

The CaMKII/MLC1 Axis Confers Ca2+-Dependence to Volume-Regulated Anion Channels (VRAC) in Astrocytes

 , , ,
, , ,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Generation of U251 Cell Lines Expressing MLC1-T17A and T17D Mutants
2.3. Immunofluorescence Staining (IF)
2.4. Pull-Down Assay and Immunoprecipitation
2.5. Protein Extract Preparation and Western Blotting (WB)
2.6. In Vitro Kinase Assay for CaMKII
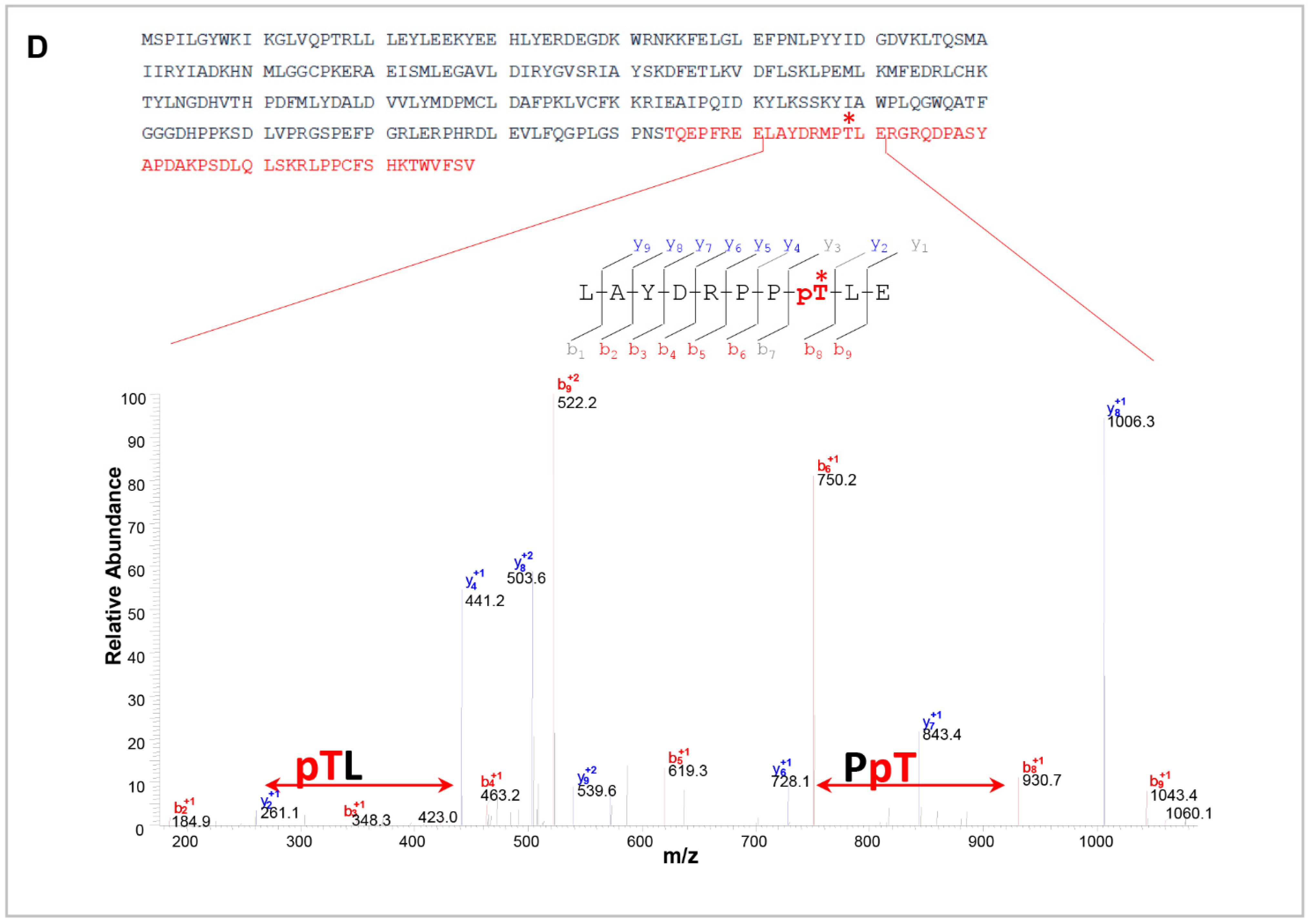
2.7. Mass Spectrometry Analysis
2.8. Electrophysiology
2.9. Fura-2-Based Ca2+ Imaging
2.10. Statistical Analysis
3. Results
3.1. Ca2+-Dependent Protein Kinases II (CaMKII) Binds MLC1 Protein in Astrocytes
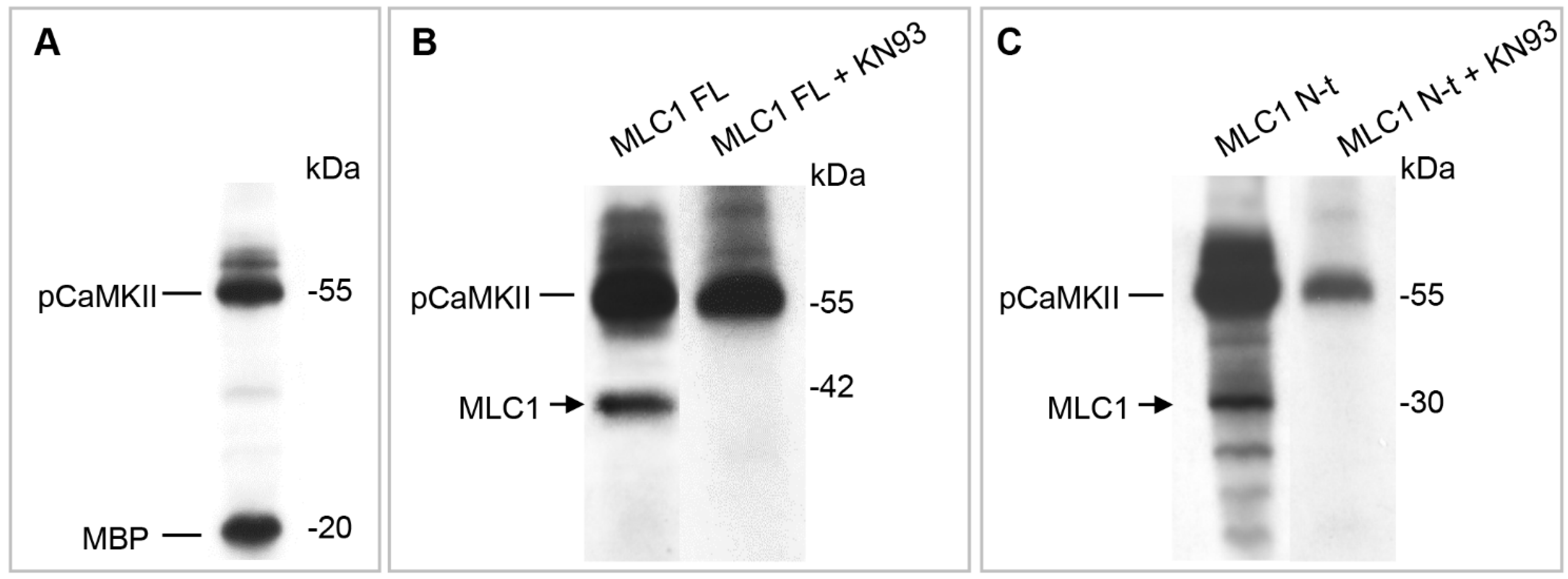
3.2. CaMKII Phosphorylates MLC1 Protein at the Amino Acid Residue Threonine 17
3.3. CaMKII-Mediated Phosphorylation of MLC1 in Response to Intracellular Ca2+ Increase Affects MLC1 Protein Assembly and Stability in U251 Cells
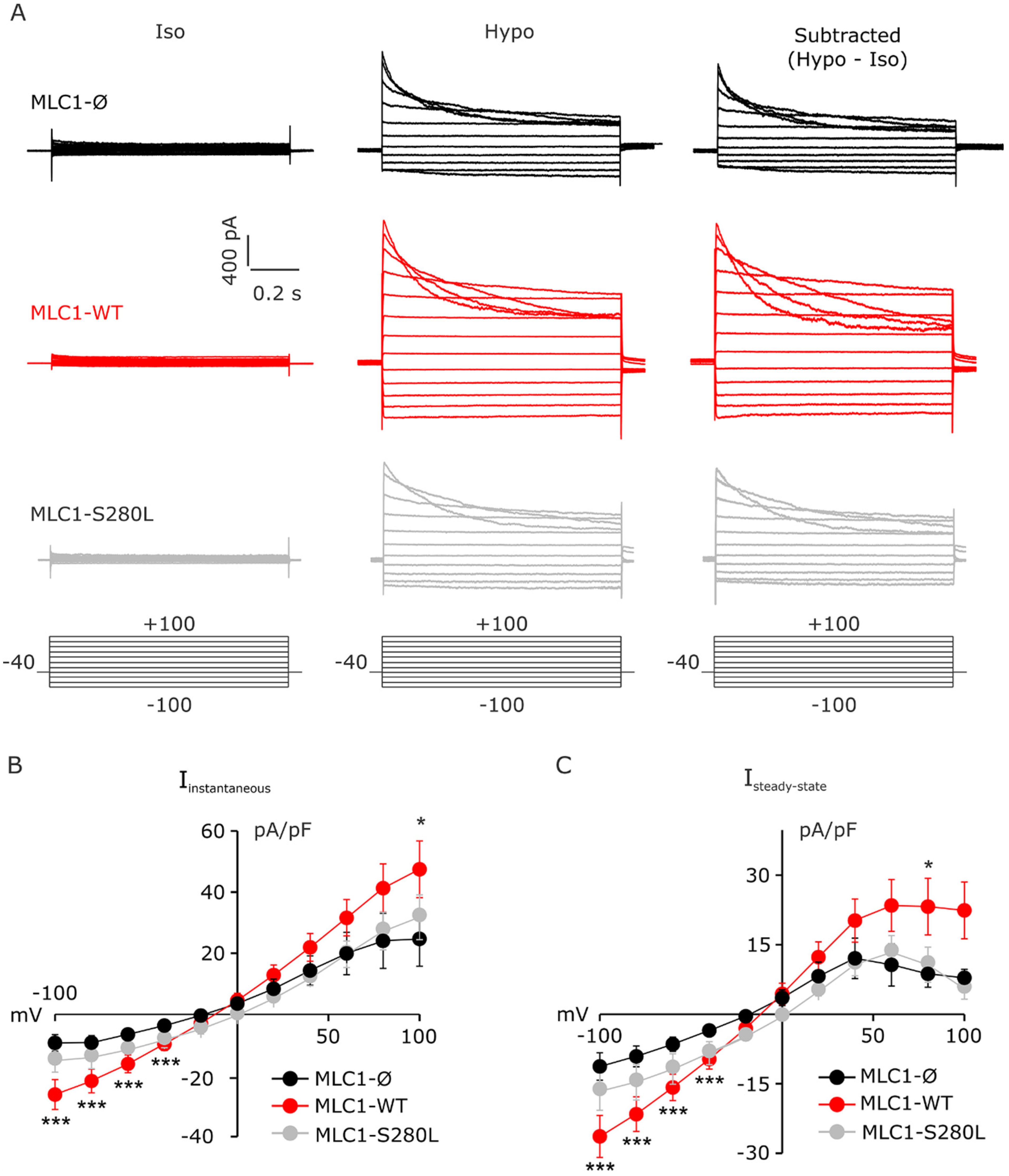
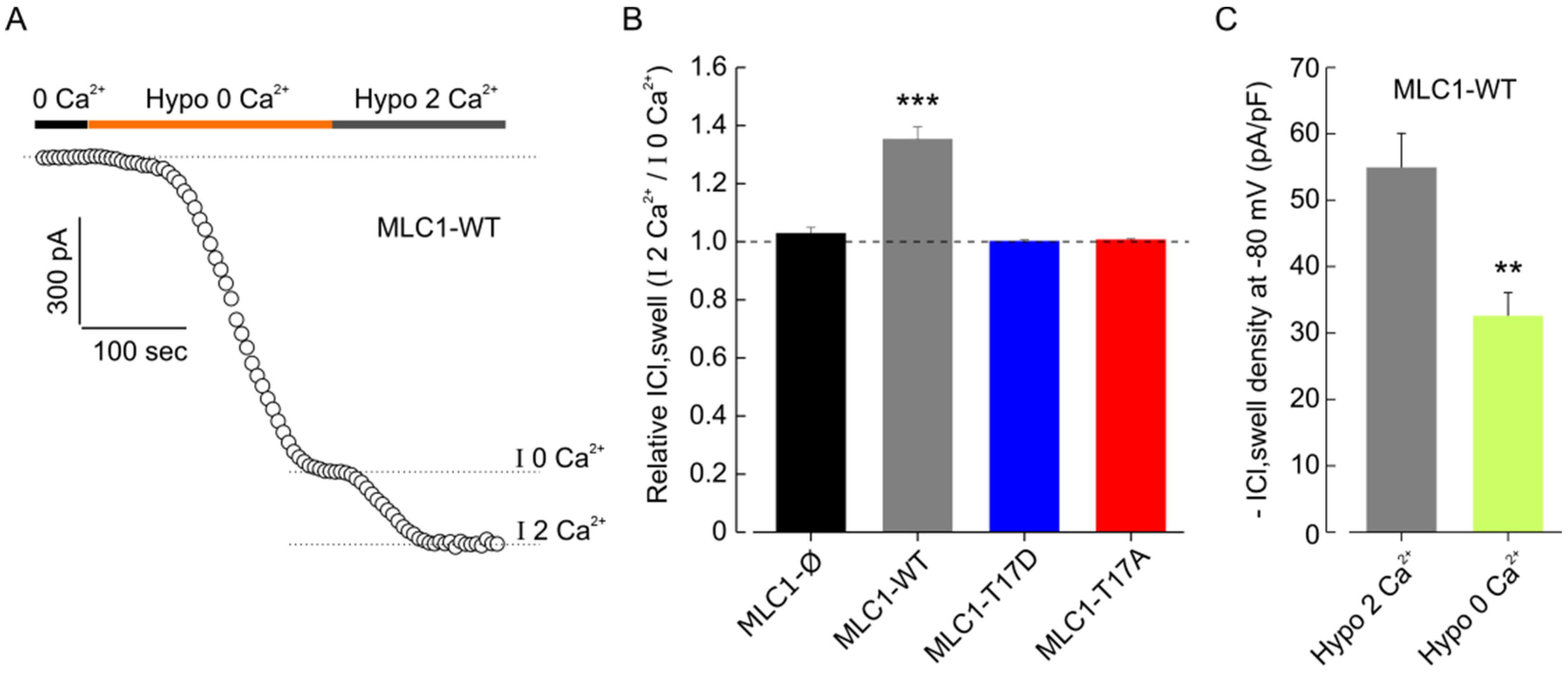
3.4. MLC1 Potentiates VRAC-Mediated ICl,swell in U251 Cells
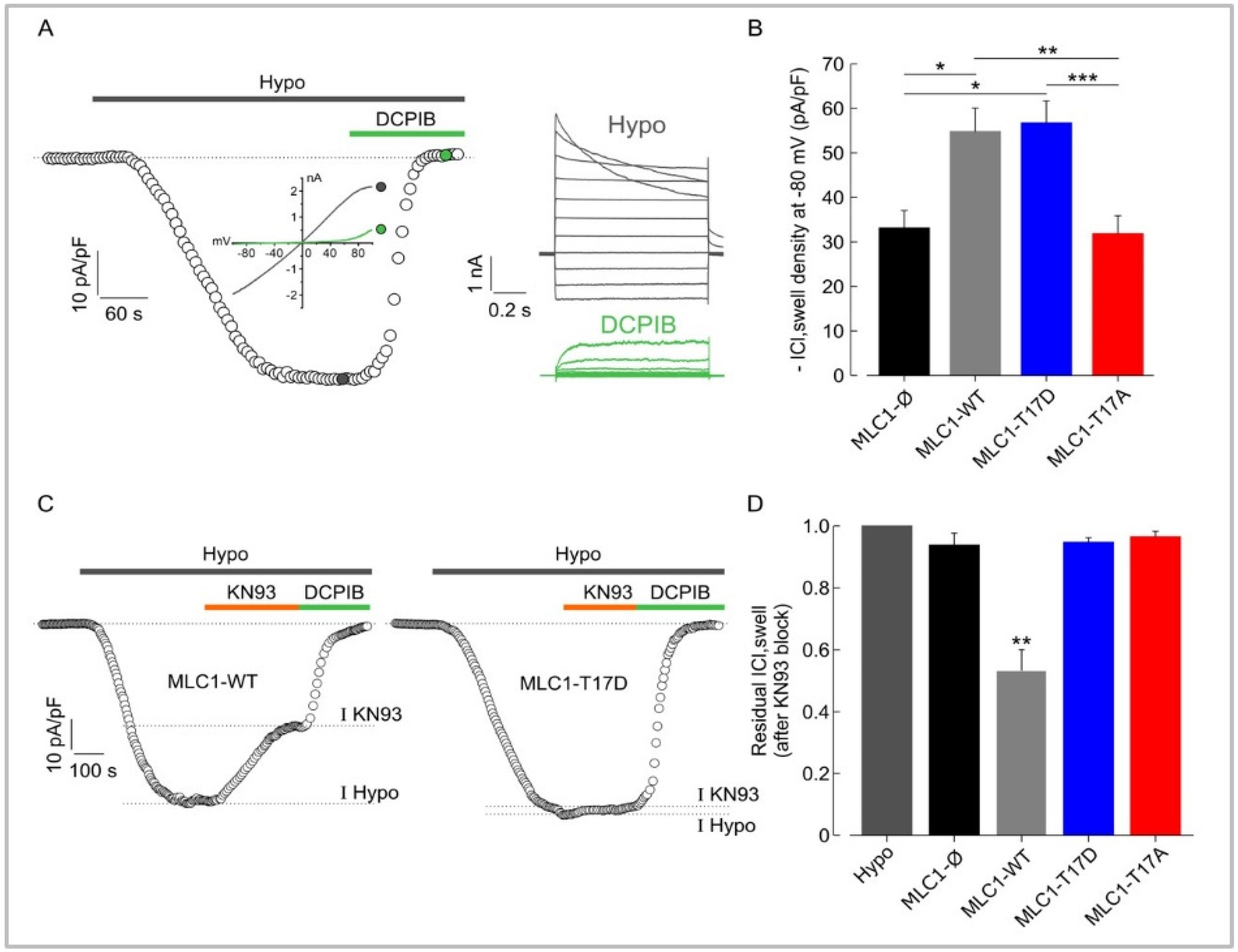
3.5. MLC1 Potentiates VRAC-Mediated ICl,swelll in U251 Cells through a CaMKII-Dependent Mechanism
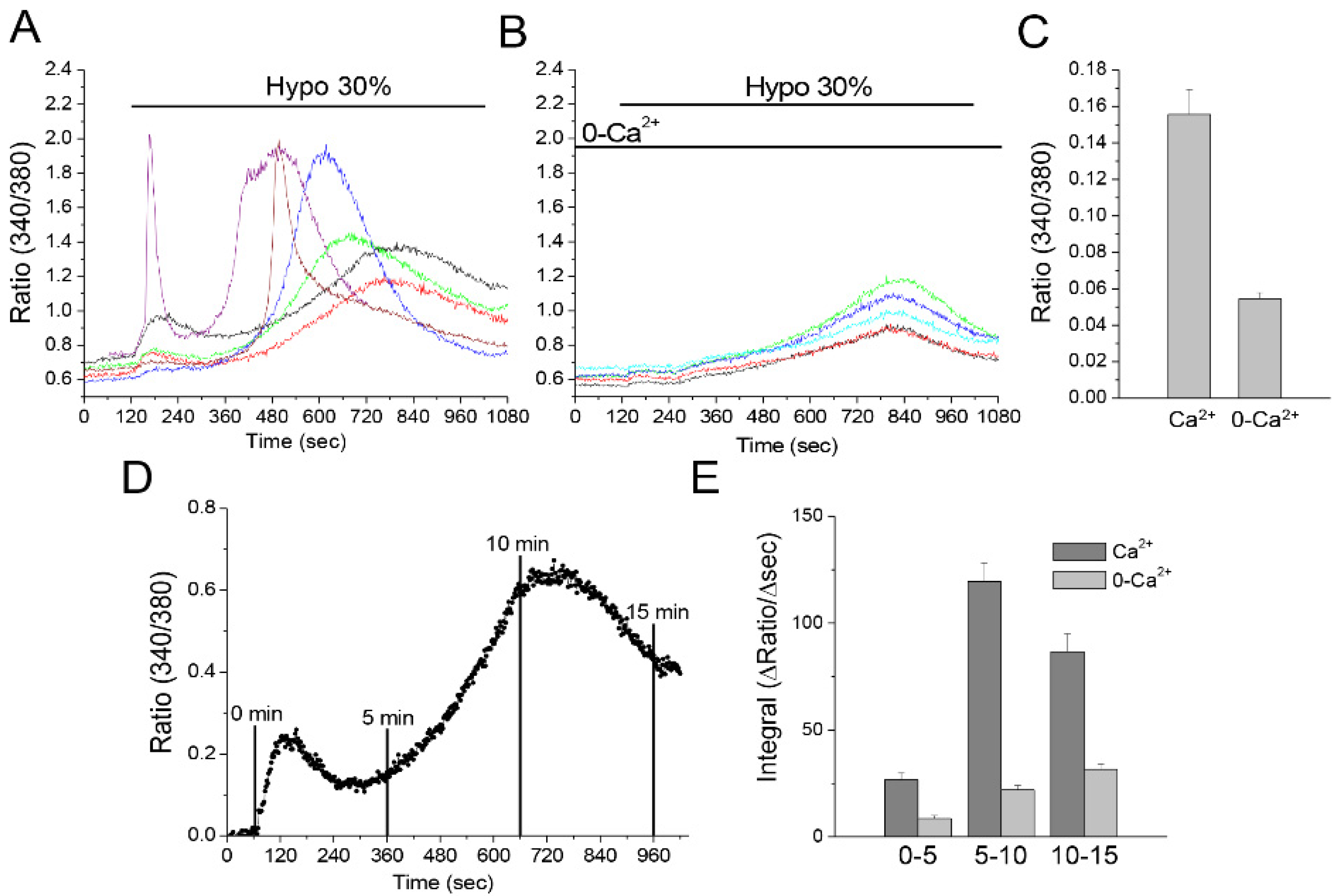
3.6. Hyposmotic Challenge Induces Ca2+ Influx and Ca2+ Release in U251 Cells
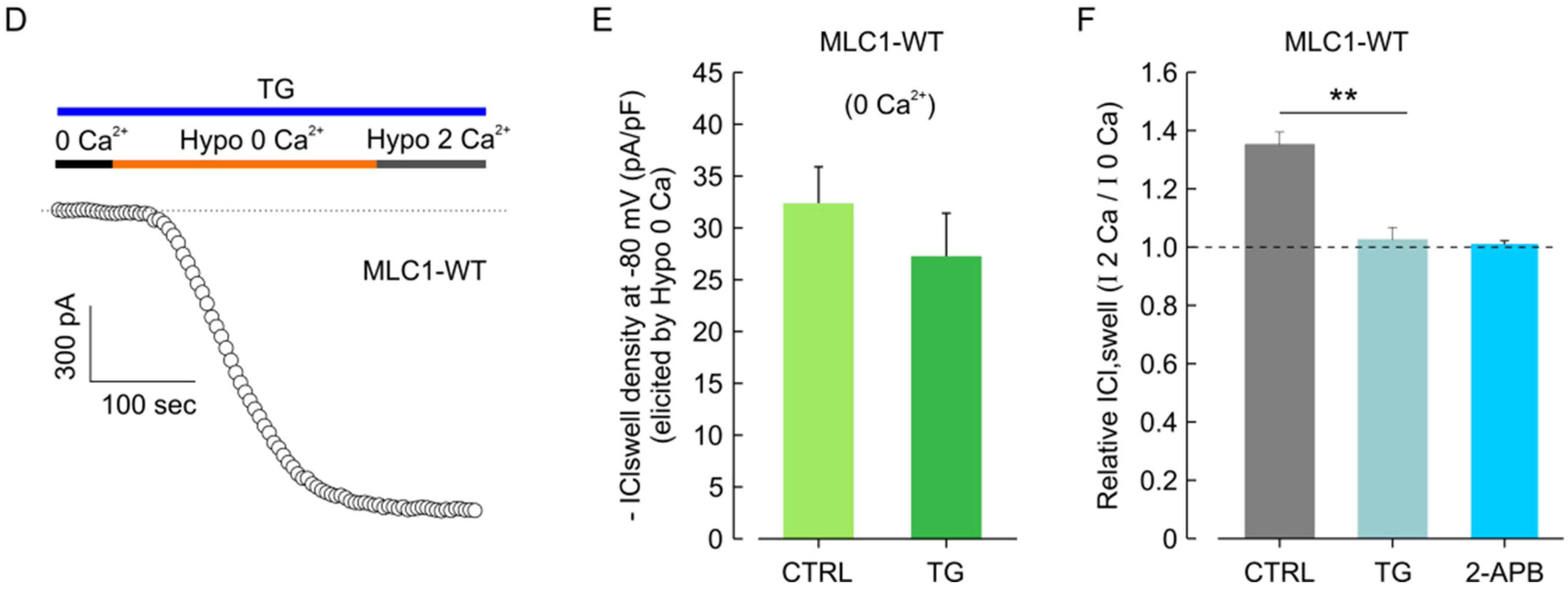
3.7. CaMKII/MLC1-Dependent Enhancement of ICl,swell Requires Release of Ca2+ from Intracellular Stores Induced by Extracellular Ca2+ Influx
4. Discussion
4.1. CamKII Binds and Phosphorylates MLC1
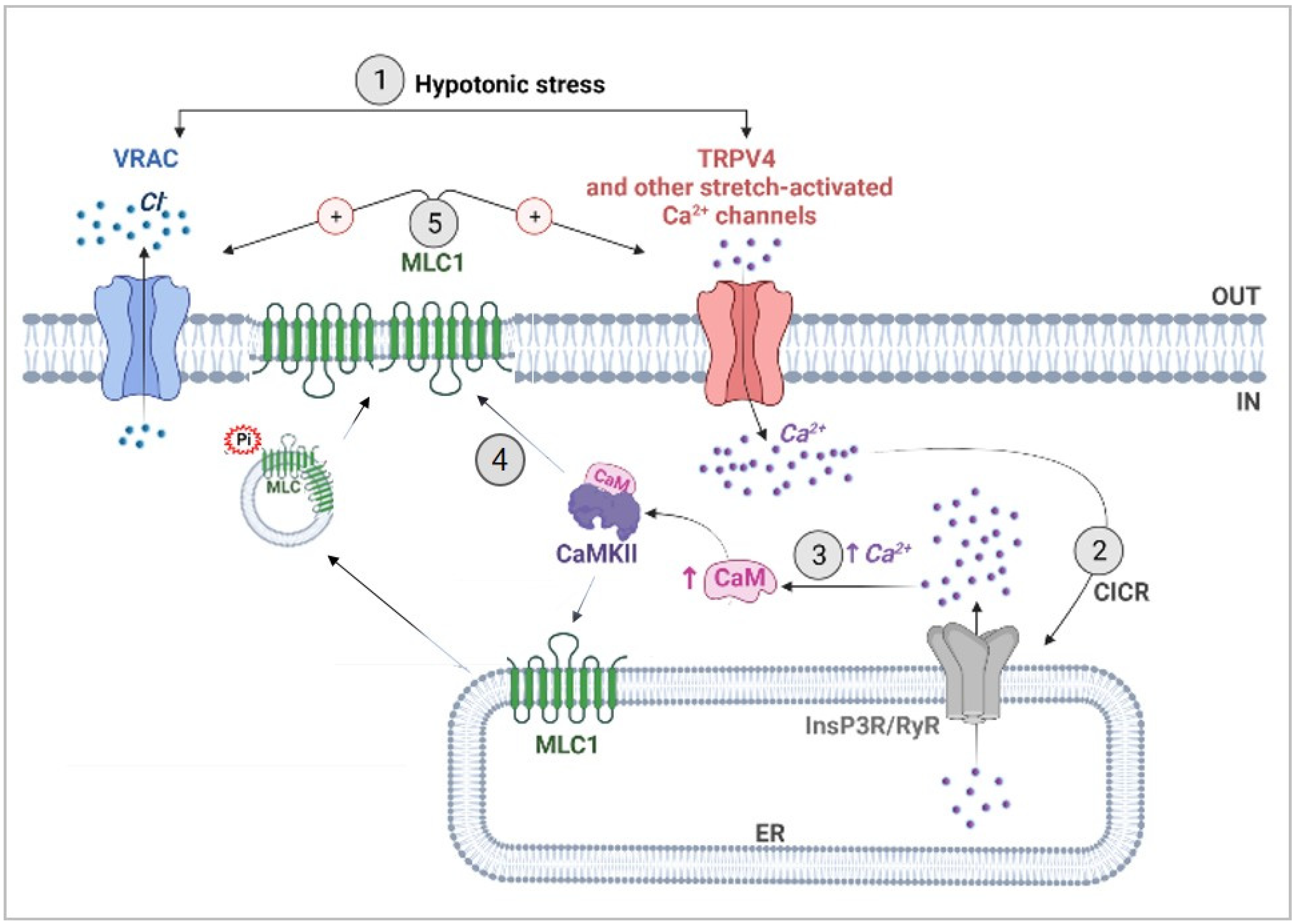
4.2. CaMKII-Mediated Phosphorylation of MLC1 Confers Ca2+-Dependence to VRAC Activation
4.3. Significance of Astrocyte Ca2+ Signaling in MLC Pathogenesis and Beyond
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verkhratsky, A.; Parpura, V.; Li, B.; Scuderi, C. Astrocytes: The Housekeepers and Guardians of the CNS. In Astrocytes in Psychiatric Disorders; Advances in Neurobiology; Li, B., Parpura, V., Verkhratsky, A., Scuderi, C., Eds.; Springer International Publishing: Cham, Switzerland, 2021; Volume 26, pp. 21–53. ISBN 978-3-030-77374-8. [Google Scholar]
- MacAulay, N. Molecular Mechanisms of Brain Water Transport. Nat. Rev. Neurosci. 2021, 22, 326–344. [Google Scholar] [CrossRef]
- Wilson, C.S.; Mongin, A.A. Cell Volume Control in Healthy Brain and Neuropathologies. Curr. Top. Membr. 2018, 81, 385–455. [Google Scholar] [CrossRef] [PubMed]
