
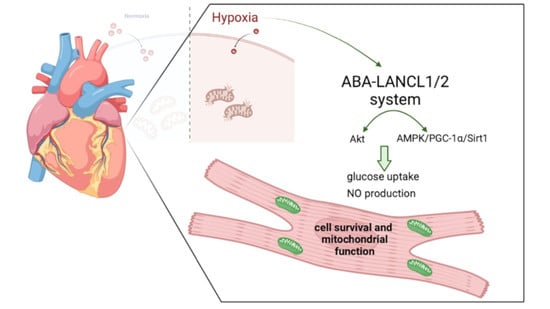
The ABA-LANCL1/2 Hormone-Receptors System Protects H9c2 Cardiomyocytes from Hypoxia-Induced Mitochondrial Injury via an AMPK- and NO-Mediated Mechanism

 , ,
, , 
 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Exposure of H9c2 to Normoxia and Hypoxia
2.3. Measurement of Glucose Uptake and of NADPH Content
2.4. O2 Consumption
2.5. Nitrite Determination
2.6. Detection of ABA
2.7. Cell Vitality Assays
2.8. Lentiviral Cell Transduction
2.9. qPCR Analysis
2.10. Western Blot
2.11. JC-1 Analysis
2.12. Statistical Analysis
3. Results
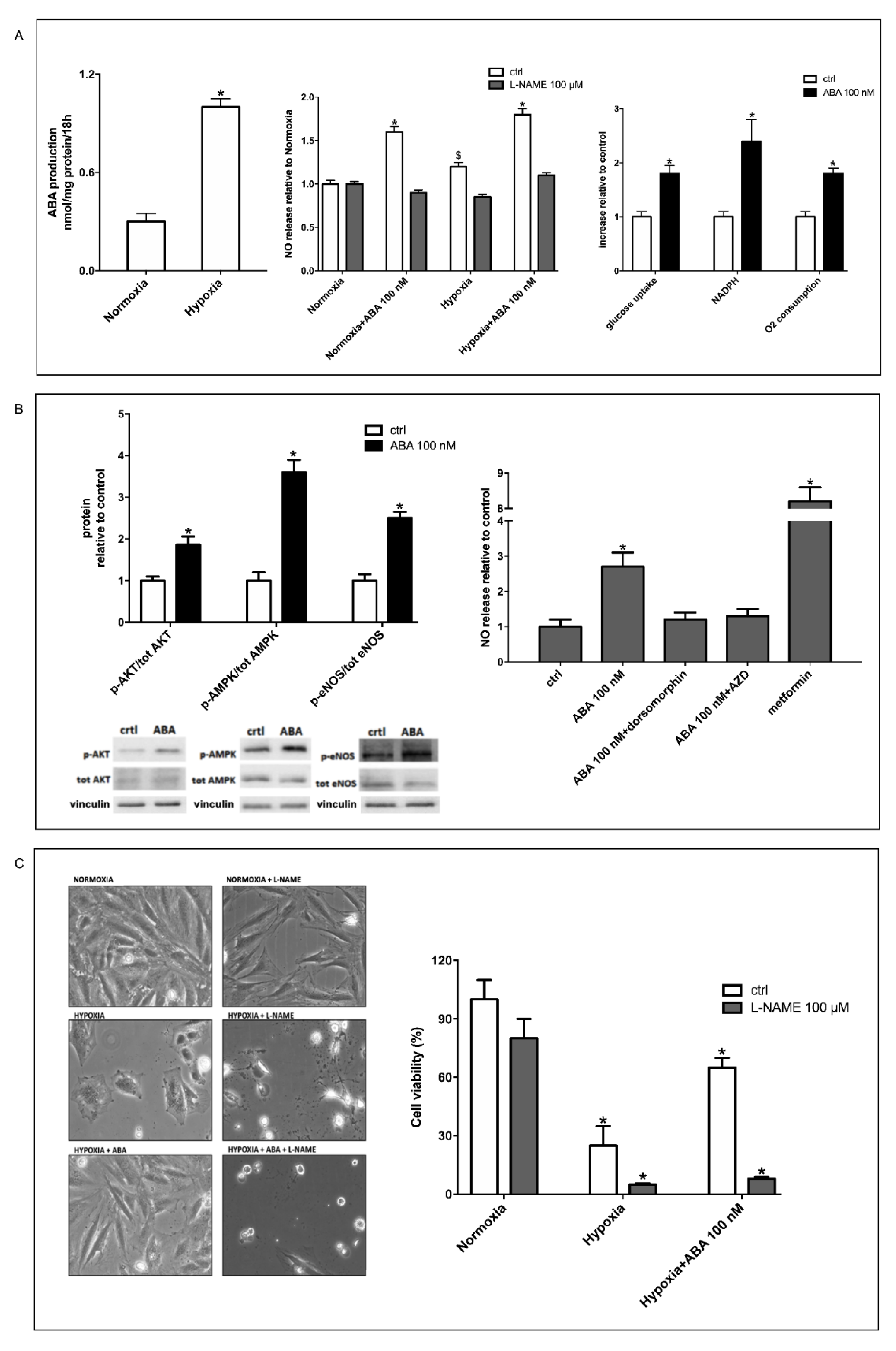
3.1. Hypoxia Stimulates ABA Release from H9c2 and ABA Stimulates NO Production, Glucose Uptake and O2 Consumption
3.2. Stimulation by ABA of NO Production in H9c2 Cells Occurs Via AMPK and Akt
3.3. The ABA-Induced Increase of NO Production Improves Survival of H9c2 under Hypoxia
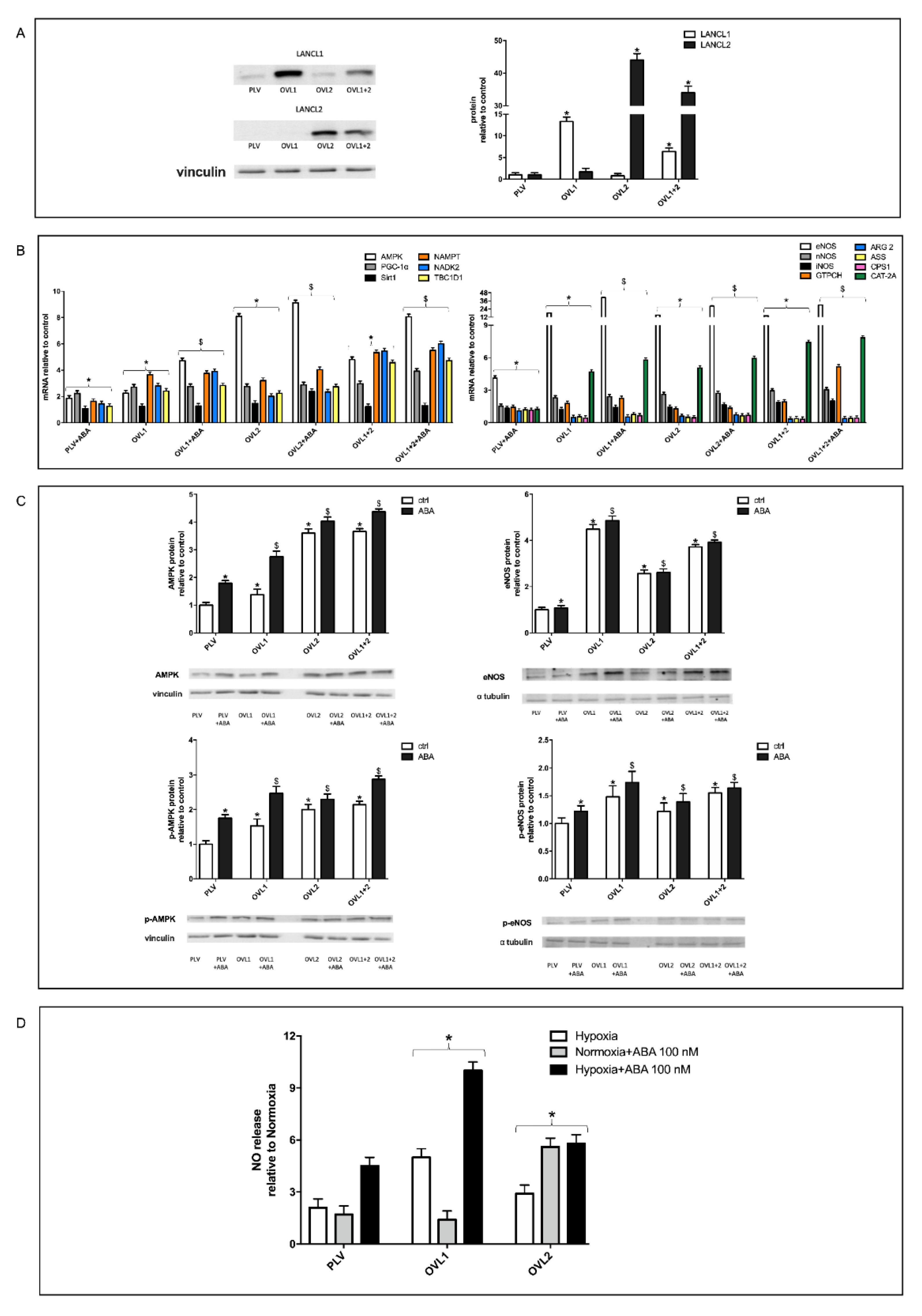
3.4. Overexpression of Either LANCL1 or LANCL2 Activates eNOS Transcription and Function in H9c2 under Both Normoxia and Hypoxia
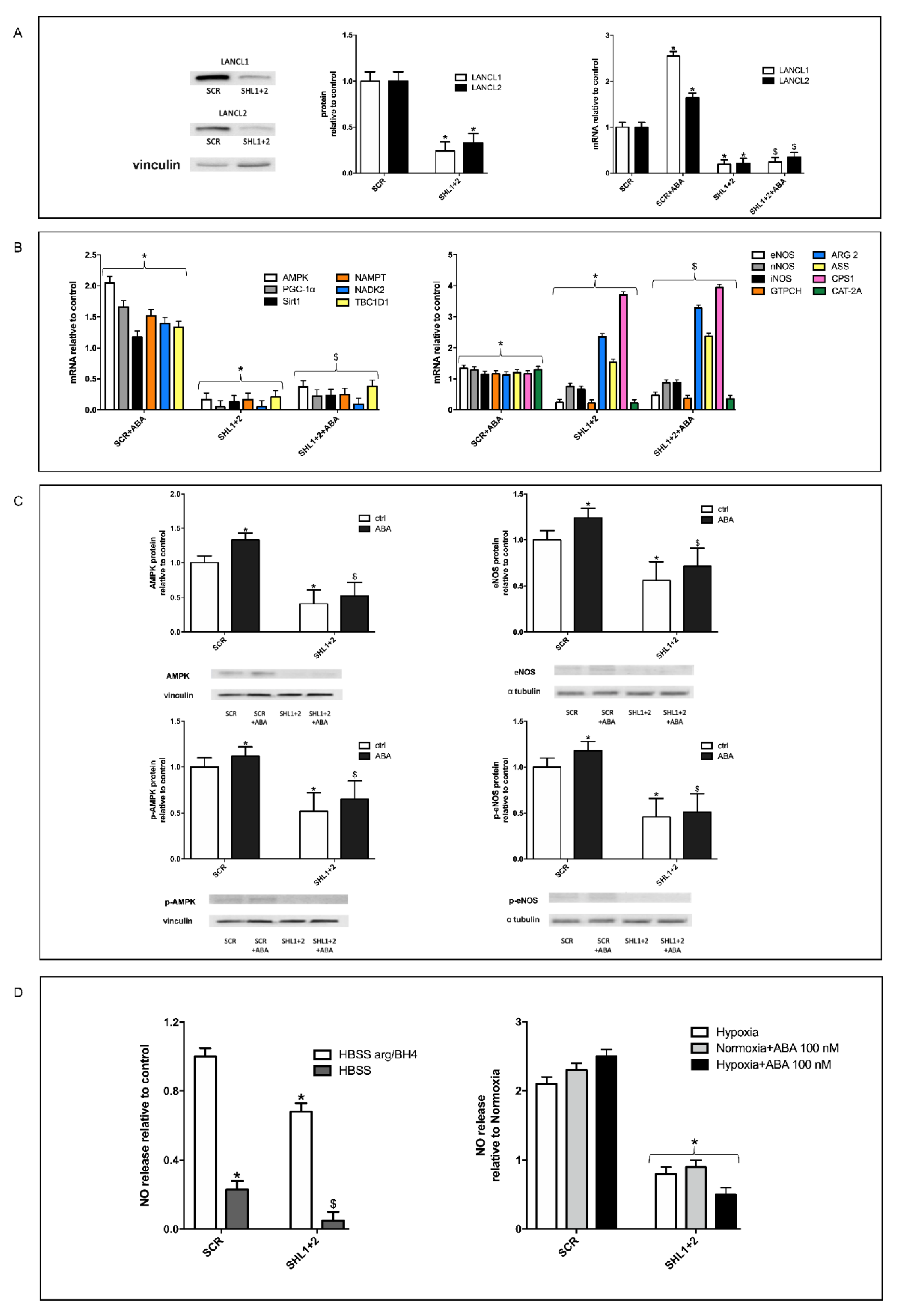
3.5. The Combined Silencing of LANCL1 and LANCL2 Reduces eNOS Transcription, Expression and Function in H9c2 under Normoxia and Hypoxia
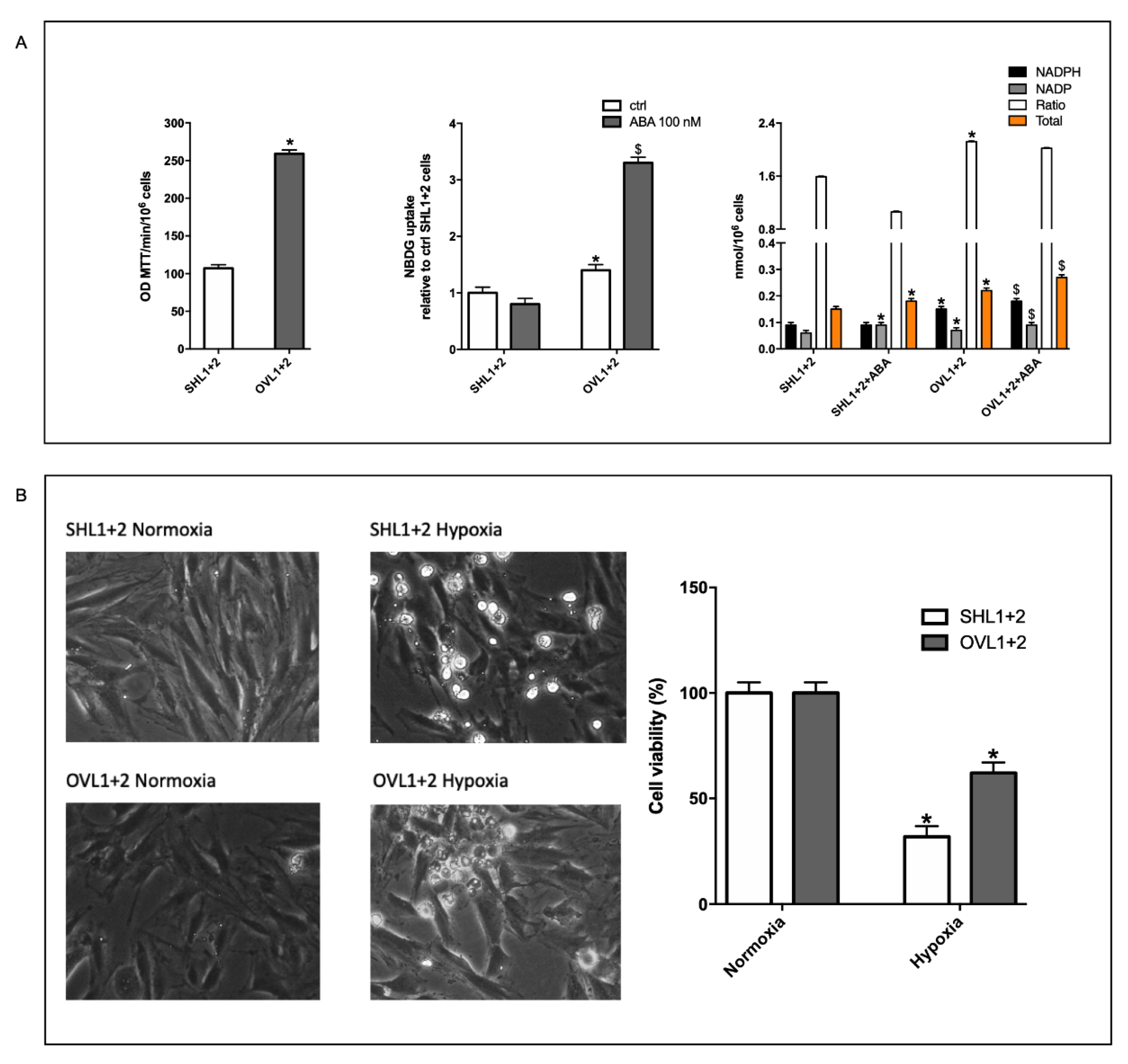
3.6. Mitochondrial Function Is Conserved in LANCL1/2-Overexpressing, but Not in LANCL1/2-Silenced Cells, Exposed to Hypoxia/Reoxygenation
3.7. The Combined LANCL1/2 Silencing Reduces, and Their Overexpression Increases, MTT Reduction, Glucose Uptake and NADP/H Content under Normoxia and Cell Vitality after Hypoxia/Reoxygenation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Olds, C.L.; Glennon, E.K.K.; Luckhart, S. Abscisic acid: New perspectives on an ancient universal stress signaling molecule. Microbes Infect. 2018, 20, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Magnone, M.; Sturla, L.; Guida, L.; Spinelli, S.; Begani, G.; Bruzzone, S.; Fresia, C.; Zocchi, E. Abscisic acid: A conserved hormone in plants and humans and a promising aid to combat prediabetes and the metabolic syndrome. Nutrients 2020, 12, 1724. