

A Single-Cell Survey of Cellular Heterogeneity in Human Great Saphenous Veins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Tissue Dissociation and Single-Cell RNA Sequencing
2.3. Bioinformatic Analysis
2.4. Functional Enrichment Analysis
2.5. Transcriptional Noise Analysis
2.6. Pseudotime Trajectory Analysis
2.7. Regulatory Analysis of Transcription Factors
2.8. Cell–Cell Interaction Analysis
2.9. Bulk RNAseq Data Validation
2.10. Data Visualization
2.11. Data Availability Statement
3. Results
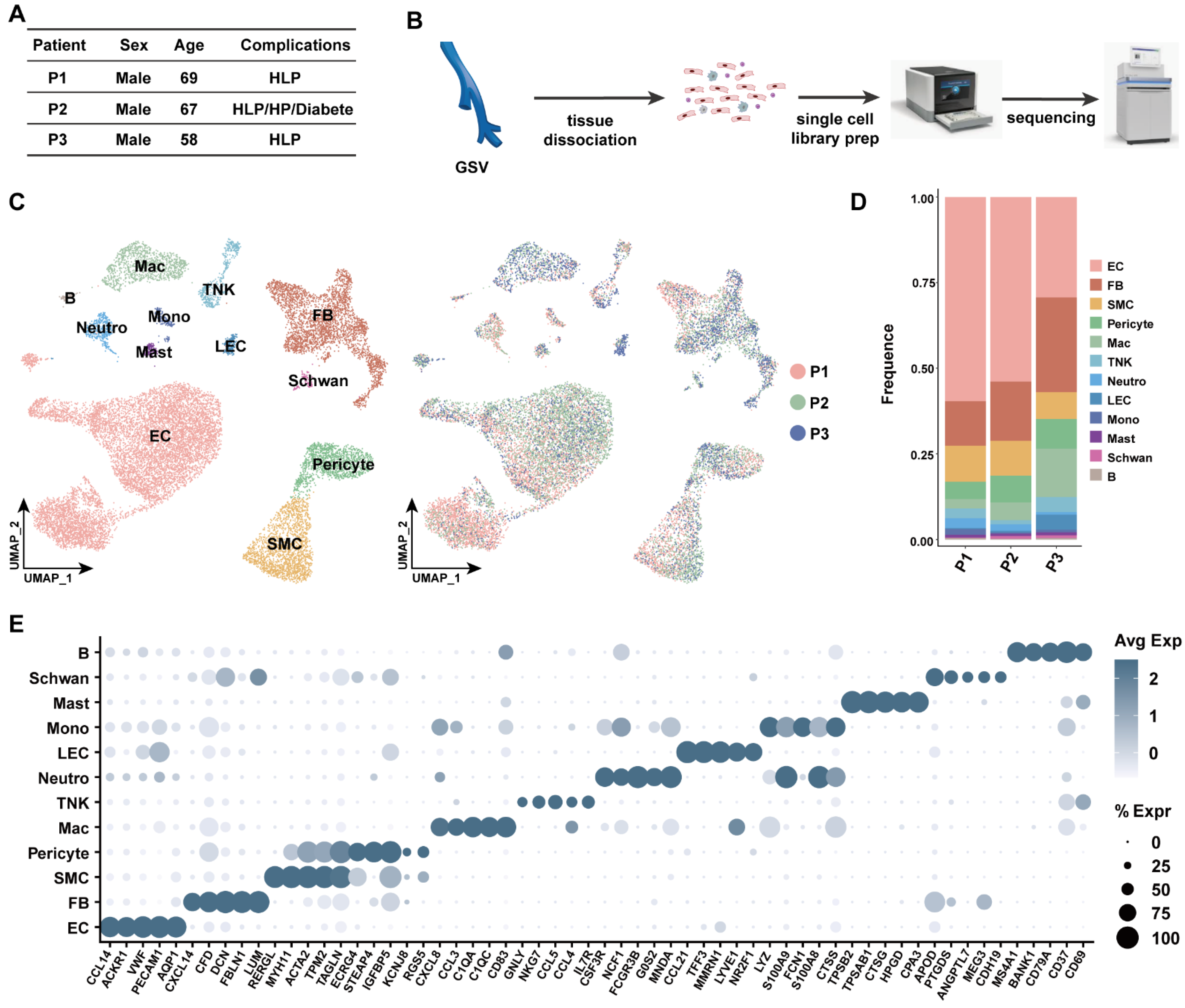
3.1. Transcriptomic Identification of Human Great Saphenous Vein Landscape
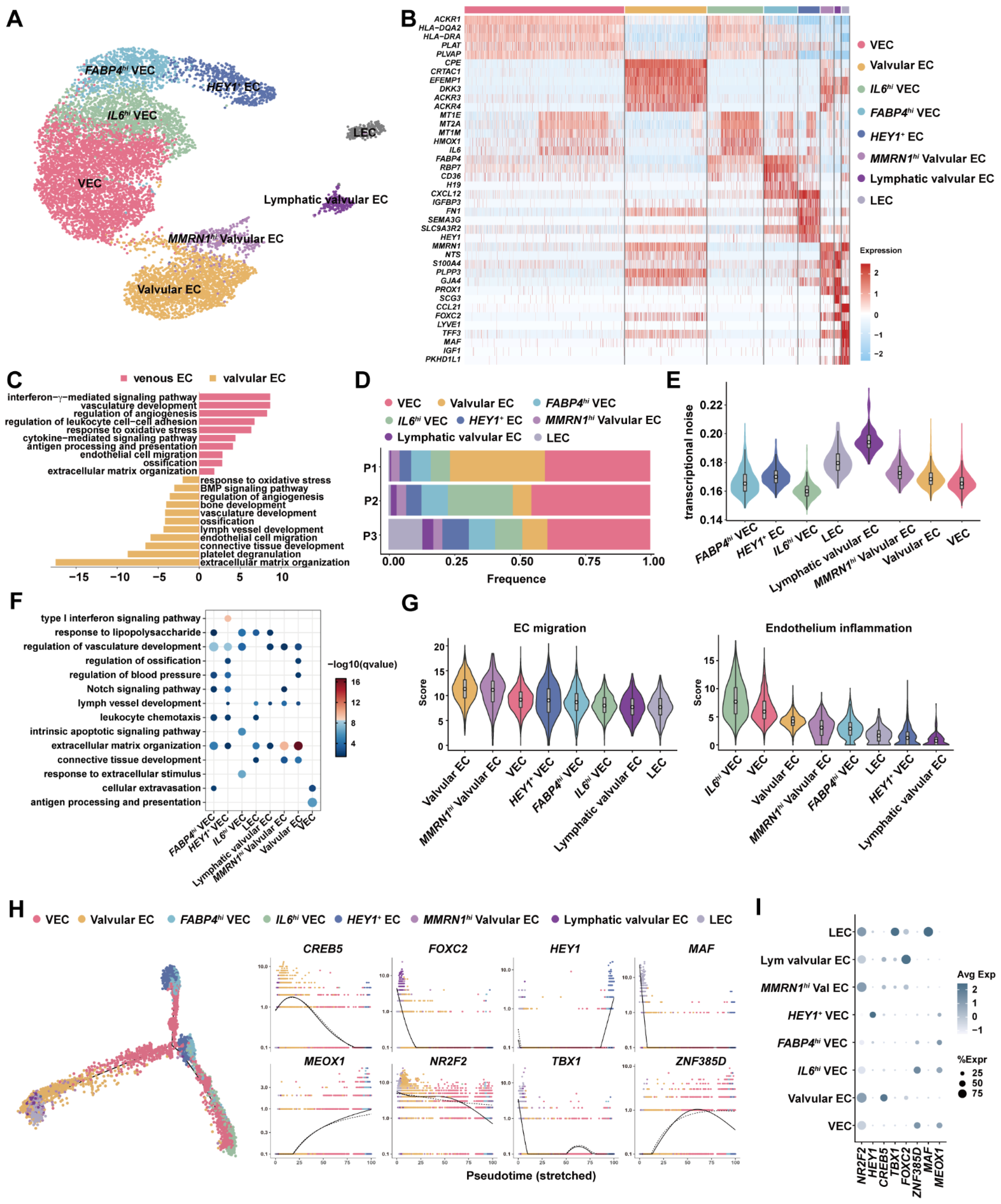
3.2. Venous ECs, Valvular ECs and Lymphatic EsC Had Distinct Expression Programs
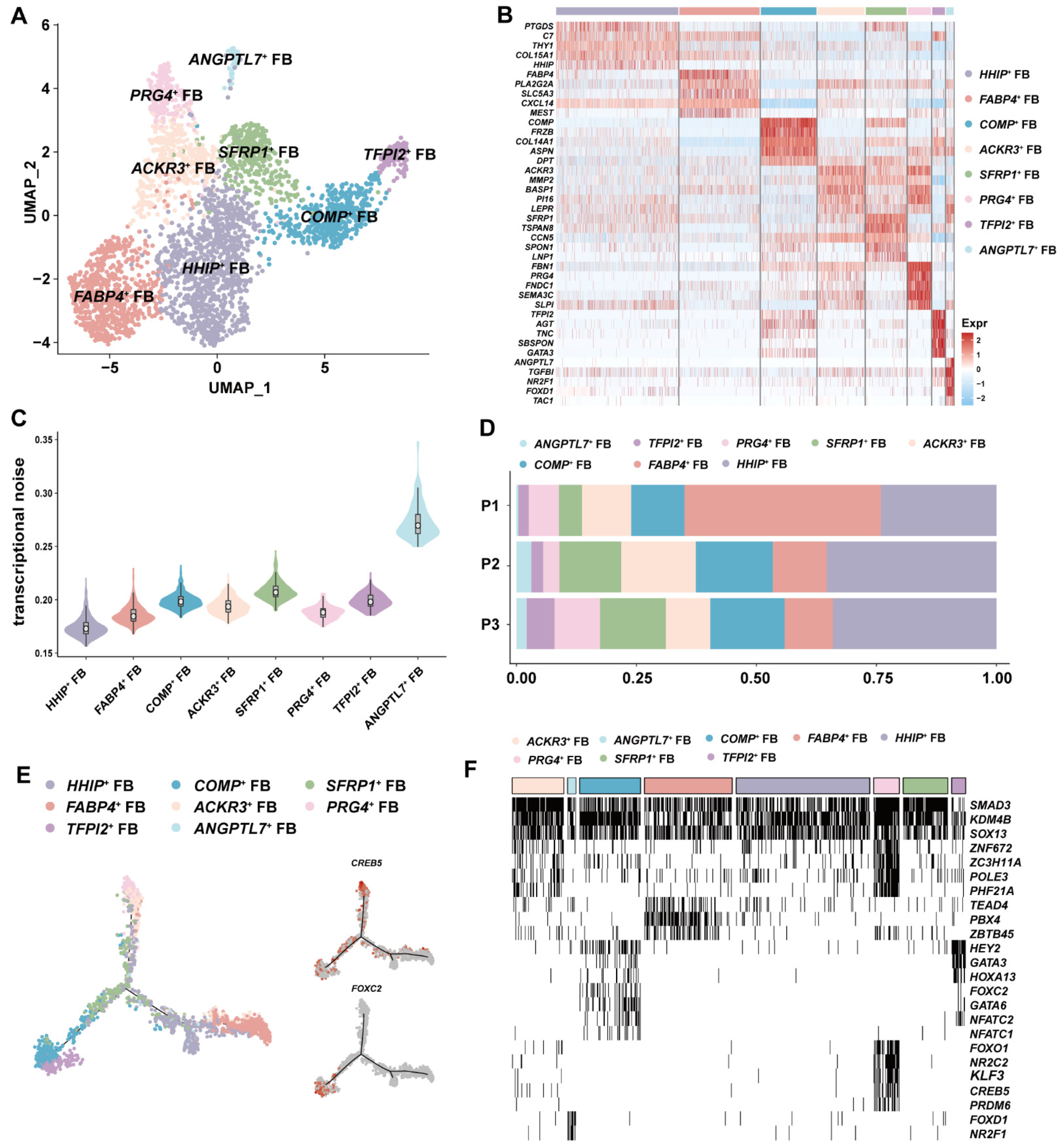
3.3. ANGPTL7+ FBs with Antiangiogenic Potential Was Identified in GSV
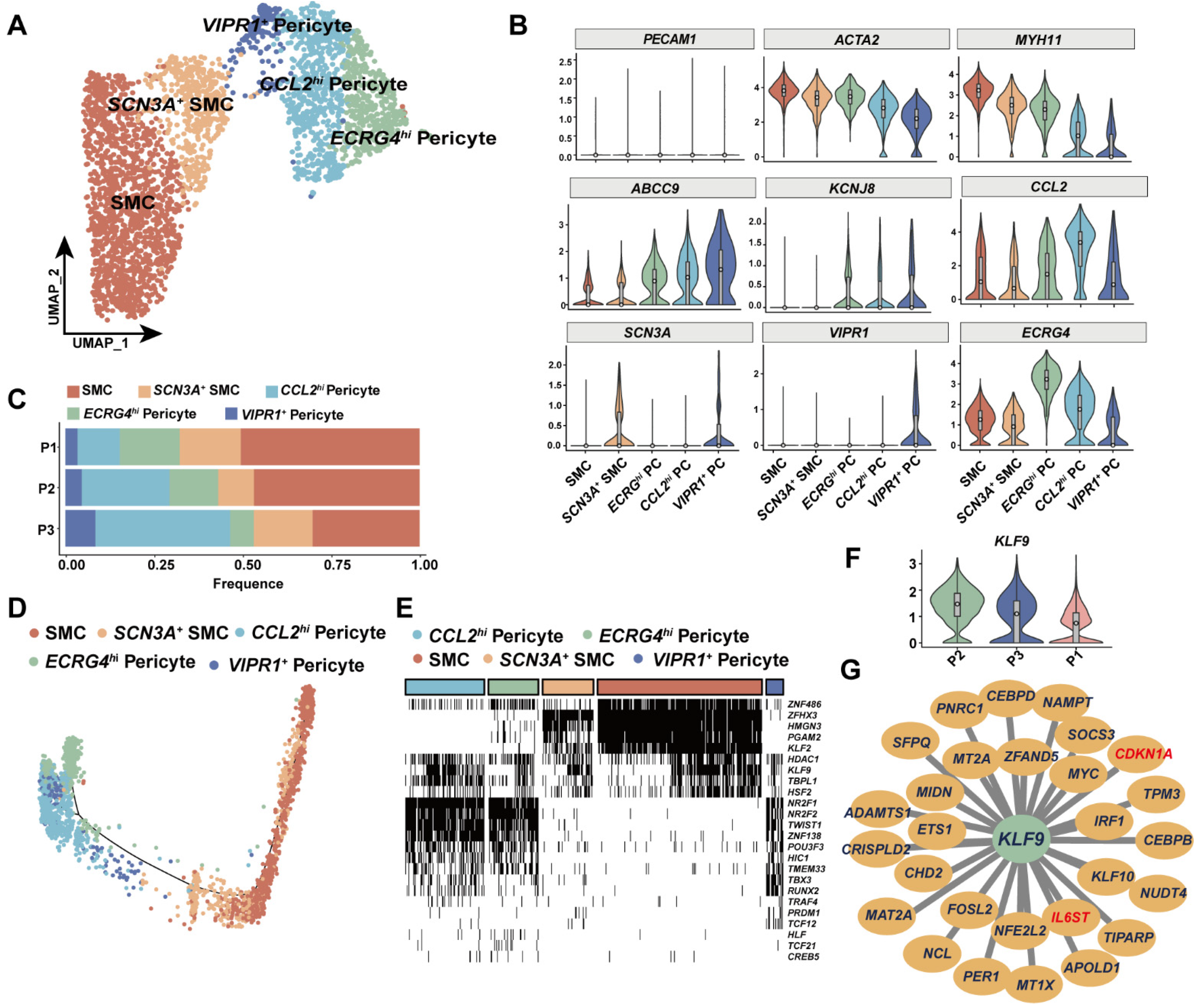
3.4. KLF9 Regulated Proinflammatory Programs in Mural Cells
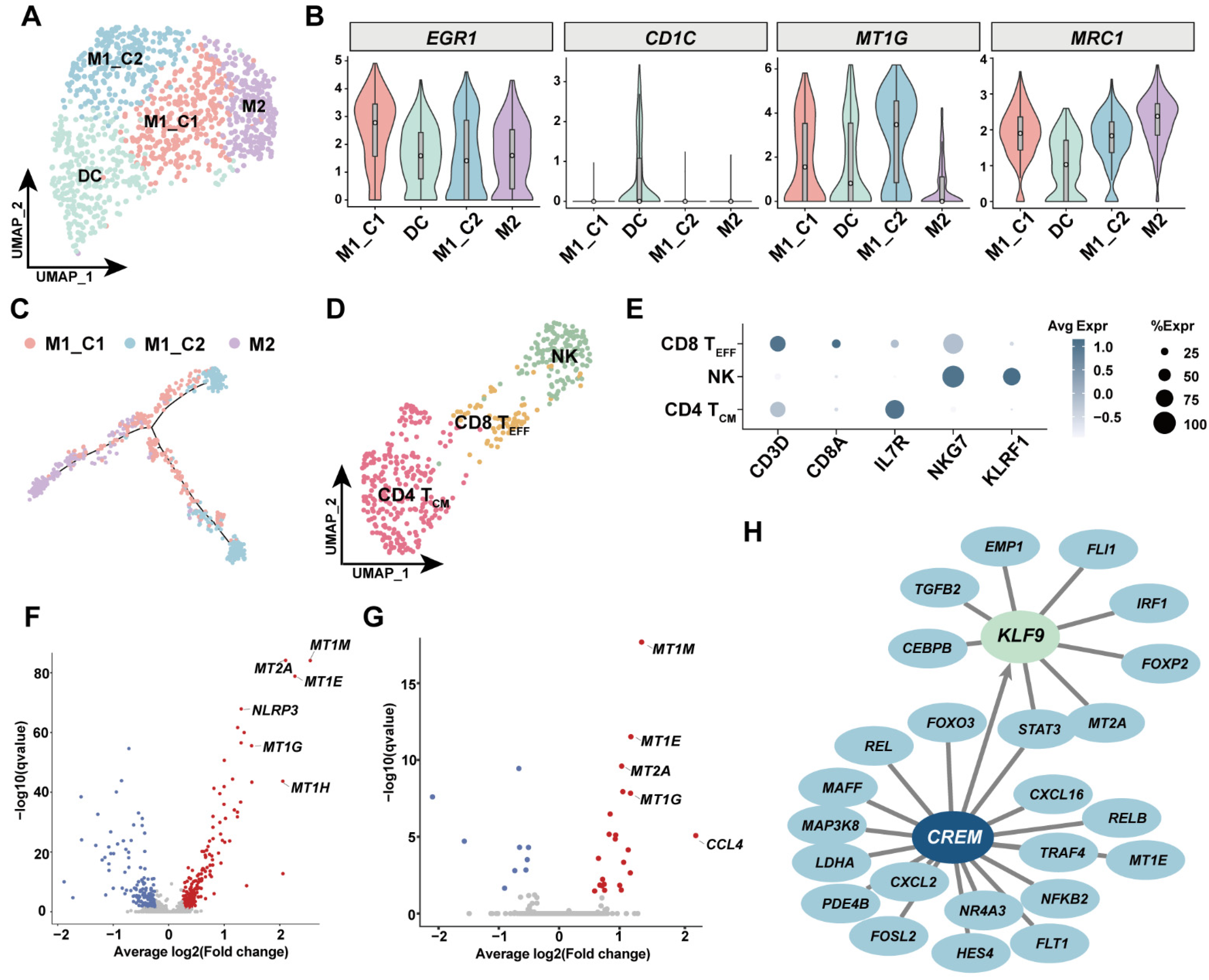
3.5. Investigation of Immune Cell Diversity in Human GSV
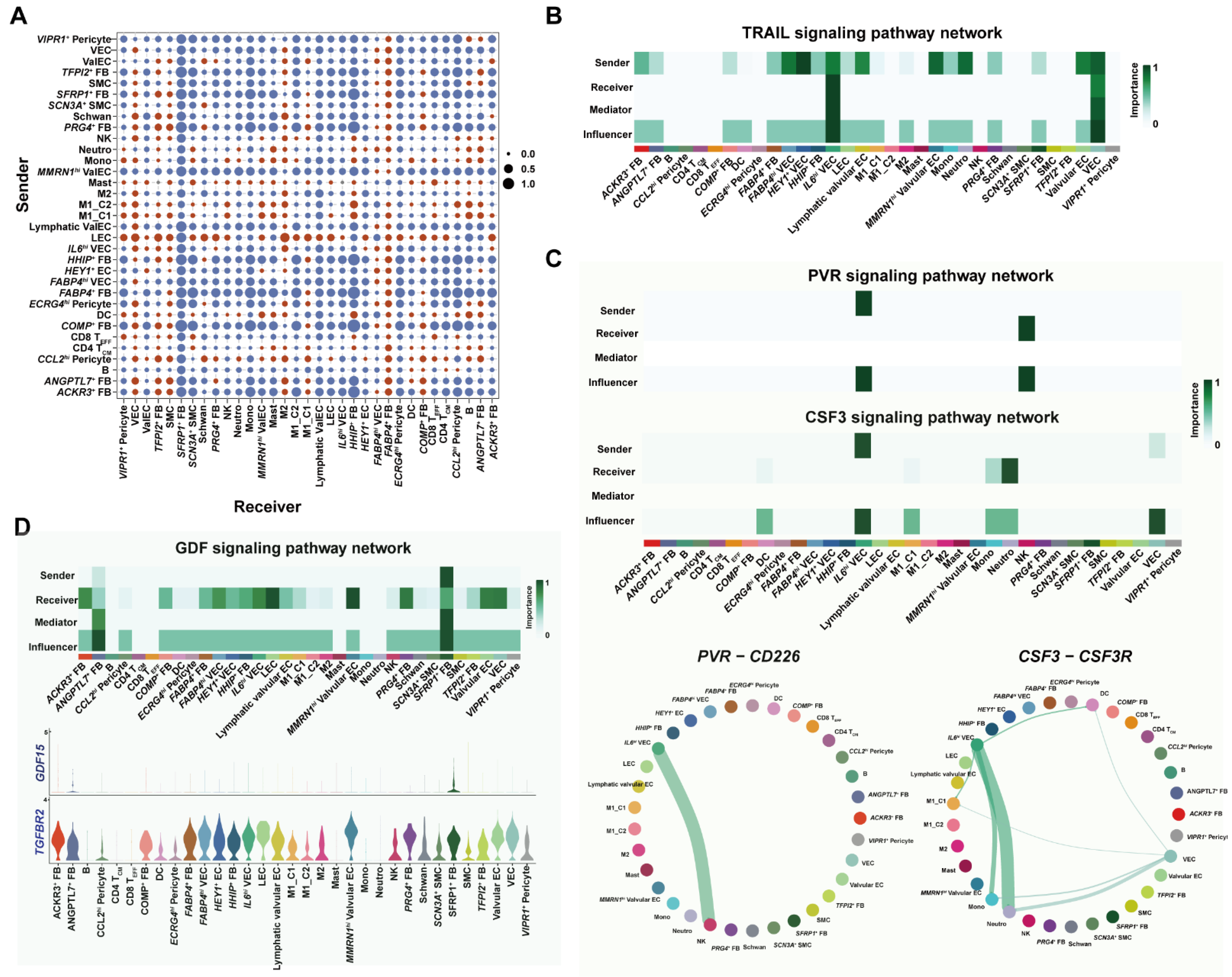
3.6. Cell–Cell Interaction Analysis Unraveled Proinflammatory Programs under Metabolic Complications
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Weintraub, W.S.; Grau-Sepulveda, M.V.; Weiss, J.M.; O’Brien, S.M.; Peterson, E.D.; Kolm, P.; Zhang, Z.; Klein, L.W.; Shaw, R.E.; McKay, C.; et al. Comparative effectiveness of revascularization strategies. N. Engl. J. Med. 2012, 366, 1467–1476. [Google Scholar] [CrossRef]