- Benga, O.; Huber, V.J. Brain Water Channel Proteins in Health and Disease. Mol. Asp. Med. 2012, 33, 562–578. [Google Scholar] [CrossRef]
- Reed, M.M.; Blazer-Yost, B. Channels and Transporters in Astrocyte Volume Regulation in Health and Disease. Cell Physiol. Biochem. 2022, 56, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Lafrenaye, A.; Simard, J. Bursting at the Seams: Molecular Mechanisms Mediating Astrocyte Swelling. IJMS 2019, 20, 330. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of Astrocyte-Mediated Cerebral Edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef]
- Jha, R.M.; Raikwar, S.P.; Mihaljevic, S.; Casabella, A.M.; Catapano, J.S.; Rani, A.; Desai, S.; Gerzanich, V.; Simard, J.M. Emerging Therapeutic Targets for Cerebral Edema. Expert Opin. Ther. Targets 2021, 25, 917–938. [Google Scholar] [CrossRef]
- Min, R.; van der Knaap, M.S. Genetic Defects Disrupting Glial Ion and Water Homeostasis in the Brain. Brain Pathol. 2018, 28, 372–387. [Google Scholar] [CrossRef]
- Hamilton, E.M.C.; Tekturk, P.; Cialdella, F.; van Rappard, D.F.; Wolf, N.I.; Yalcinkaya, C.; Çetinçelik, Ü.; Rajaee, A.; Kariminejad, A.; Paprocka, J.; et al. Megalencephalic Leukoencephalopathy with Subcortical Cysts: Characterization of Disease Variants. Neurology 2018, 90, e1395–e1403. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Barth, P.G.; Vrensen, G.F.J.M.; Valk, J. Histopathology of an Infantile-Onset Spongiform Leukoencephalopathy with a Discrepantly Mild Clinical Course. Acta Neuropathol. 1996, 92, 206–212. [Google Scholar] [CrossRef]
- Leegwater, P.A.J.; Yuan, B.Q.; van der Steen, J.; Mulders, J.; Könst, A.A.M.; Boor, P.K.I.; Mejaski-Bosnjak, V.; van der Maarel, S.M.; Frants, R.R.; Oudejans, C.B.M.; et al. Mutations of MLC1 (KIAA0027), Encoding a Putative Membrane Protein, Cause Megalencephalic Leukoencephalopathy with Subcortical Cysts. Am. J. Hum. Genet. 2001, 68, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Estévez, R. Megalencephalic Leukoencephalopathy: Insights Into Pathophysiology and Perspectives for Therapy. Front. Cell. Neurosci. 2020, 14, 627887. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.K.I.; de Groot, K.; Waisfisz, Q.; Kamphorst, W.; Oudejans, C.B.M.; Powers, J.M.; Pronk, J.C.; Scheper, G.C.; van der Knaap, M.S. MLC1: A Novel Protein in Distal Astroglial Processes. J. Neuropathol. Exp. Neurol. 2005, 64, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Ridder, M.C.; Boor, I.; Lodder, J.C.; Postma, N.L.; Capdevila-Nortes, X.; Duarri, A.; Brussaard, A.B.; Estévez, R.; Scheper, G.C.; Mansvelder, H.D.; et al. MegalencephalicLeucoencephalopathy with Cysts: Defect in Chloride Currents and Cell Volume Regulation. Brain 2011, 134, 3342–3354. [Google Scholar] [CrossRef] [PubMed]
