
Noradrenaline and Movement Initiation Disorders in Parkinson’s Disease: A Pharmacological Functional MRI Study with Clonidine

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Drug Design
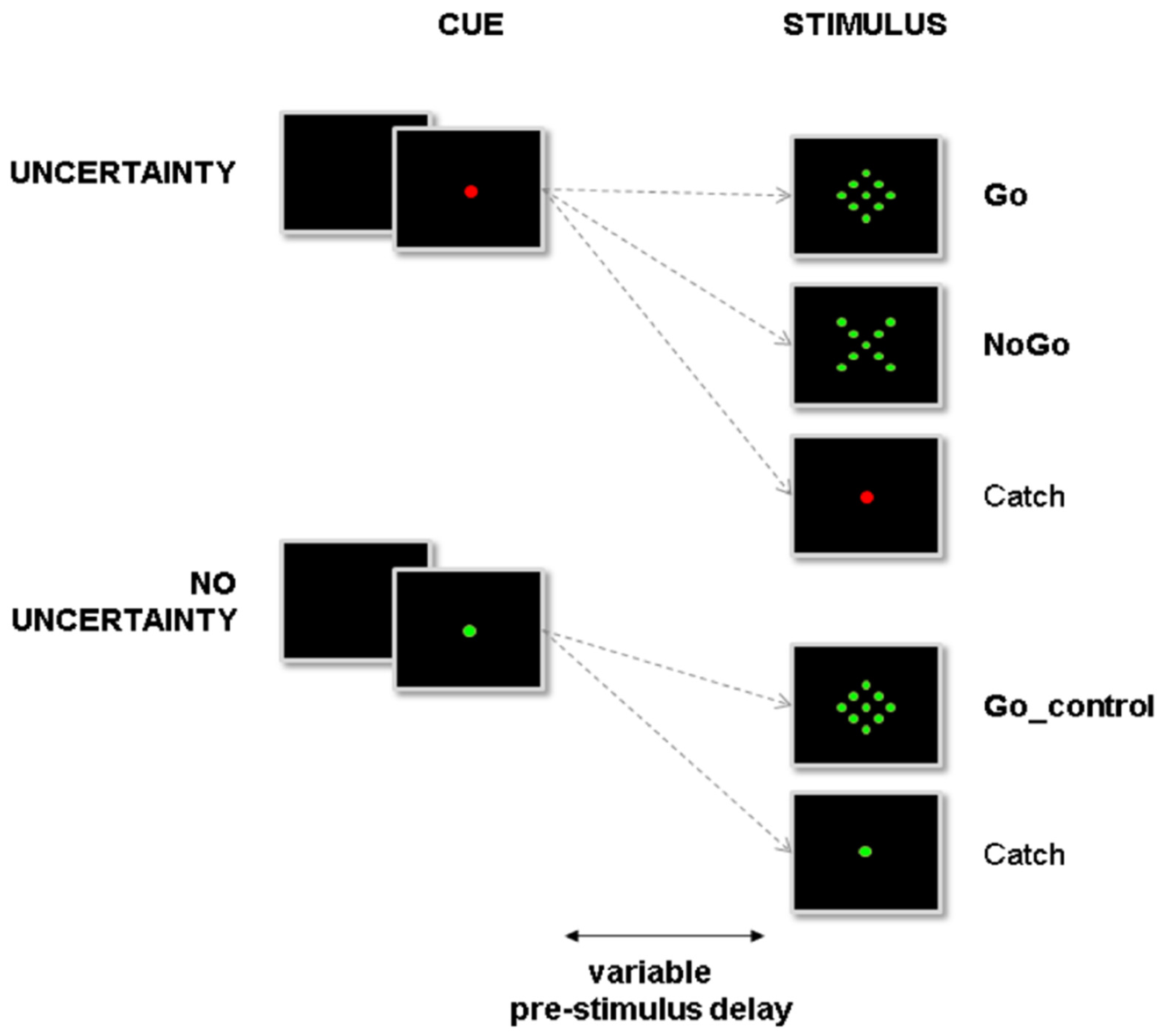
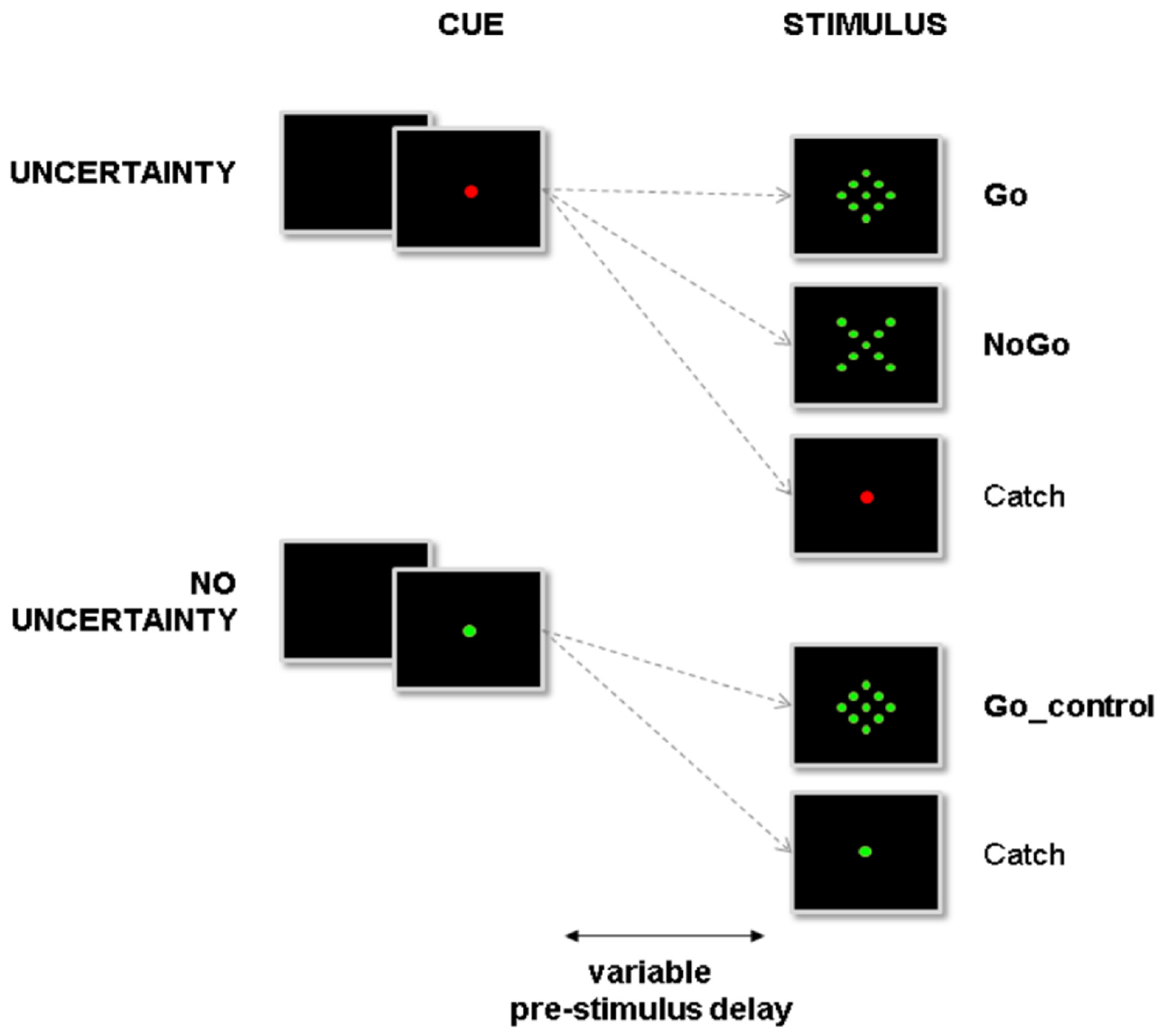
2.3. Experimental Design and Apparatus
2.4. Data Processing
2.5. Statistical Analysis
3. Results
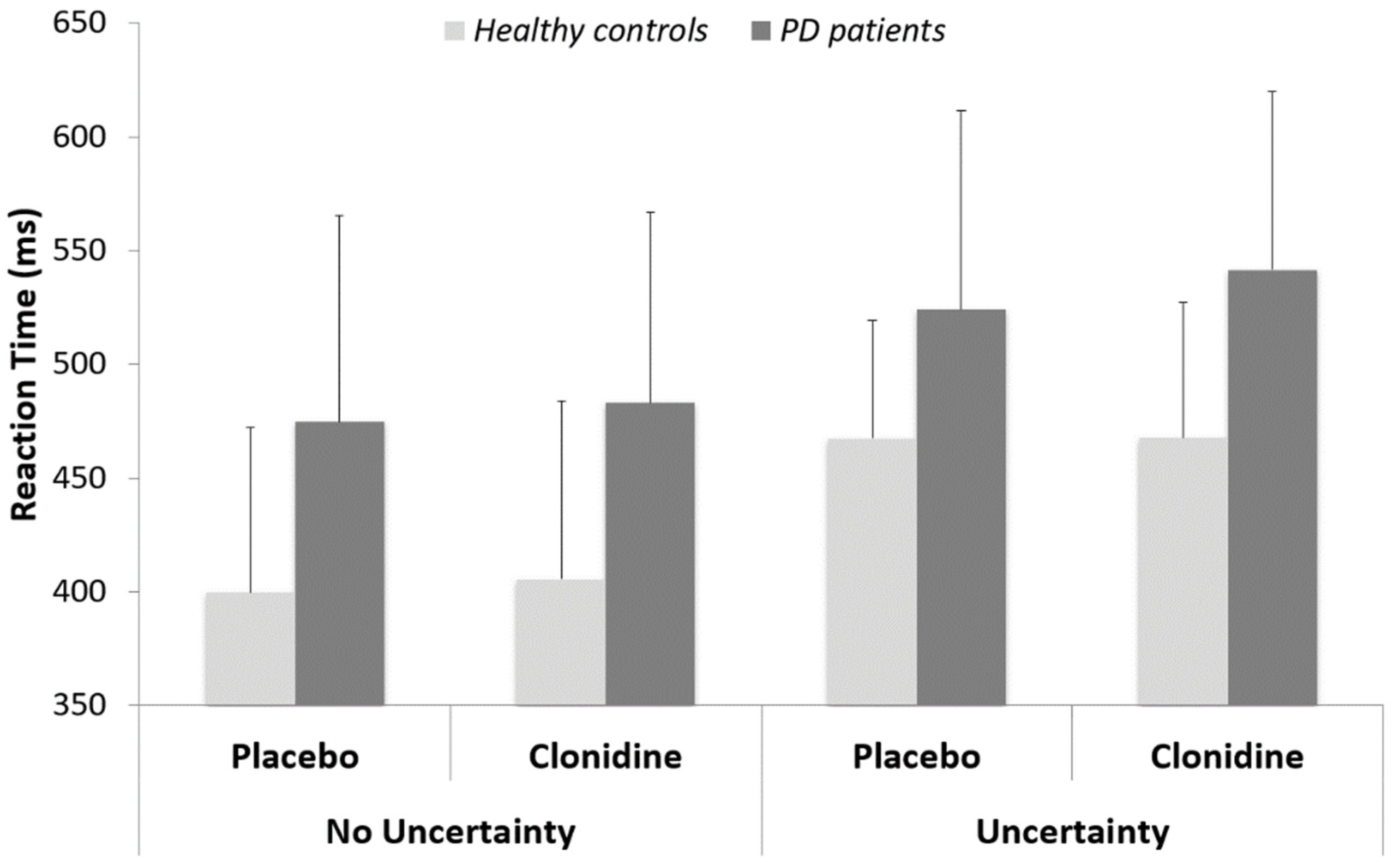
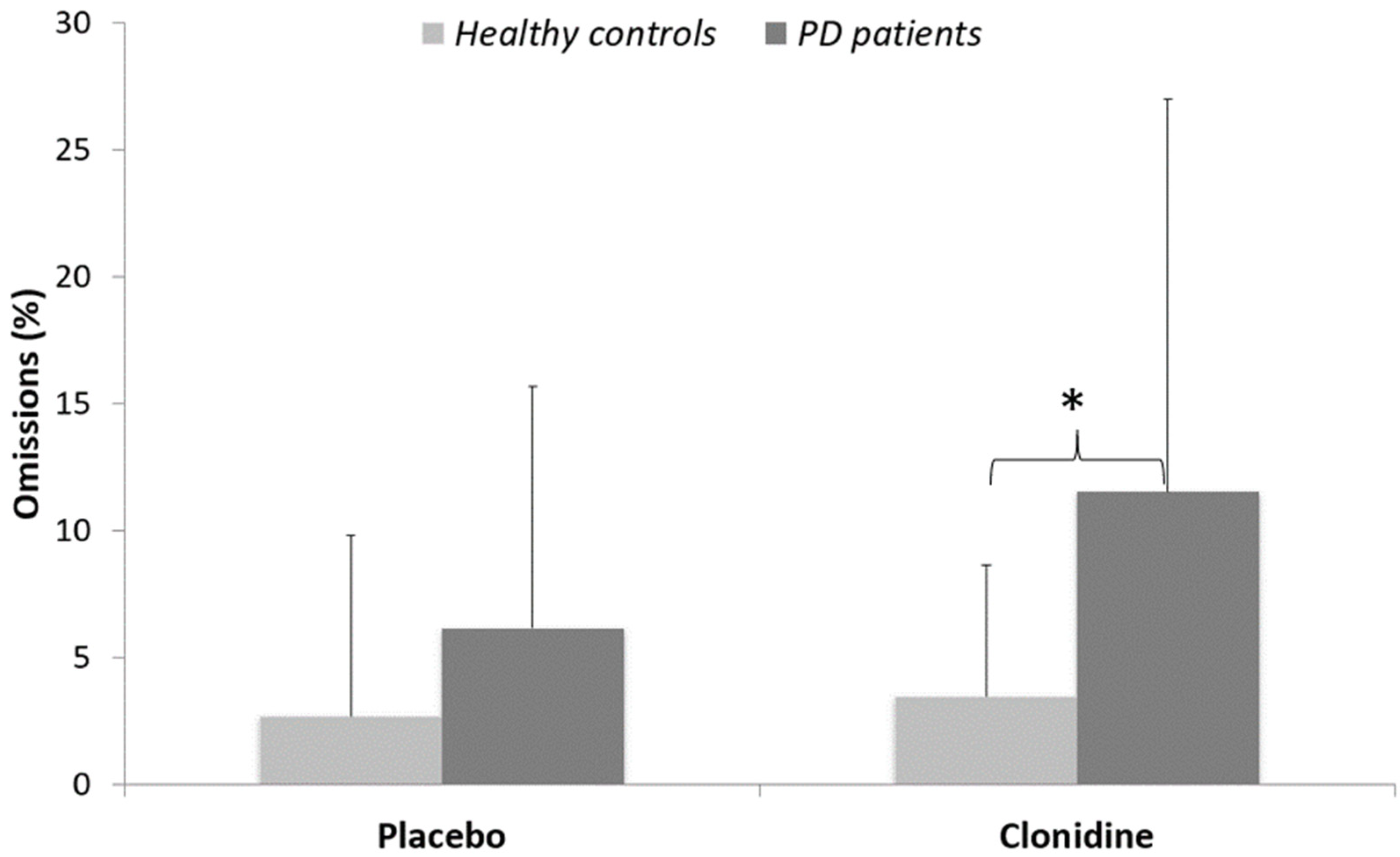
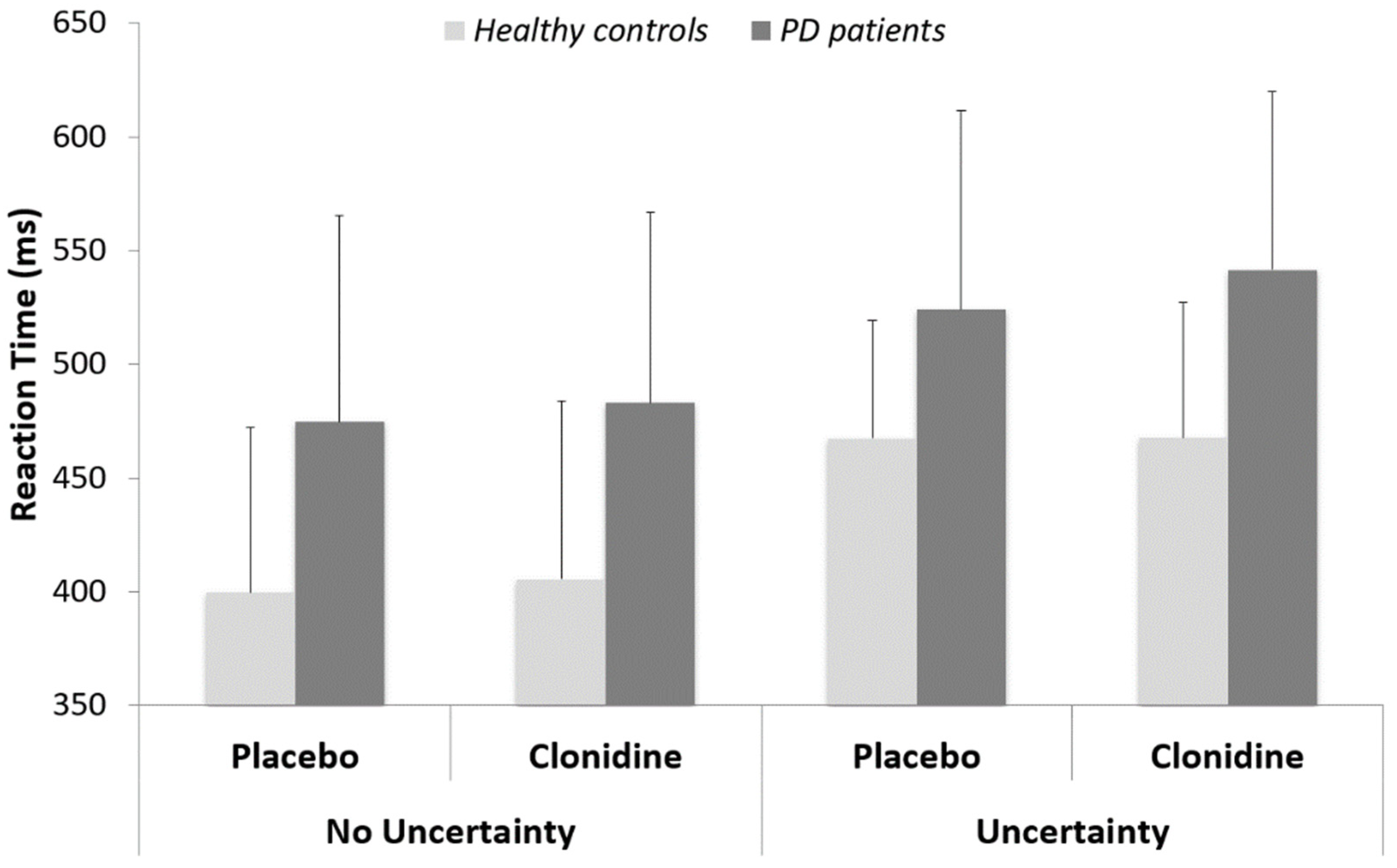
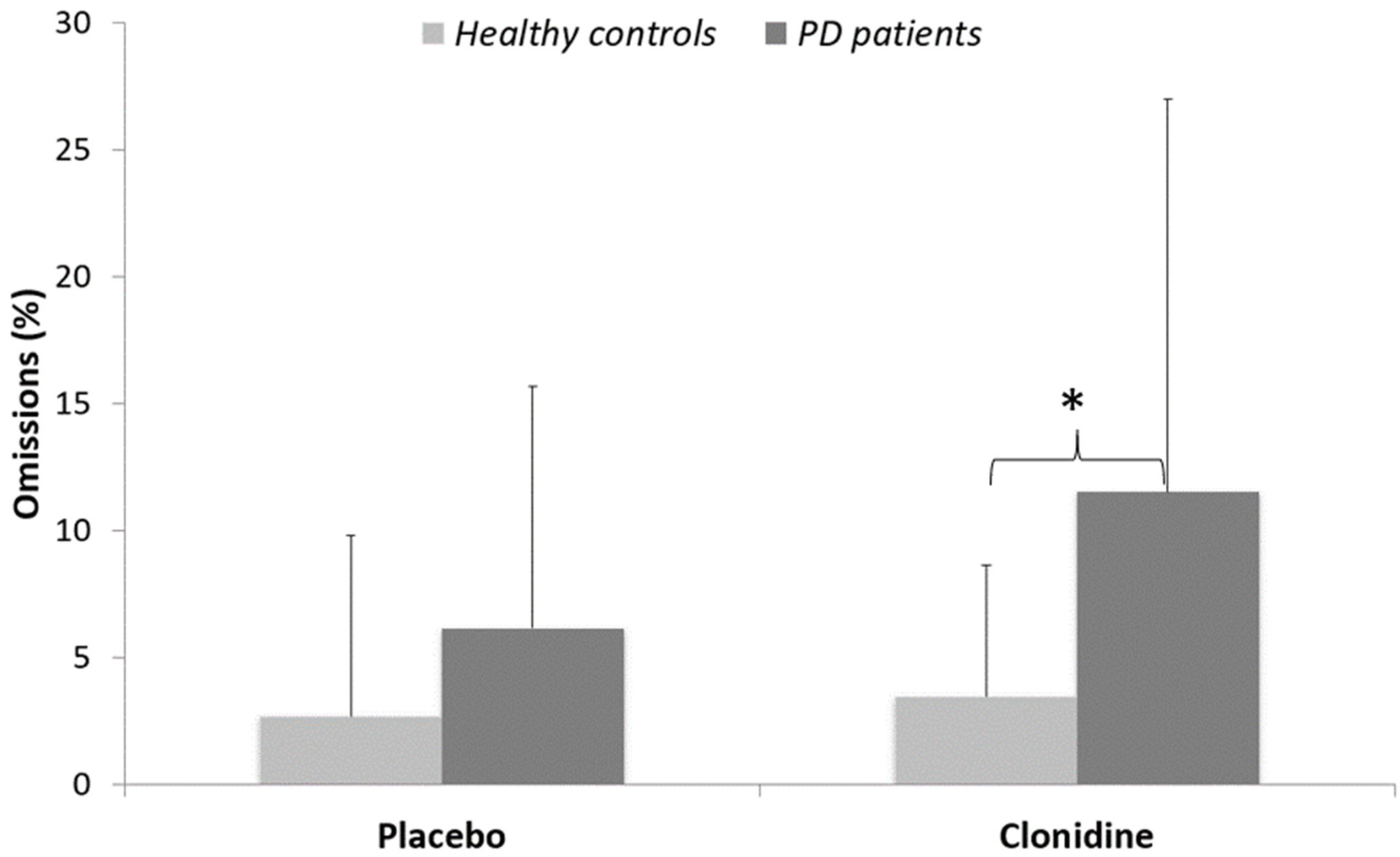
3.1. Behavioral Data
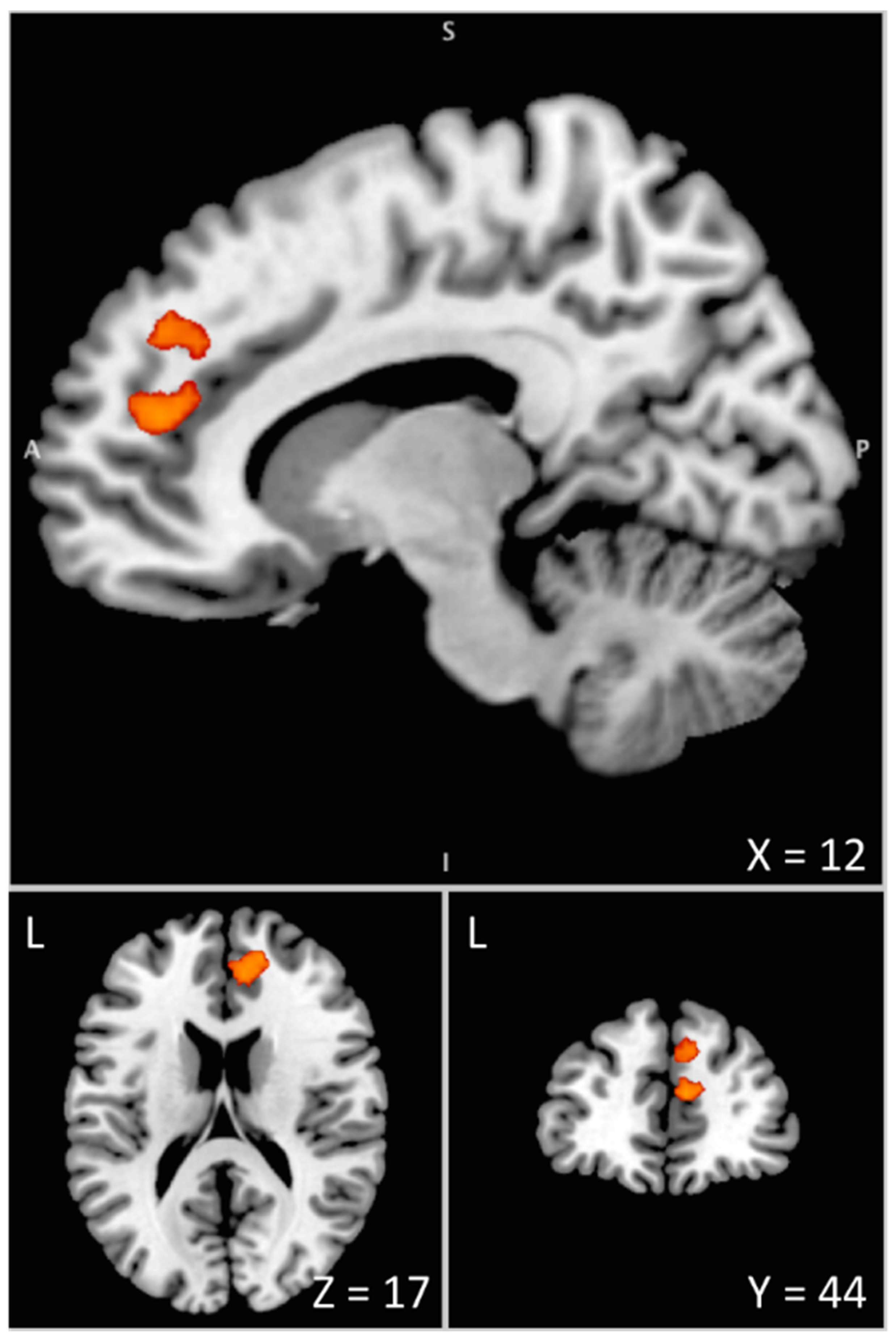
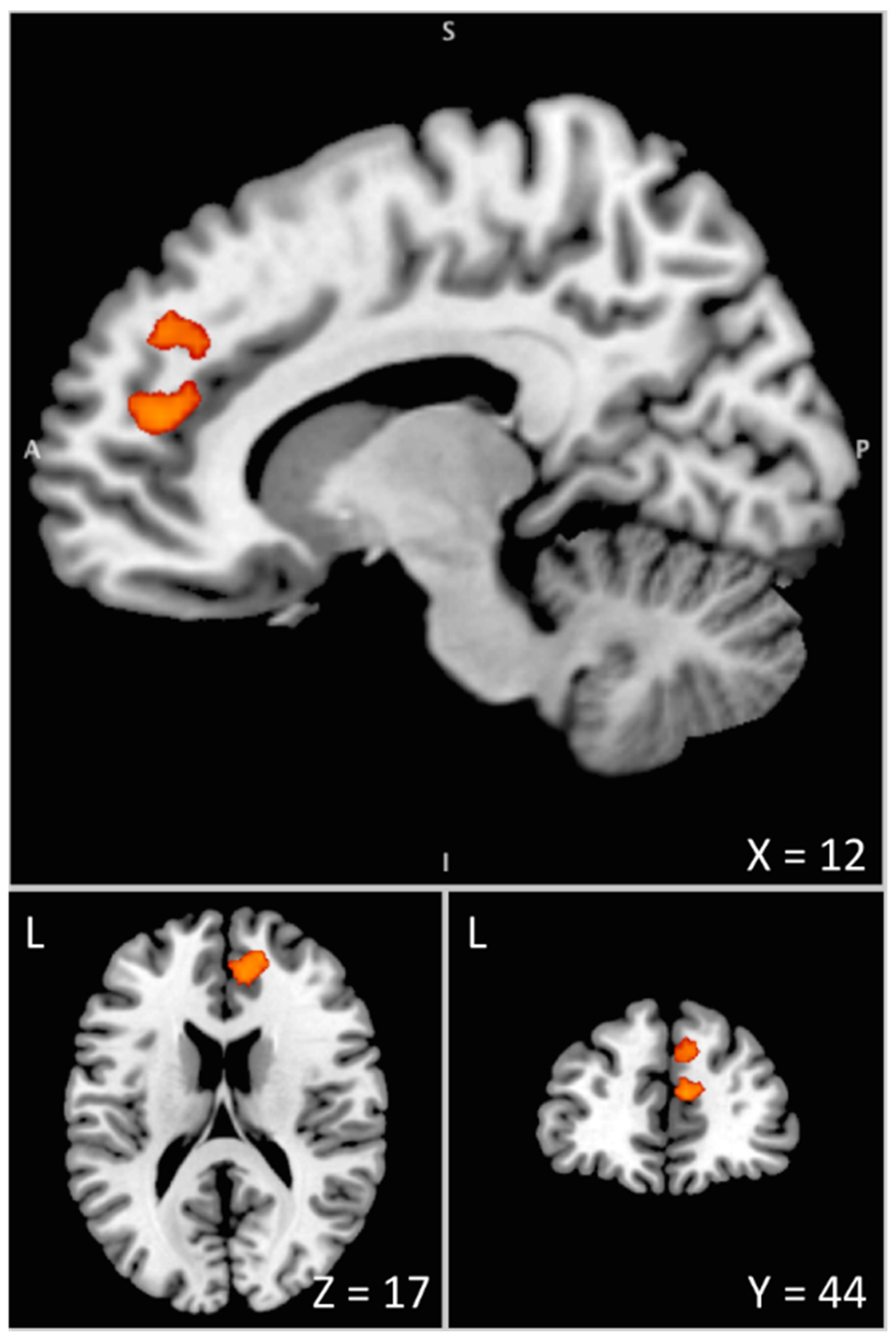
3.2. Imaging Data
4. Discussion
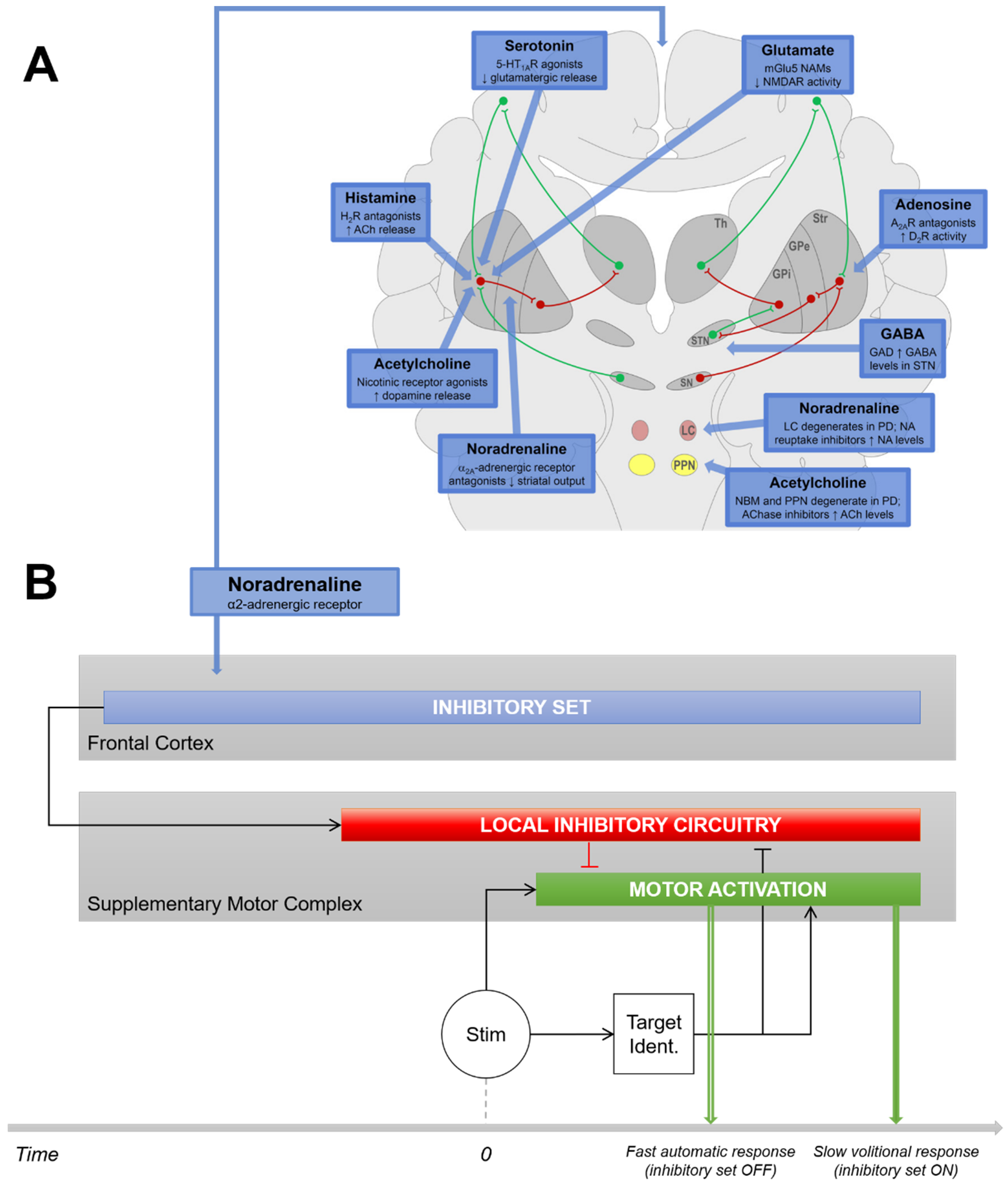
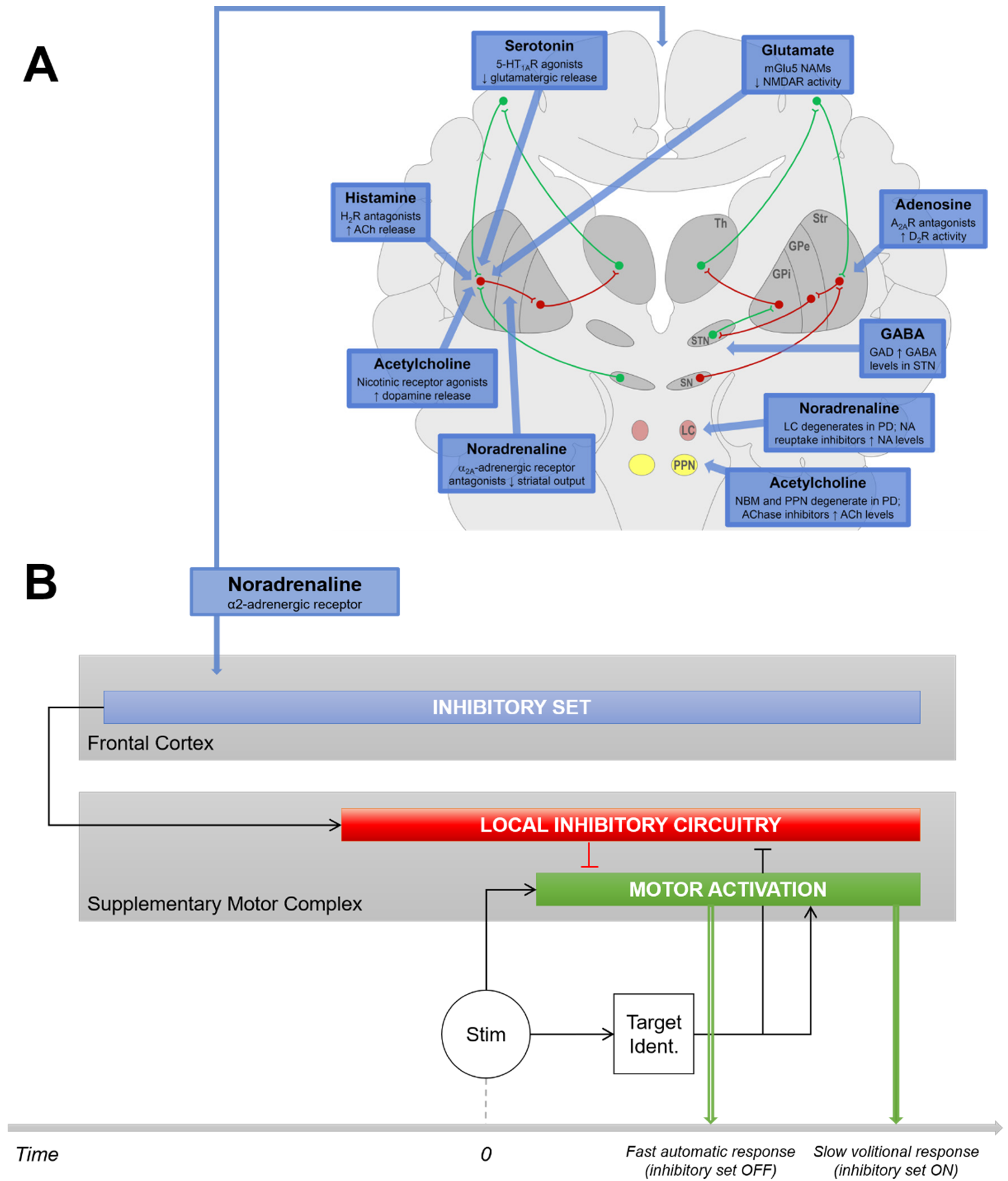
4.1. Noradrenergic Modulation of Movement Initiation Control
4.2. Noradrenergic Modulation of the Proactive Inhibitory Network
4.3. Relevance to a Noradrenergic Approach to Current Medical Therapies in PD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalia, L.V.; Brotchie, J.M.; Fox, S.H. Novel Nondopaminergic Targets for Motor Features of Parkinson’s Disease: Review of Recent Trials: Nondopaminergic Targets for Motor Features of PD. Mov. Disord. 2013, 28, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Latapi, P.; Bhowmick, S.S.; Saranza, G.; Fox, S.H. Non-Dopaminergic Treatments for Motor Control in Parkinson’s Disease: An Update. CNS Drugs 2020, 34, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H. Non-Dopaminergic Treatments for Motor Control in Parkinson’s Disease. Drugs 2013, 73, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, D.G.; Reyes, N.G.D.; Fox, S.H. Newly Approved and Investigational Drugs for Motor Symptom Control in Parkinson’s Disease. Drugs 2022, 82, 1027–1053. [Google Scholar] [CrossRef] [PubMed]