- Goldman, S.; Zadina, K.; Moritz, T.; Ovitt, T.; Sethi, G.; Copeland, J.G.; Thottapurathu, L.; Krasnicka, B.; Ellis, N.; Anderson, R.J.; et al. Long-term patency of saphenous vein and left internal mammary artery grafts after coronary artery bypass surgery: Results from a Department of Veterans Affairs Cooperative Study. J. Am. Coll. Cardiol. 2004, 44, 2149–2156. [Google Scholar] [CrossRef]
- Hess, C.N.; Lopes, R.D.; Gibson, C.M.; Hager, R.; Wojdyla, D.M.; Englum, B.R.; Mack, M.J.; Califf, R.M.; Kouchoukos, N.T.; Peterson, E.D.; et al. Saphenous vein graft failure after coronary artery bypass surgery: Insights from PREVENT IV. Circulation 2014, 130, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, B.; Algarni, K.D.; Singh, S.K.; Deb, S.; Vincent, J.; Elituv, R.; Desai, N.D.; Rajamani, K.; McManus, B.M.; Liu, P.P.; et al. Clinical, biochemical, and genetic predictors of coronary artery bypass graft failure. J. Thorac. Cardiovasc. Surg. 2014, 148, 515–520.e512. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.S.; Bandyk, D.F.; Clowes, A.W.; Moneta, G.L.; Namini, H.; Seely, L. Risk factors, medical therapies and perioperative events in limb salvage surgery: Observations from the PREVENT III multicenter trial. J. Vasc. Surg. 2005, 42, 456–464, discussion 464–455. [Google Scholar] [CrossRef]
- Christenson, J.T. Preoperative lipid control with simvastatin reduces the risk for graft failure already 1 year after myocardial revascularization. Cardiovasc. Surg. 2001, 9, 33–43. [Google Scholar] [CrossRef]
- Efird, J.T.; O’Neal, W.T.; Gouge, C.A.; Kindell, L.C.; Kennedy, W.L.; Bolin, P., Jr.; O’Neal, J.B.; Anderson, C.A.; Rodriguez, E.; Ferguson, T.B.; et al. Implications of Hemodialysis in Patients Undergoing Coronary Artery Bypass Grafting. Int. J. Cardiovasc. Res. 2013, 2, 1000154. [Google Scholar] [CrossRef]