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, E.; Carpaneto, A.; Cerrano, C.; Bavestrello, G.; Giovine, M.; Bruzzone, S.; Guida, L.; Franco, L.; Usai, C. The temperature-signaling cascade in sponges involves a heat-gated cation channel, abscisic acid, and cyclic ADP-ribose. Proc. Natl. Acad. Sci. USA 2001, 98, 14859–14864. [Google Scholar] [CrossRef] [PubMed]
- Puce, S.; Basile, G.; Bavestrello, G.; Bruzzone, S.; Cerrano, C.; Giovine, M.; Arillo, A.; Zocchi, E. Abscisic acid signaling through cyclic ADP-ribose in hydroid regeneration. J. Biol. Chem. 2004, 279, 39783–39788. [Google Scholar] [CrossRef]
- Bharath, P.; Gahir, S.; Raghavendra, A.S. Abscisic acid-induced stomatal closure: An important component of plant defense against abiotic and biotic stress. Front. Plant Sci. 2021, 12, 615114. [Google Scholar] [CrossRef]
- González-Guzmán, M.; Gómez-Cadenas, A.; Arbona, V. Abscisic acid as an emerging modulator of the responses of plants to low oxygen conditions. Front. Plant Sci. 2021, 12, 661789. [Google Scholar] [CrossRef]
- Sun, L.R.; Yue, C.M.; Hao, F.S. Update on roles of nitric oxide in regulating stomatal closure. Plant Signal. Behav. 2019, 14, e1649569. [Google Scholar] [CrossRef]
- Bruzzone, S.; Moreschi, I.; Usai, C.; Guida, L.; Damonte, G.; Salis, A.; Scarfì, S.; Millo, E.; de Flora, A.; Zocchi, E. Abscisic acid is an endogenous cytokine in human granulocytes with cyclic ADP-ribose as second messenger. Proc. Natl. Acad. Sci. USA 2007, 104, 5759–5764. [Google Scholar] [CrossRef]
- Bruzzone, S.; Basile, G.; Mannino, E.; Sturla, L.; Magnone, M.; Grozio, A.; Salis, A.; Fresia, C.; Vigliarolo, T.; Guida, L.; et al. Autocrine abscisic acid mediates the UV-B-induced inflammatory response in human granulocytes and keratinocytes. J. Cell. Physiol. 2012, 227, 2502–2510. [Google Scholar] [CrossRef]
- Bodrato, N.; Franco, L.; Fresia, C.; Guida, L.; Usai, C.; Salis, A.; Moreschi, I.; Ferraris, C.; Verderio, C.; Basile, G.; et al. Abscisic acid activates the murine microglial cell line N9 through the second messenger cyclic ADP-ribose. J. Biol. Chem. 2009, 284, 14777–14787. [Google Scholar] [CrossRef]