- Hoegg-Beiler, M.B.; Sirisi, S.; Orozco, I.J.; Ferrer, I.; Hohensee, S.; Auberson, M.; Gödde, K.; Vilches, C.; de Heredia, M.L.; Nunes, V.; et al. Disrupting MLC1 and GlialCAM and ClC-2 Interactions in Leukodystrophy Entails Glial Chloride Channel Dysfunction. Nat. Commun. 2014, 5, 3475. [Google Scholar] [CrossRef] [PubMed]
- Sirisi, S.; Elorza-Vidal, X.; Arnedo, T.; Armand-Ugón, M.; Callejo, G.; Capdevila-Nortes, X.; López-Hernández, T.; Schulte, U.; Barrallo-Gimeno, A.; Nunes, V.; et al. Depolarization Causes the Formation of a Ternary Complex between GlialCAM, MLC1 and ClC-2 in Astrocytes: Implications in Megalencephalic Leukoencephalopathy. Hum. Mol. Genet. 2017, 26, 2436–2450. [Google Scholar] [CrossRef]
- Formaggio, F.; Saracino, E.; Mola, M.G.; Rao, S.B.; Amiry-Moghaddam, M.; Muccini, M.; Zamboni, R.; Nicchia, G.P.; Caprini, M.; Benfenati, V. LRRC8A Is Essential for Swelling-activated Chloride Current and for Regulatory Volume Decrease in Astrocytes. FASEB J. 2019, 33, 101–113. [Google Scholar] [CrossRef]
- Brignone, M.S.; Lanciotti, A.; Macioce, P.; Macchia, G.; Gaetani, M.; Aloisi, F.; Petrucci, T.C.; Ambrosini, E. The Β1 Subunit of the Na,K-ATPase Pump Interacts with Megalencephalic Leucoencephalopathy with Subcortical Cysts Protein 1 (MLC1) in Brain Astrocytes: New Insights into MLC Pathogenesis. Hum. Mol. Genet. 2011, 20, 90–103. [Google Scholar] [CrossRef]
- Lanciotti, A.; Brignone, M.S.; Molinari, P.; Visentin, S.; De Nuccio, C.; Macchia, G.; Aiello, C.; Bertini, E.; Aloisi, F.; Petrucci, T.C.; et al. Megalencephalic Leukoencephalopathy with Subcortical Cysts Protein 1 Functionally Cooperates with the TRPV4 Cation Channel to Activate the Response of Astrocytes to Osmotic Stress: Dysregulation by Pathological Mutations. Hum. Mol. Genet. 2012, 21, 2166–2180. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, A.; Brignone, M.S.; Camerini, S.; Serafini, B.; Macchia, G.; Raggi, C.; Molinari, P.; Crescenzi, M.; Musumeci, M.; Sargiacomo, M.; et al. MLC1 Trafficking and Membrane Expression in Astrocytes: Role of Caveolin-1 and Phosphorylation. Neurobiol. Dis. 2010, 37, 581–595. [Google Scholar] [CrossRef]