- De Vries, M.R.; Simons, K.H.; Jukema, J.W.; Braun, J.; Quax, P.H. Vein graft failure: From pathophysiology to clinical outcomes. Nat. Rev. Cardiol. 2016, 13, 451–470. [Google Scholar] [CrossRef]
- Caliskan, E.; De Souza, D.R.; Böning, A.; Liakopoulos, O.J.; Choi, Y.H.; Pepper, J.; Gibson, C.M.; Perrault, L.P.; Wolf, R.K.; Kim, K.B.; et al. Saphenous vein grafts in contemporary coronary artery bypass graft surgery. Nat. Rev. Cardiol. 2020, 17, 155–169. [Google Scholar] [CrossRef]
- Tian, M.; Wang, X.; Sun, H.; Feng, W.; Song, Y.; Lu, F.; Wang, L.; Wang, Y.; Xu, B.; Wang, H.; et al. No-Touch Versus Conventional Vein Harvesting Techniques at 12 Months After Coronary Artery Bypass Grafting Surgery: Multicenter Randomized, Controlled Trial. Circulation 2021, 144, 1120–1129. [Google Scholar] [CrossRef]
- Garcia, F.J.; Sun, N.; Lee, H.; Godlewski, B.; Galani, K.; Zhou, B.; Mantero, J.; Bennett, D.A.; Sahin, M.; Kellis, M.; et al. Single-cell dissection of the human brain vasculature. Nature 2022, 603, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Stanley, G.; Sinha, R.; D’Amato, G.; Das, S.; Rhee, S.; Chang, A.H.; Poduri, A.; Raftrey, B.; Dinh, T.T.; et al. Single-cell analysis of early progenitor cells that build coronary arteries. Nature 2018, 559, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e720. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, W.; Hua, X.; Chen, X.; Chang, Y.; Hu, Y.; Xu, Z.; Song, J. Single-Cell Transcriptomic Atlas of Different Human Cardiac Arteries Identifies Cell Types Associated With Vascular Physiology. Arter. Thromb. Vasc. Biol. 2021, 41, 1408–1427. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330.e314. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Schupp, J.C.; Adams, T.S.; Cosme, C., Jr.; Raredon, M.S.B.; Yuan, Y.; Omote, N.; Poli, S.; Chioccioli, M.; Rose, K.A.; Manning, E.P.; et al. Integrated Single-Cell Atlas of Endothelial Cells of the Human Lung. Circulation 2021, 144, 286–302. [Google Scholar] [CrossRef]
- Francois, M.; Harvey, N.L.; Hogan, B.M. The transcriptional control of lymphatic vascular development. Physiology 2011, 26, 146–155. [Google Scholar] [CrossRef]
- Srinivasan, R.S.; Dillard, M.E.; Lagutin, O.V.; Lin, F.J.; Tsai, S.; Tsai, M.J.; Samokhvalov, I.M.; Oliver, G. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 2007, 21, 2422–2432. [Google Scholar] [CrossRef] [Green Version]
- Bazigou, E.; Lyons, O.T.; Smith, A.; Venn, G.E.; Cope, C.; Brown, N.A.; Makinen, T. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J. Clin. Investig. 2011, 121, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Mellor, R.H.; Brice, G.; Stanton, A.W.; French, J.; Smith, A.; Jeffery, S.; Levick, J.R.; Burnand, K.G.; Mortimer, P.S. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation 2007, 115, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Sabine, A.; Bovay, E.; Demir, C.S.; Kimura, W.; Jaquet, M.; Agalarov, Y.; Zangger, N.; Scallan, J.P.; Graber, W.; Gulpinar, E.; et al. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Investig. 2015, 125, 3861–3877. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Cebo, M.; Dittrich, K.; Fu, X.; Manke, M.C.; Emschermann, F.; Rheinlaender, J.; von Eysmondt, H.; Ferreirós, N.; Sudmann-Innerhofer, J.; Witte, A.; et al. Platelet ACKR3/CXCR7 Favors Anti-Platelet Lipids over an Atherothrombotic Lipidome and Regulates Thrombo-inflammation. Blood 2021, 139, 1722–1742. [Google Scholar] [CrossRef]