- Spay, C.; Meyer, G.; Welter, M.-L.; Lau, B.; Boulinguez, P.; Ballanger, B. Functional Imaging Correlates of Akinesia in Parkinson’s Disease: Still Open Issues. NeuroImage Clin. 2018, 21, 101644. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M. Clinical Neurophysiology of Akinesia. Rev. Neurol. 1990, 146, 585–590. [Google Scholar]
- Alexander, G.E.; Crutcher, M.D. Functional Architecture of Basal Ganglia Circuits: Neural Substrates of Parallel Processing. Trends Neurosci. 1990, 13, 266–271. [Google Scholar] [CrossRef]
- Alexander, G.E.; Crutcher, M.D.; DeLong, M.R. Basal Ganglia-Thalamocortical Circuits: Parallel Substrates for Motor, Oculomotor, “Prefrontal” and “Limbic” Functions. Prog. Brain Res. 1990, 85, 119–146. [Google Scholar]
- Canavan, A.G.M.; Nixon, P.D.; Passingham, R.E. Motor Learning in Monkeys (Macaca Fascicularis) with Lesions in Motor Thalamus. Exp. Brain Res. 1989, 77, 113–126. [Google Scholar] [CrossRef]
- Marsden, C.D.; Obeso, J.A. The Functions of the Basal Ganglia and the Paradox of Stereotaxic Surgery in Parkinson’s Disease. Brain J. Neurol. 1994, 117, 877–897. [Google Scholar] [CrossRef]
- Ballanger, B.; Gil, R.; Audiffren, M.; Desmurget, M. Perceptual Factors Contribute to Akinesia in Parkinson’s Disease. Exp. Brain Res. 2007, 179, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Favre, E.; Ballanger, B.; Thobois, S.; Broussolle, E.; Boulinguez, P. Deep Brain Stimulation of the Subthalamic Nucleus, but Not Dopaminergic Medication, Improves Proactive