- Ambrosini, E.; Serafini, B.; Lanciotti, A.; Tosini, F.; Scialpi, F.; Psaila, R.; Raggi, C.; Di Girolamo, F.; Petrucci, T.C.; Aloisi, F. Biochemical Characterization of MLC1 Protein in Astrocytes and Its Association with the Dystrophin-Glycoprotein Complex. Mol. Cell. Neurosci. 2008, 37, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Brignone, M.S.; Lanciotti, A.; Camerini, S.; De Nuccio, C.; Petrucci, T.C.; Visentin, S.; Ambrosini, E. MLC1 Protein: A Likely Link between Leukodystrophies and Brain Channelopathies. Front. Cell. Neurosci. 2015, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Weerth, S.H.; Holtzclaw, L.A.; Russell, J.T. Signaling Proteins in Raft-like Microdomains Are Essential for Ca2+ Wave Propagation in Glial Cells. Cell Calcium. 2007, 41, 155–167. [Google Scholar] [CrossRef]
- Petrini, S.; Minnone, G.; Coccetti, M.; Frank, C.; Aiello, C.; Cutarelli, A.; Ambrosini, E.; Lanciotti, A.; Brignone, M.S.; D’Oria, V.; et al. Monocytes and Macrophages as Biomarkers for the Diagnosis of Megalencephalic Leukoencephalopathy with Subcortical Cysts. Mol. Cell. Neurosci. 2013, 56, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, A.; Brignone, M.S.; Visentin, S.; De Nuccio, C.; Catacuzzeno, L.; Mallozzi, C.; Petrini, S.; Caramia, M.; Veroni, C.; Minnone, G.; et al. Megalencephalic Leukoencephalopathy with Subcortical Cysts Protein-1 Regulates Epidermal Growth Factor Receptor Signaling in Astrocytes. Hum. Mol. Genet. 2016, 25, 1543–1558. [Google Scholar] [CrossRef]
- Cardin, V.; Lezama, R.; Torres-Márquez, M.E.; Pasantes-Morales, H. Potentiation of the Osmosensitive Taurine Release and Cell Volume Regulation by Cytosolic Ca2+ Rise in Cultured Cerebellar Astrocytes. Glia 2003, 44, 119–128. [Google Scholar] [CrossRef]
- Eleuteri, C.; Olla, S.; Veroni, C.; Umeton, R.; Mechelli, R.; Romano, S.; Buscarinu, M.; Ferrari, F.; Calò, G.; Ristori, G.; et al. A Staged Screening of Registered Drugs Highlights Remyelinating Drug Candidates for Clinical Trials. Sci. Rep. 2017, 7, 45780. [Google Scholar] [CrossRef]
- Sforna, L.; Cenciarini, M.; Belia, S.; Michelucci, A.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. Hypoxia Modulates the Swelling-Activated Cl Current in Human Glioblastoma Cells: Role in Volume Regulation and Cell Survival: Hypoxia-Activated Cl Current in Glioblastoma. J. Cell. Physiol. 2017, 232, 91–100. [Google Scholar] [CrossRef]
- Arnedo, T.; López-Hernández, T.; Jeworutzki, E.; Capdevila-Nortes, X.; Sirisi, S.; Pusch, M.; Estévez, R. Functional Analyses of Mutations in HEPACAM Causing Megalencephalic Leukoencephalopathy. Hum. Mutat. 2014, 35, 1175–1178. [Google Scholar] [CrossRef]