- Tossi, V.; Cassia, R.; Bruzzone, S.; Zocchi, E.; Lamattina, L. ABA says NO to UV-B: A universal response? Trends Plant Sci. 2012, 17, 510–517. [Google Scholar] [CrossRef]
- Bruzzone, S.; Ameri, P.; Briatore, L.; Mannino, E.; Basile, G.; Andraghetti, G.; Grozio, A.; Magnone, M.; Guida, L.; Scarfì, S.; et al. The plant hormone abscisic acid increases in human plasma after hyperglycemia and stimulates glucose consumption by adipocytes and myoblasts. FASEB J. 2012, 26, 1251–1260. [Google Scholar] [CrossRef]
- Magnone, M.; Ameri, P.; Salis, A.; Andraghetti, G.; Emionite, L.; Murialdo, G.; de Flora, A.; Zocchi, E. Microgram amounts of abscisic acid in fruit extracts improve glucose tolerance and reduce insulinemia in rats and in humans. FASEB J. 2015, 29, 4783–4793. [Google Scholar] [CrossRef]
- Magnone, M.; Emionite, L.; Guida, L.; Vigliarolo, T.; Sturla, L.; Spinelli, S.; Buschiazzo, A.; Marini, C.; Sambuceti, G.; de Flora, A.; et al. Insulin-independent stimulation of skeletal muscle glucose uptake by low-dose abscisic acid via AMPK activation. Sci. Rep. 2020, 10, 1454. [Google Scholar] [CrossRef]
- Spinelli, S.; Begani, G.; Guida, L.; Magnone, M.; Galante, D.; D’Arrigo, C.; Scotti, C.; Iamele, L.; de Jonge, H.; Zocchi, E.; et al. LANCL1 binds abscisic acid and stimulates glucose transport and mitochondrial respiration in muscle cells via the AMPK/PGC-1α/Sirt1 pathway. Mol. Metab. 2021, 53, 101263. [Google Scholar] [CrossRef]
- He, C.; Zeng, M.; Dutta, D.; Koh, T.H.; Chen, J.; van der Donk, W.A. LanCL proteins are not involved in lanthionine synthesis in mammals. Sci. Rep. 2017, 7, 40980. [Google Scholar] [CrossRef]
- Fresia, C.; Vigliarolo, T.; Guida, L.; Booz, V.; Bruzzone, S.; Sturla, L.; di Bona, M.; Pesce, M.; Usai, C.; de Flora, A.; et al. G-protein coupling and nuclear translocation of the human abscisic acid receptor LANCL2. Sci. Rep. 2016, 6, 26658. [Google Scholar] [CrossRef]