Inhibitory Control of Movement Initiation in Parkinson’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2013, 10, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkinson, N.; Nandi, D.; Oram, R.; Stein, J.F.; Aziz, T.Z. Pedunculopontine Nucleus Electric Stimulation Alleviates Akinesia Independently of Dopaminergic Mechanisms. NeuroReport 2006, 17, 639–641. [Google Scholar] [CrossRef]
- Jahanshahi, M.; Rothwell, J.C. Inhibitory Dysfunction Contributes to Some of the Motor and Non-Motor Symptoms of Movement Disorders and Psychiatric Disorders. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160198. [Google Scholar] [CrossRef] [PubMed]
- Criaud, M.; Poisson, A.; Thobois, S.; Metereau, E.; Redouté, J.; Ibarrola, D.; Baraduc, P.; Broussolle, E.; Strafella, A.P.; Ballanger, B.; et al. Slowness in Movement Initiation Is Associated with Proactive Inhibitory Network Dysfunction in Parkinson’s Disease. J. Park. Dis. 2016, 6, 433–440. [Google Scholar] [CrossRef]
- Albares, M.; Thobois, S.; Favre, E.; Broussolle, E.; Polo, G.; Domenech, P.; Boulinguez, P.; Ballanger, B. Interaction of Noradrenergic Pharmacological Manipulation and Subthalamic Stimulation on Movement Initiation Control in Parkinson’s Disease. Brain Stimulat. 2015, 8, 27–35. [Google Scholar] [CrossRef]
- Jaffard, M.; Benraiss, A.; Longcamp, M.; Velay, J.-L.; Boulinguez, P. Cueing Method Biases in Visual Detection Studies. Brain Res. 2007, 1179, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Jaffard, M.; Longcamp, M.; Velay, J.-L.; Anton, J.-L.; Roth, M.; Nazarian, B.; Boulinguez, P. Proactive Inhibitory Control of Movement Assessed by Event-Related FMRI. NeuroImage 2008, 42, 1196–1206. [Google Scholar] [CrossRef]