- Brignone, M.S.; Lanciotti, A.; Serafini, B.; Mallozzi, C.; Sbriccoli, M.; Veroni, C.; Molinari, P.; Elorza-Vidal, X.; Petrucci, T.C.; Estévez, R.; et al. Megalencephalic Leukoencephalopathy with Subcortical Cysts Protein-1 (MLC1) Counteracts Astrocyte Activation in Response to Inflammatory Signals. Mol. Neurobiol. 2019, 56, 8237–8254. [Google Scholar] [CrossRef]
- Cherubini, F.; Serio, D.; Guccini, I.; Fortuni, S.; Arcuri, G.; Condò, I.; Rufini, A.; Moiz, S.; Camerini, S.; Crescenzi, M.; et al. Src Inhibitors Modulate Frataxin Protein Levels. Hum. Mol. Genet. 2015, 24, 4296–4305. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Eng, J.K.; McCormack, A.L.; Schieltz, D. Method to Correlate Tandem Mass Spectra of Modified Peptides to Amino Acid Sequences in the Protein Database. Anal. Chem. 1995, 67, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Sforna, L.; Michelucci, A.; Morena, F.; Argentati, C.; Franciolini, F.; Vassalli, M.; Martino, S.; Catacuzzeno, L. Piezo1 Controls Cell Volume and Migration by Modulating Swelling-Activated Chloride Current through Ca2+ Influx. J. Cell. Physiol. 2022, 237, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, L.; Prevarskaya, N.; Mazurier, J.; Shuba, Y.; Skryma, R. 2-APB inhibits volume-regulated anion channels independently from intracellular calcium signaling modulation. FEBS Lett. 2004, 556, 121–126. [Google Scholar] [CrossRef]
- Swulius, M.T.; Waxham, M.N. Ca2+/Calmodulin-Dependent Protein Kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef]
- Leslie, S.N.; Nairn, A.C. CAMP Regulation of Protein Phosphatases PP1 and PP2A in Brain. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 64–73. [Google Scholar] [CrossRef]
- Kim, K.-R.; Jeong, H.-J.; Kim, Y.; Lee, S.Y.; Kim, Y.; Kim, H.-J.; Lee, S.-H.; Cho, H.; Kang, J.-S.; Ho, W.-K. Calbindin Regulates Kv4.1 Trafficking and Excitability in Dentate Granule Cells via CaMKII-Dependent Phosphorylation. Exp. Mol. Med. 2021, 53, 1134–1147. [Google Scholar] [CrossRef]
- Li, S.; Jiang, B.; Lu, H.; Huang, S.; Fan, B.; Chen, Z.; Li, Y. Inhibition of CAMK II Modulates Water Permeability by Reducing AQP4 Expression in Astrocytes after Oxygen-Glucose Deprivation. Neuroreport 2021, 32, 1058–1064. [Google Scholar] [CrossRef]
- Hartmann, J.; Verkhratsky, A. Relations between Intracellular Ca2+ Stores and Store-Operated Ca2+ Entry in Primary Cultured Human Glioblastoma Cells. J. Physiol. 1998, 513 Pt 2, 411–424. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Sun, X.; Yang, J.; Yang, Z.; Shen, H.; Li, X.; Liu, Y.; Chen, G. Critical Role for Annexin A7 in Secondary Brain Injury Mediated by Its Phosphorylation after Experimental Intracerebral Hemorrhage in Rats. Neurobiol. Dis. 2018, 110, 82–92. [Google Scholar] [CrossRef]