- Kramer, E.M. How far can a molecule of weak acid travel in the apoplast or xylem? Plant Physiol. 2006, 141, 1233–1236. [Google Scholar] [CrossRef]
- Vigliarolo, T.; Guida, L.; Millo, E.; Fresia, C.; Turco, E.; de Flora, A.; Zocchi, E. Abscisic acid transport in human erythrocytes. J. Biol. Chem. 2015, 290, 13042–13052. [Google Scholar] [CrossRef]
- Vigliarolo, T.; Zocchi, E.; Fresia, C.; Booz, V.; Guida, L. Abscisic acid influx into human nucleated cells occurs through the anion exchanger AE2. Int. J. Biochem. Cell Biol. 2016, 75, 99–103. [Google Scholar] [CrossRef]
- Shemarova, I.; Nesterov, V.; Emelyanova, L.; Korotkov, S. Mitochondrial mechanisms by which gasotransmitters (H2S, NO and CO) protect cardiovascular system against hypoxia. Front. Biosci. 2021, 13, 105–130. [Google Scholar]
- Bruzzone, S.; Fruscione, F.; Morando, S.; Ferrando, T.; Poggi, A.; Garuti, A.; D’Urso, A.; Selmo, M.; Benvenuto, F.; Cea, M.; et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS ONE 2009, 4, e7897. [Google Scholar] [CrossRef]
- Misko, T.P.; Schilling, R.J.; Salvemini, D.; Moore, W.M.; Currie, M.G. A fluorometric assay for the measurement of nitrite in biological samples. Anal. Biochem. 1993, 214, 11–16. [Google Scholar] [CrossRef]
- Chen, G.; Yang, Y.; Xu, C.; Gao, S. A Flow cytometry-based assay for measuring mitochondrial membrane potential in cardiac myocytes after hypoxia/reoxygenation. J. Vis. Exp. 2018, 137, 57725. [Google Scholar] [CrossRef]
- Strijdom, H.; Jacobs, S.; Hattingh, S.; Page, C.; Lochner, A. Nitric oxide production is higher in rat cardiac microvessel endothelial cells than ventricular cardiomyocytes in baseline and hypoxic conditions: A comparative study. FASEB J. 2006, 20, 314–316. [Google Scholar] [CrossRef]
- Liang, X.X.; Wang, R.Y.; Guo, Y.Z.; Cheng, Z.; Lv, D.Y.; Luo, M.H.; He, A.; Luo, S.X.; Xia, Y. Phosphorylation of Akt at Thr308 regulates p-eNOS Ser1177 during physiological conditions. FEBS Open Bio. 2021, 11, 1953–1964. [Google Scholar] [CrossRef]