- Criaud, M.; Longcamp, M.; Anton, J.-L.; Nazarian, B.; Roth, M.; Sescousse, G.; Strafella, A.P.; Ballanger, B.; Boulinguez, P. Testing the Physiological Plausibility of Conflicting Psychological Models of Response Inhibition: A Forward Inference FMRI Study. Behav. Brain Res. 2017, 333, 192–202. [Google Scholar] [CrossRef]
- Criaud, M.; Anton, J.-L.; Nazarian, B.; Longcamp, M.; Metereau, E.; Boulinguez, P.; Ballanger, B. The Human Basal Ganglia Mediate the Interplay between Reactive and Proactive Control of Response through Both Motor Inhibition and Sensory Modulation. Brain Sci. 2021, 11, 560. [Google Scholar] [CrossRef]
- Criaud, M.; Wardak, C.; Ben Hamed, S.; Ballanger, B.; Boulinguez, P. Proactive Inhibitory Control of Response as the Default State of Executive Control. Front. Psychol. 2012, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Isoda, M.; Hikosaka, O. Switching from Automatic to Controlled Action by Monkey Medial Frontal Cortex. Nat. Neurosci. 2007, 10, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Hikosaka, O.; Isoda, M. Switching from Automatic to Controlled Behavior: Cortico-Basal Ganglia Mechanisms. Trends Cogn. Sci. 2010, 14, 154–161. [Google Scholar] [CrossRef]
- Chamberlain, S.R.; Robbins, T.W. Noradrenergic Modulation of Cognition: Therapeutic Implications. J. Psychopharmacol. 2013, 27, 694–718. [Google Scholar] [CrossRef]
- Bari, A.; Robbins, T.W. Noradrenergic versus Dopaminergic Modulation of Impulsivity, Attention and Monitoring Behaviour in Rats Performing the Stop-Signal Task: Possible Relevance to ADHD. Psychopharmacology 2013, 230, 89–111. [Google Scholar] [CrossRef]
