetes Development in Obese Subjects. Cell. Physiol. Biochem. 2016, 38, 1257–1266. [Google Scholar] [CrossRef]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [PubMed]
- Carpino, P.A.; Goodwin, B. Diabetes area participation analysis: A review of companies and targets described in the 2008–2010 patent literature. Expert Opin. Ther. Pat. 2010, 20, 1627–1651. [Google Scholar] [CrossRef]
- Osborn, O.; Brownell, S.E.; Sanchez-Alavez, M.; Salomon, D.; Gram, H.; Bartfai, T. Treatment with an Interleukin 1 beta antibody improves glycemic control in diet-induced obesity. Cytokine 2008, 44, 141–148. [Google Scholar] [CrossRef]
- Mao, Y.; Mohan, R.; Zhang, S.; Tang, X. MicroRNAs as pharmacological targets in diabetes. Pharmacol. Res. 2013, 75, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Alipourfard, I.; Datukishvili, N.; Mikeladze, D. TNF-alpha Downregulation Modifies Insulin Receptor Substrate 1 (IRS-1) in Metabolic Signaling of Diabetic Insulin-Resistant Hepatocytes. Mediat. Inflamm. 2019, 2019, 3560819. [Google Scholar] [CrossRef] [PubMed]
- Burska, A.N.; Sakthiswary, R.; Sattar, N. Effects of Tumour Necrosis Factor Antagonists on Insulin Sensitivity/Resistance in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0128889. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Feldmann, M. Anti-TNF biologic agents: Still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [Google Scholar] [CrossRef]
- Wang, Q.; Li, H.; Xiao, Y.; Li, S.; Li, B.; Zhao, X.; Ye, L.; Guo, B.; Chen, X.; Ding, Y.; et al. Locally controlled delivery of TNFalpha antibody from a novel glucose-sensitive scaffold enhances alveolar bone healing in diabetic conditions. J. Control. Release 2015, 206, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Paquot, N.; Castillo, M.J.; Lefebvre, P.J.; Scheen, A.J. No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J. Clin. Endocrinol. Metab. 2000, 85, 1316–1319. [Google Scholar] [PubMed]
- Ofei, F.; Hurel, S.; Newkirk, J.; Sopwith, M.; Taylor, R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 1996, 45, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Jager, J.; Gremeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef]
- Dinarello, C.A. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 2011, 41, 1203–1217. [Google Scholar] [CrossRef]
- Schultz, O.; Oberhauser, F.; Saech, J.; Rubbert-Roth, A.; Hahn, M.; Krone, W.; Laudes, M. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS ONE 2010, 5, e14328. [Google Scholar] [CrossRef]
- Dagdeviren, S.; Jung, D.Y.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Inashima, K.; Tran, D.A.; Hu, X.; et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017, 31, 701–710. [Google Scholar] [CrossRef]
- De Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Xie, J.; Ameres, S.L.; Friedline, R.; Hung, J.H.; Zhang, Y.; Xie, Q.; Zhong, L.; Su, Q.; He, R.; Li, M.; et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods 2012, 9, 403–409. [Google Scholar] [CrossRef]
- Vester, B.; Wengel, J. LNA (locked nucleic acid): High-affinity targeting of complementary RNA and DNA. Biochemistry 2004, 43, 13233–13241. [Google Scholar] [CrossRef]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Varghese, B.; Widmer, W.R.; Breur, G.J.; Vlashi, E.; Low, P.S. Folate-targeted immunotherapy effectively treats established adjuvant and collagen-induced arthritis. Arthritis Res. Ther. 2006, 8, R77. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Hilgenbrink, A.R.; Matteson, E.L.; Lockwood, M.B.; Cheng, J.X.; Low, P.S. A functional folate receptor is induced during macrophage activation and can be used to target drugs to activated macrophages. Blood 2009, 113, 438–446. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Meng, Y.; He, S.; Tan, X.; Zhang, Y.; Zhang, X.; Wang, L.; Zheng, W. Macrophages, Chronic Inflammation, and Insulin Resistance. Cells 2022, 11, 3001. https://doi.org/10.3390/cells11193001
Li H, Meng Y, He S, Tan X, Zhang Y, Zhang X, Wang L, Zheng W. Macrophages, Chronic Inflammation, and Insulin Resistance. Cells. 2022; 11(19):3001. https://doi.org/10.3390/cells11193001
Chicago/Turabian StyleLi, He, Ya Meng, Shuwang He, Xiaochuan Tan, Yujia Zhang, Xiuli Zhang, Lulu Wang, and Wensheng Zheng. 2022. "Macrophages, Chronic Inflammation, and Insulin Resistance" Cells 11, no. 19: 3001. https://doi.org/10.3390/cells11193001
APA StyleLi, H., Meng, Y., He, S., Tan, X., Zhang, Y., Zhang, X., Wang, L., & Zheng, W. (2022). Macrophages, Chronic Inflammation, and Insulin Resistance. Cells, 11(19), 3001. https://doi.org/10.3390/cells11193001