- Lee, H.J.; Koh, G.Y. Shear stress activates Tie2 receptor tyrosine kinase in human endothelial cells. Biochem. Biophys. Res. Commun. 2003, 304, 399–404. [Google Scholar] [CrossRef]
- Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [CrossRef]
- Rao, M.; Wang, X.; Guo, G.; Wang, L.; Chen, S.; Yin, P.; Chen, K.; Chen, L.; Zhang, Z.; Chen, X.; et al. Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res. Cardiol. 2021, 116, 55. [Google Scholar] [CrossRef]
- Potente, M.; Mäkinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef]
- Hansen, A.; Henderson, S.; Lagos, D.; Nikitenko, L.; Coulter, E.; Roberts, S.; Gratrix, F.; Plaisance, K.; Renne, R.; Bower, M.; et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 2010, 24, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.O.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Gur-Cohen, S.; Yang, H.; Baksh, S.C.; Miao, Y.; Levorse, J.; Kataru, R.P.; Liu, X.; de la Cruz-Racelis, J.; Mehrara, B.J.; Fuchs, E. Stem cell-driven lymphatic remodeling coordinates tissue regeneration. Science 2019, 366, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Gordon-Weeks, A.; Allen, D.; Kersemans, V.; Beech, J.; Smart, S.; Muschel, R.J. Cd11b(+) myeloid cells support hepatic metastasis through down-regulation of angiopoietin-like 7 in cancer cells. Hepatology 2015, 62, 521–533. [Google Scholar] [CrossRef]
- Xu, T.; Xu, L.; Meng, P.; Ma, X.; Yang, X.; Zhou, Y.; Feng, M. Angptl7 promotes insulin resistance and type 2 diabetes mellitus by multiple mechanisms including SOCS3-mediated IRS1 degradation. FASEB J. 2020, 34, 13548–13560. [Google Scholar] [CrossRef]