- Borodovitsyna, O.; Flamini, M.; Chandler, D. Noradrenergic Modulation of Cognition in Health and Disease. Neural Plast. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Berridge, C.W.; Waterhouse, B.D. The Locus Coeruleus–Noradrenergic System: Modulation of Behavioral State and State-Dependent Cognitive Processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef]
- Bouret, S.; Sara, S.J. Network Reset: A Simplified Overarching Theory of Locus Coeruleus Noradrenaline Function. Trends Neurosci. 2005, 28, 574–582. [Google Scholar] [CrossRef]
- Ramos, B.P.; Arnsten, A.F.T. Adrenergic Pharmacology and Cognition: Focus on the Prefrontal Cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef]
- Tait, D.S.; Brown, V.J.; Farovik, A.; Theobald, D.E.; Dalley, J.W.; Robbins, T.W. Lesions of the Dorsal Noradrenergic Bundle Impair Attentional Set-Shifting in the Rat. Eur. J. Neurosci. 2007, 25, 3719–3724. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Cohen, J.D. Adaptive Gain and the Role of the Locus Coeruleus-Norepinephrine System in Optimal Performance. J. Comp. Neurol. 2005, 493, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Spay, C.; Albares, M.; Lio, G.; Thobois, S.; Broussolle, E.; Lau, B.; Ballanger, B.; Boulinguez, P. Clonidine Modulates the Activity of the Subthalamic-Supplementary Motor Loop: Evidence from a Pharmacological Study Combining Deep Brain Stimulation and Electroencephalography Recordings in Parkinsonian Patients. J. Neurochem. 2018, 146, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Ballanger, B.; van Eimeren, T.; Moro, E.; Lozano, A.M.; Hamani, C.; Boulinguez, P.; Pellecchia, G.; Houle, S.; Poon, Y.Y.; Lang, A.E.; et al. Stimulation of the Subthalamic Nucleus and Impulsivity: Release Your Horses. Ann. Neurol. 2009, 66, 817–824. [Google Scholar] [CrossRef]