- Mola, M.G.; Saracino, E.; Formaggio, F.; Amerotti, A.G.; Barile, B.; Posati, T.; Cibelli, A.; Frigeri, A.; Palazzo, C.; Zamboni, R.; et al. Cell Volume Regulation Mechanisms in Differentiated Astrocytes. Cell. Physiol. Biochem. 2021, 55, 196–212. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, E.; Kimelberg, H. Role of Calcium in Astrocyte Volume Regulation and in the Release of Ions and Amino Acids. J. Neurosci. 1993, 13, 2638–2650. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Boor, I.; Estévez, R. Megalencephalic Leukoencephalopathy with Subcortical Cysts: Chronic White Matter Oedema Due to a Defect in Brain Ion and Water Homoeostasis. Lancet Neurol. 2012, 11, 973–985. [Google Scholar] [CrossRef]
- Goswami, C.; Kuhn, J.; Heppenstall, P.A.; Hucho, T. Importance of Non-Selective Cation Channel TRPV4 Interaction with Cytoskeleton and Their Reciprocal Regulations in Cultured Cells. PLoS ONE 2010, 5, e11654. [Google Scholar] [CrossRef] [PubMed]
- Saberbaghi, T.; Wong, R.; Rutka, J.T.; Wang, G.-L.; Feng, Z.-P.; Sun, H.-S. Role of Cl- Channels in Primary Brain Tumour. Cell Calcium 2019, 81, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.; Chien, L.-T.; Hartzell, H.C. Drosophila Bestrophin-1 Currents Are Regulated by Phosphorylation via a CaMKII Dependent Mechanism. PLoS ONE 2013, 8, e58875. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Sontheimer, H. Molecular Interaction and Functional Regulation of ClC-3 by Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) in Human Malignant Glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Lutter, D.; Planells-Cases, R.; Ullrich, F.; Voss, F.K. VRAC: Molecular Identification as LRRC8 Heteromers with Differential Functions. Pflügers Arch. Eur. J. Physiol. 2016, 468, 385–393. [Google Scholar] [CrossRef]
- Bertelli, S.; Remigante, A.; Zuccolini, P.; Barbieri, R.; Ferrera, L.; Picco, C.; Gavazzo, P.; Pusch, M. Mechanisms of Activation of LRRC8 Volume Regulated Anion Channels. Cell. Physiol. Biochem. 2021, 55, 41–56. [Google Scholar] [CrossRef]
- Capdevila-Nortes, X.; López-Hernández, T.; Apaja, P.M.; López de Heredia, M.; Sirisi, S.; Callejo, G.; Arnedo, T.; Nunes, V.; Lukacs, G.L.; Gasull, X.; et al. Insights into MLC Pathogenesis: GlialCAM Is an MLC1 Chaperone Required for Proper Activation of Volume-Regulated Anion Currents. Hum. Mol. Genet. 2013, 22, 4405–4416. [Google Scholar] [CrossRef] [Green Version]