- Comes, N.; Buie, L.K.; Borrás, T. Evidence for a role of angiopoietin-like 7 (ANGPTL7) in extracellular matrix formation of the human trabecular meshwork: Implications for glaucoma. Genes Cells 2011, 16, 243–259. [Google Scholar] [CrossRef]
- Sun, M.; Liu, W.; Zhou, M. ANGPTL7 is transcriptionally regulated by SP1 and modulates glucocorticoid-induced cross-linked actin networks in trabecular meshwork cells via the RhoA/ROCK pathway. Cell Death Discov. 2022, 8, 50. [Google Scholar] [CrossRef]
- Chen, J.H.; Yip, C.Y.; Sone, E.D.; Simmons, C.A. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am. J. Pathol. 2009, 174, 1109–1119. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.S.; Kenny, C.J.; Ganesh, V.; Jang, A.; Borges-Monroy, R.; Partlow, J.N.; Hill, R.S.; Shin, T.; Chen, A.Y.; Doan, R.N.; et al. Sodium Channel SCN3A (Na(V)1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development. Neuron 2018, 99, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Luchtefeld, M.; Schunkert, H.; Stoll, M.; Selle, T.; Lorier, R.; Grote, K.; Sagebiel, C.; Jagavelu, K.; Tietge, U.J.; Assmus, U.; et al. Signal transducer of inflammation gp130 modulates atherosclerosis in mice and man. J. Exp. Med. 2007, 204, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, M.; Antoniades, C.; Benedetto, U.; Deb, S.; Di Franco, A.; Di Giammarco, G.; Fremes, S.; Glineur, D.; Grau, J.; He, G.W.; et al. Mechanisms, Consequences, and Prevention of Coronary Graft Failure. Circulation 2017, 136, 1749–1764. [Google Scholar] [CrossRef]