- Meyer, G.M.; Spay, C.; Beliakova, A.; Gaugain, G.; Pezzoli, G.; Cilia, R. Inhibitory Control Dysfunction in Parkinsonian Impulse Control Disorders. Brain 2020, 143, 3734–3747. [Google Scholar] [CrossRef] [PubMed]
- Anavekar, S.N.; Jarrott, B.; Toscano, M.; Louis, W.J. Pharmacokinetic and Pharmacodynamic Studies of Oral Clonidine in Normotensive Subjects. Eur. J. Clin. Pharmacol. 1982, 23, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.L.; Stowe, R.; Patel, S.; Rick, C.; Gray, R.; Clarke, C.E. Systematic Review of Levodopa Dose Equivalency Reporting in Parkinson’s Disease: Systematic Review of LED Reporting in PD. Mov. Disord. 2010, 25, 2649–2653. [Google Scholar] [CrossRef] [PubMed]
- Boulinguez, P.; Blouin, J.; Nougier, V. The Gap Effect for Eye and Hand Movements in Double-Step Pointing. Exp. Brain Res. 2001, 138, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Criaud, M.; Boulinguez, P. Have We Been Asking the Right Questions When Assessing Response Inhibition in Go/No-Go Tasks with FMRI? A Meta-Analysis and Critical Review. Neurosci. Biobehav. Rev. 2013, 37, 11–23. [Google Scholar] [CrossRef]
- Chiu, Y.-C.; Aron, A.R. Unconsciously Triggered Response Inhibition Requires an Executive Setting. J. Exp. Psychol. Gen. 2014, 143, 56–61. [Google Scholar] [CrossRef]
- Tzourio-Mazoyer, N.; Landeau, B.; Papathanassiou, D.; Crivello, F.; Etard, O.; Delcroix, N.; Mazoyer, B.; Joliot, M. Automated Anatomical Labeling of Activations in SPM Using a Macroscopic Anatomical Parcellation of the MNI MRI Single-Subject Brain. NeuroImage 2002, 15, 273–289. [Google Scholar] [CrossRef]