- Catacuzzeno, L.; Michelucci, A.; Sforna, L.; Aiello, F.; Sciaccaluga, M.; Fioretti, B.; Castigli, E.; Franciolini, F. Identification of Key Signaling Molecules Involved in the Activation of the Swelling-Activated Chloride Current in Human Glioblastoma Cells. J. Membr. Biol. 2014, 247, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Mongin, A.A.; Kimelberg, H.K. ATP Regulates Anion Channel-Mediated Organic Osmolyte Release from Cultured Rat Astrocytes via Multiple Ca 2+ -Sensitive Mechanisms. Am. J. Physiol.-Cell Physiol. 2005, 288, C204–C213. [Google Scholar] [CrossRef] [PubMed]
- Voss, F.K.; Ullrich, F.; Münch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Elorza-Vidal, X.; Sirisi, S.; Gaitán-Peñas, H.; Pérez-Rius, C.; Alonso-Gardón, M.; Armand-Ugón, M.; Lanciotti, A.; Brignone, M.S.; Prat, E.; Nunes, V.; et al. GlialCAM/MLC1 modulates LRRC8/VRAC currents in an indirect manner: Implications for megalencephalic leukoencephalopathy. Neurobiol. Dis. 2018, 119, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.E.; Kwon, J.; Won, J.; An, H.; Jang, M.W.; Woo, J.; Lee, J.S.; Park, M.G.; Yoon, B.E.; Lee, S.E.; et al. Tweety-homolog (Ttyh) Family Encodes the Pore-forming Subunits of the Swelling-dependent Volume-regulated Anion Channel (VRACswell) in the Brain. Exp. Neurobiol. 2019, 28, 183–215. [Google Scholar] [CrossRef]
- Schaeffer, S.; Iadecola, C. Revisiting the Neurovascular Unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Semyanov, A. Spatiotemporal Pattern of Calcium Activity in Astrocytic Network. Cell Calcium 2019, 78, 15–25. [Google Scholar] [CrossRef]
- Fujii, Y.; Maekawa, S.; Morita, M. Astrocyte Calcium Waves Propagate Proximally by Gap Junction and Distally by Extracellular Diffusion of ATP Released from Volume-Regulated Anion Channels. Sci. Rep. 2017, 7, 13115. [Google Scholar] [CrossRef]
- Lanciotti, A.; Brignone, M.S.; Belfiore, M.; Columba-Cabezas, S.; Mallozzi, C.; Vincentini, O.; Molinari, P.; Petrucci, T.C.; Visentin, S.; Ambrosini, E. Megalencephalic Leukoencephalopathy with Subcortical Cysts Disease-Linked MLC1 Protein Favors Gap-Junction Intercellular Communication by Regulating Connexin 43 Trafficking in Astrocytes. Cells 2020, 9, 1425. [Google Scholar] [CrossRef]