- Chen, Y.; Ba, L.; Huang, W.; Liu, Y.; Pan, H.; Mingyao, E.; Shi, P.; Wang, Y.; Li, S.; Qi, H.; et al. Role of carvacrol in cardioprotection against myocardial ischemia/reperfusion injury in rats through activation of MAPK/ERK and Akt/eNOS signaling pathways. Eur. J. Pharmacol. 2017, 796, 90–100. [Google Scholar] [CrossRef]
- Hu, L.; Zhou, L.; Wu, X.; Liu, C.; Fan, Y.; Li, Q. Hypoxic preconditioning protects cardiomyocytes against hypoxia/reoxygenation injury through AMPK/eNOS/PGC-1α signaling pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 7378–7388. [Google Scholar] [PubMed]
- Zhang, W.; Wang, Q.; Wu, Y.; Moriasi, C.; Liu, Z.; Dai, X.; Wang, Q.; Liu, W.; Yuan, Z.Y.; Zou, M.H. Endothelial cell-specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation 2014, 129, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; de Montellano, P.R.O.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef]
- Espelage, L.; Al-Hasani, H.; Chadt, A. RabGAPs in skeletal muscle function and exercise. J. Mol. Endocrinol. 2020, 64, R1–R19. [Google Scholar] [CrossRef]
- Barbeau, P.A.; Houad, J.M.; Huber, J.S.; Paglialunga, S.; Snook, L.A.; Herbst, E.A.F.; Dennis, K.M.J.H.; Simpson, J.A.; Holloway, G.P. Ablating the Rab-GTPase activating protein TBC1D1 predisposes rats to high-fat diet-induced cardiomyopathy. J Physiol. 2020, 598, 683–697. [Google Scholar] [CrossRef]
- Koo, B.H.; Lee, J.; Jin, Y.; Lim, H.K.; Ryoo, S. Arginase inhibition by rhaponticin increases L-arginine concentration that contributes to Ca2+-dependent eNOS activation. BMB Rep. 2021, 54, 516–521. [Google Scholar] [CrossRef]
- Channon, K.M. Tetrahydrobiopterin and nitric oxide synthase recouplers. Handb. Exp. Pharmacol. 2021, 264, 339–352. [Google Scholar]
- Chacon, E.; Acosta, D.; Lemasters, J.J. Primary cultures of cardiac myocytes as in vitro models for pharmacological and toxicological assessments. In In Vitro Methods in Pharmaceutical Research; Academic Press: Cambridge, MA, USA, 1997; Volume 9, pp. 209–223. [Google Scholar]