- Keren, N.I.; Lozar, C.T.; Harris, K.C.; Morgan, P.S.; Eckert, M.A. In Vivo Mapping of the Human Locus Coeruleus. NeuroImage 2009, 47, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Kirilina, E.; Otaduy, M.C.G.; Ivanov, D.; Acosta-Cabronero, J.; Callaghan, M.F.; Lambert, C.; Cardenas-Blanco, A.; Pine, K.; Passamonti, L.; et al. Locus Coeruleus Imaging as a Biomarker for Noradrenergic Dysfunction in Neurodegenerative Diseases. Brain 2019, 142, 2558–2571. [Google Scholar] [CrossRef] [PubMed]
- Kochunov, P.; Lancaster, J.; Thompson, P.; Toga, A.W.; Brewer, P.; Hardies, J.; Fox, P. An Optimized Individual Target Brain in the Talairach Coordinate System. NeuroImage 2002, 17, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Riekkinen, M.; Kejonen, K.; Jäkälä, P.; Soininen, H.; Riekkinen, P. Reduction of Noradrenaline Impairs Attention and Dopamine Depletion Slows Responses in Parkinson’s Disease: Noradrenaline, Dopamine and Attention in Parkinson’s Disease. Eur. J. Neurosci. 1998, 10, 1429–1435. [Google Scholar] [CrossRef]
- Nieuwenhuis, S.; Aston-Jones, G.; Cohen, J.D. Decision Making, the P3, and the Locus Coeruleus--Norepinephrine System. Psychol. Bull. 2005, 131, 510–532. [Google Scholar] [CrossRef]
- Bouret, S. Locus Coeruleus, Noradrenaline, and Behavior: Network Effect, Network Effects? Neuron 2019, 103, 554–556. [Google Scholar] [CrossRef]
- Chandler, D.J.; Jensen, P.; McCall, J.G.; Pickering, A.E.; Schwarz, L.A.; Totah, N.K. Redefining Noradrenergic Neuromodulation of Behavior: Impacts of a Modular Locus Coeruleus Architecture. J. Neurosci. 2019, 39, 8239–8249. [Google Scholar] [CrossRef]