- Sarkar, S.; Melchior, J.T.; Henry, H.R.; Syed, F.; Mirmira, R.G.; Nakayasu, E.S.; Metz, T.O. GDF15: A potential therapeutic target for type 1 diabetes. Expert Opin. Ther. Targets 2022, 26, 57–67. [Google Scholar] [CrossRef]
- Ceelen, D.; Voors, A.A.; Tromp, J.; van Veldhuisen, D.J.; Dickstein, K.; de Boer, R.A.; Lang, C.C.; Anker, S.D.; Ng, L.L.; Metra, M.; et al. Pathophysiological pathways related to high plasma growth differentiation factor 15 concentrations in patients with heart failure. Eur. J. Heart Fail. 2022, 24, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Keipert, S.; Ost, M. Stress-induced FGF21 and GDF15 in obesity and obesity resistance. Trends Endocrinol. Metab. 2021, 32, 904–915. [Google Scholar] [CrossRef]
- Wang, D.; Day, E.A.; Townsend, L.K.; Djordjevic, D.; Jørgensen, S.B.; Steinberg, G.R. GDF15: Emerging biology and therapeutic applications for obesity and cardiometabolic disease. Nat. Rev. Endocrinol. 2021, 17, 592–607. [Google Scholar] [CrossRef]
- Gaudino, M.; Benedetto, U.; Fremes, S.; Biondi-Zoccai, G.; Sedrakyan, A.; Puskas, J.D.; Angelini, G.D.; Buxton, B.; Frati, G.; Hare, D.L.; et al. Radial-Artery or Saphenous-Vein Grafts in Coronary-Artery Bypass Surgery. N. Engl. J. Med. 2018, 378, 2069–2077. [Google Scholar] [CrossRef]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef] [PubMed]
- Raguraman, R.; Srivastava, A.; Munshi, A.; Ramesh, R. Therapeutic approaches targeting molecular signaling pathways common to diabetes, lung diseases and cancer. Adv. Drug Deliv. Rev. 2021, 178, 113918. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef]