- Zhao, C.; Wang, Y.; Chan, K.X.; Marchant, D.B.; Franks, P.J.; Randall, D.; Tee, E.E.; Chen, G.; Ramesh, S.; Phua, S.Y.; et al. Evolution of chloroplast retrograde signaling facilitates green plant adaptation to land. Proc. Natl. Acad. Sci. USA 2019, 116, 5015–5020. [Google Scholar] [CrossRef]
- Foresi, N.; Mayta, M.L.; Lodeyro, A.F.; Scuffi, D.; Correa-Aragunde, N.; García-Mata, C.; Casalongué, C.; Carrillo, N.; Lamattina, L. Expression of the tetrahydrofolate-dependent nitric oxide synthase from the green alga Ostreococcus tauri increases tolerance to abiotic stresses and influences stomatal development in Arabidopsis. Plant J. 2015, 82, 806–821. [Google Scholar] [CrossRef]
- Zhang, Y.H. Nitric oxide signalling and neuronal nitric oxide synthase in the heart under stress. F1000Res. 2017, 6, 742. [Google Scholar] [CrossRef]
- Bertrand, L.; Auquier, J.; Renguet, E.; Angé, M.; Cumps, J.; Horman, S.; Beauloye, C. Glucose transporters in cardiovascular system in health and disease. Pflug. Arch. 2020, 472, 1385–1399. [Google Scholar] [CrossRef]
- Boycott, H.E.; Nguyen, M.N.; Vrellaku, B.; Gehmlich, K.; Robinson, P. Nitric oxide and mechano-electrical transduction in cardiomyocytes. Front. Physiol. 2020, 11, 606740. [Google Scholar] [CrossRef]
- Carlström, M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef]
- Pisarenko, O.; Studneva, I. Modulating the bioactivity of nitric oxide as a therapeutic strategy in cardiac surgery. J. Surg. Res. 2021, 257, 178–188. [Google Scholar] [CrossRef]
- Eid, R.A.; Bin-Meferij, M.M.; El-Kott, A.F.; Eleawa, S.M.; Zaki, M.S.A.; Al-Shraim, M.; El-Sayed, F.; Eldeen, M.A.; Alkhateeb, M.A.; Alharbi, S.A.; et al. Exendin-4 protects against myocardial ischemia-reperfusion injury by upregulation of SIRT1 and SIRT3 and activation of AMPK. J. Cardiovasc. Transl. Res. 2020, 14, 619–635. [Google Scholar] [CrossRef]
- Hsu, C.P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, K.; Li, P.; Wu, Z.; Zhang, K.; Li, P.; Wu, Z. Down-regulating miR-217-5p protects cardiomyocytes against ischemia/reperfusion injury by restoring mitochondrial function via targeting SIRT1. Inflammation 2021, 44, 383–396. [Google Scholar] [CrossRef]