- Lind, B.L.; Jessen, S.B.; Lønstrup, M.; Joséphine, C.; Bonvento, G.; Lauritzen, M. Fast Ca 2+ Responses in Astrocyte End-Feet and Neurovascular Coupling in Mice. Glia 2018, 66, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Eilert-Olsen, M.; Hjukse, J.B.; Thoren, A.E.; Tang, W.; Enger, R.; Jensen, V.; Pettersen, K.H.; Nagelhus, E.A. Astroglial Endfeet Exhibit Distinct Ca 2+ Signals during Hypoosmotic Conditions. Glia 2019, 67, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.; Elorza-Vidal, X.; Rancillac, A.; Chagnot, A.; Yetim, M.; Hingot, V.; Deffieux, T.; Boulay, A.-C.; Alvear-Perez, R.; Cisternino, S.; et al. Megalencephalic Leukoencephalopathy with Subcortical Cysts Is a Developmental Disorder of the Gliovascular Unit. eLife 2021, 10, e71379. [Google Scholar] [CrossRef]
- Morales, J.E.; De, A.; Miller, A.A.; Chen, Z.; McCarty, J.H. Mlc1-Expressing Perivascular Astrocytes Promote Blood-Brain Barrier Integrity. J. Neurosci. 2022, 42, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.R. The Role of Brain Vasculature in Glioblastoma. Mol. Neurobiol. 2019, 56, 6645–6653. [Google Scholar] [CrossRef]
- Verkhratsky, A. Astroglial Calcium Signaling in Aging and Alzheimer’s Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a035188. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brignone, M.S.; Lanciotti, A.; Michelucci, A.; Mallozzi, C.; Camerini, S.; Catacuzzeno, L.; Sforna, L.; Caramia, M.; D’Adamo, M.C.; Ceccarini, M.; et al. The CaMKII/MLC1 Axis Confers Ca2+-Dependence to Volume-Regulated Anion Channels (VRAC) in Astrocytes. Cells 2022, 11, 2656. https://doi.org/10.3390/cells11172656
Brignone MS, Lanciotti A, Michelucci A, Mallozzi C, Camerini S, Catacuzzeno L, Sforna L, Caramia M, D’Adamo MC, Ceccarini M, et al. The CaMKII/MLC1 Axis Confers Ca2+-Dependence to Volume-Regulated Anion Channels (VRAC) in Astrocytes. Cells. 2022; 11(17):2656. https://doi.org/10.3390/cells11172656
Chicago/Turabian StyleBrignone, Maria Stefania, Angela Lanciotti, Antonio Michelucci, Cinzia Mallozzi, Serena Camerini, Luigi Catacuzzeno, Luigi Sforna, Martino Caramia, Maria Cristina D’Adamo, Marina Ceccarini, and et al. 2022. "The CaMKII/MLC1 Axis Confers Ca2+-Dependence to Volume-Regulated Anion Channels (VRAC) in Astrocytes" Cells 11, no. 17: 2656. https://doi.org/10.3390/cells11172656
APA StyleBrignone, M. S., Lanciotti, A., Michelucci, A., Mallozzi, C., Camerini, S., Catacuzzeno, L., Sforna, L., Caramia, M., D’Adamo, M. C., Ceccarini, M., Molinari, P., Macioce, P., Macchia, G., Petrucci, T. C., Pessia, M., Visentin, S., & Ambrosini, E. (2022). The CaMKII/MLC1 Axis Confers Ca2+-Dependence to Volume-Regulated Anion Channels (VRAC) in Astrocytes. Cells, 11(17), 2656. https://doi.org/10.3390/cells11172656