- Fetting, J.L.; Guay, J.A.; Karolak, M.J.; Iozzo, R.V.; Adams, D.C.; Maridas, D.E.; Brown, A.C.; Oxburgh, L. FOXD1 promotes nephron progenitor differentiation by repressing decorin in the embryonic kidney. Development 2014, 141, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Molecular functions of metallothionein and its role in hematological malignancies. J. Hematol. Oncol. 2012, 5, 41. [Google Scholar] [CrossRef]
- Vijay, J.; Gauthier, M.F.; Biswell, R.L.; Louiselle, D.A.; Johnston, J.J.; Cheung, W.A.; Belden, B.; Pramatarova, A.; Biertho, L.; Gibson, M.; et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2020, 2, 97–109. [Google Scholar] [CrossRef]
- Cui, A.; Fan, H.; Zhang, Y.; Zhang, Y.; Niu, D.; Liu, S.; Liu, Q.; Ma, W.; Shen, Z.; Shen, L.; et al. Dexamethasone-induced Krüppel-like factor 9 expression promotes hepatic gluconeogenesis and hyperglycemia. J. Clin. Investig. 2019, 129, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Hu, X.; Zhang, K.; Rao, M.; Yin, P.; Dong, R. A Single-Cell Survey of Cellular Heterogeneity in Human Great Saphenous Veins. Cells 2022, 11, 2711. https://doi.org/10.3390/cells11172711
Sun Y, Hu X, Zhang K, Rao M, Yin P, Dong R. A Single-Cell Survey of Cellular Heterogeneity in Human Great Saphenous Veins. Cells. 2022; 11(17):2711. https://doi.org/10.3390/cells11172711
Chicago/Turabian StyleSun, Yiping, Xueqing Hu, Kui Zhang, Man Rao, Pengbin Yin, and Ran Dong. 2022. "A Single-Cell Survey of Cellular Heterogeneity in Human Great Saphenous Veins" Cells 11, no. 17: 2711. https://doi.org/10.3390/cells11172711
APA StyleSun, Y., Hu, X., Zhang, K., Rao, M., Yin, P., & Dong, R. (2022). A Single-Cell Survey of Cellular Heterogeneity in Human Great Saphenous Veins. Cells, 11(17), 2711. https://doi.org/10.3390/cells11172711