- Sturla, L.; Mannino, E.; Scarfì, S.; Bruzzone, S.; Magnone, M.; Sociali, G.; Booz, V.; Guida, L.; Vigliarolo, T.; Fresia, C.; et al. Abscisic acid enhances glucose disposal and induces brown fat activity in adipocytes in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 131–144. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Zhang, J.; He, Z.; Fedorova, J.; Logan, C.; Bates, L.; Davitt, K.; Le, V.; Murphy, J.; Li, M.; Wang, M.; et al. Alterations in mitochondrial dynamics with age-related Sirtuin1/Sirtuin3 deficiency impair cardiomyocyte contractility. Aging Cell 2021, 20, e13419. [Google Scholar] [CrossRef]
- Gan, L.; Xie, D.; Liu, J.; Bond Lau, W.; Christopher, T.A.; Lopez, B.; Zhang, L.; Gao, E.; Koch, W.; Ma, X.L.; et al. Small Extracellular microvesicles mediated pathological communications between dysfunctional adipocytes and cardiomyocytes as a novel mechanism exacerbating ischemia/reperfusion injury in diabetic mice. Circulation 2020, 141, 968–983. [Google Scholar] [CrossRef]
- Luo, G.; Jian, Z.; Zhu, Y.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef]
- Tian, L.; Cao, W.; Yue, R.; Yuan, Y.; Guo, X.; Qin, D.; Xing, J.; Wang, X. Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. J. Pharmacol. Sci. 2019, 139, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, X.; Yang, D.; Lu, Y.; Wei, G.; Yu, W.; Liu, X.; Zheng, Q.; Ying, J.; Hua, F. Relieving cellular energy stress in aging, neurodegenerative, and metabolic diseases, SIRT1 as a therapeutic and promising node. Front. Aging Neurosci. 2021, 13, 738686. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Ameri, P.; Bruzzone, S.; Mannino, E.; Sociali, G.; Andraghetti, G.; Salis, A.; Ponta, M.L.; Briatore, L.; Adami, G.F.; Ferraiolo, A.; et al. Impaired increase of plasma abscisic acid in response to oral glucose load in type 2 diabetes and in gestational diabetes. PLoS ONE 2015, 10, e0115992. [Google Scholar] [CrossRef]
- Magnone, M.; Leoncini, G.; Vigliarolo, T.; Emionite, L.; Sturla, L.; Zocchi, E.; Murialdo, G. Chronic intake of micrograms of abscisic acid improves glycemia and lipidemia in a human study and in high-glucose fed mice. Nutrients 2018, 10, 1495. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P.; D’Angelo, A.; Preti, P.; Tenore, G.; Novellino, E. Abscisic acid treatment in patients with prediabetes. Nutrients 2020, 12, 2931. [Google Scholar] [CrossRef]
- Magnone, M.; Spinelli, S.; Begani, G.; Guida, L.; Sturla, L.; Emionite, L.; Zocchi, E. Abscisic acid improves insulin action on glycemia in insulin-deficient mouse models of type 1 diabetes. Metabolites 2022, 12, 523. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinelli, S.; Guida, L.; Vigliarolo, T.; Passalacqua, M.; Begani, G.; Magnone, M.; Sturla, L.; Benzi, A.; Ameri, P.; Lazzarini, E.; et al. The ABA-LANCL1/2 Hormone-Receptors System Protects H9c2 Cardiomyocytes from Hypoxia-Induced Mitochondrial Injury via an AMPK- and NO-Mediated Mechanism. Cells 2022, 11, 2888. https://doi.org/10.3390/cells11182888
Spinelli S, Guida L, Vigliarolo T, Passalacqua M, Begani G, Magnone M, Sturla L, Benzi A, Ameri P, Lazzarini E, et al. The ABA-LANCL1/2 Hormone-Receptors System Protects H9c2 Cardiomyocytes from Hypoxia-Induced Mitochondrial Injury via an AMPK- and NO-Mediated Mechanism. Cells. 2022; 11(18):2888. https://doi.org/10.3390/cells11182888
Chicago/Turabian StyleSpinelli, Sonia, Lucrezia Guida, Tiziana Vigliarolo, Mario Passalacqua, Giulia Begani, Mirko Magnone, Laura Sturla, Andrea Benzi, Pietro Ameri, Edoardo Lazzarini, and et al. 2022. "The ABA-LANCL1/2 Hormone-Receptors System Protects H9c2 Cardiomyocytes from Hypoxia-Induced Mitochondrial Injury via an AMPK- and NO-Mediated Mechanism" Cells 11, no. 18: 2888. https://doi.org/10.3390/cells11182888
APA StyleSpinelli, S., Guida, L., Vigliarolo, T., Passalacqua, M., Begani, G., Magnone, M., Sturla, L., Benzi, A., Ameri, P., Lazzarini, E., Bearzi, C., Rizzi, R., & Zocchi, E. (2022). The ABA-LANCL1/2 Hormone-Receptors System Protects H9c2 Cardiomyocytes from Hypoxia-Induced Mitochondrial Injury via an AMPK- and NO-Mediated Mechanism. Cells, 11(18), 2888. https://doi.org/10.3390/cells11182888