- Bari, A.; Eagle, D.M.; Mar, A.C.; Robinson, E.S.J.; Robbins, T.W. Dissociable Effects of Noradrenaline, Dopamine, and Serotonin Uptake Blockade on Stop Task Performance in Rats. Psychopharmacology 2009, 205, 273–283. [Google Scholar] [CrossRef]
- Eagle, D.M.; Bari, A.; Robbins, T.W. The Neuropsychopharmacology of Action Inhibition: Cross-Species Translation of the Stop-Signal and Go/No-Go Tasks. Psychopharmacology 2008, 199, 439–456. [Google Scholar] [CrossRef]
- Chamberlain, S.R.; Hampshire, A.; Müller, U.; Rubia, K.; del Campo, N.; Craig, K.; Regenthal, R.; Suckling, J.; Roiser, J.P.; Grant, J.E.; et al. Atomoxetine Modulates Right Inferior Frontal Activation During Inhibitory Control: A Pharmacological Functional Magnetic Resonance Imaging Study. Biol. Psychiatry 2009, 65, 550–555. [Google Scholar] [CrossRef]
- Kehagia, A.A.; Housden, C.R.; Regenthal, R.; Barker, R.A.; Müller, U.; Rowe, J.; Sahakian, B.J.; Robbins, T.W. Targeting Impulsivity in Parkinson’s Disease Using Atomoxetine. Brain 2014, 137, 1986–1997. [Google Scholar] [CrossRef] [PubMed]
- Aron, A.R. From Reactive to Proactive and Selective Control: Developing a Richer Model for Stopping Inappropriate Responses. Biol. Psychiatry 2011, 69, e55–e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poe, G.R.; Foote, S.; Eschenko, O.; Johansen, J.P.; Bouret, S.; Aston-Jone, G.; Harley, C.W.; Manahan-Vaughan, D.; Weinshenker, D.; Valentino, R.; et al. Locus Coeruleus: A New Look at the Blue Spot. Nat. Rev. Neurosci. 2020, 21, 644–659. [Google Scholar] [CrossRef] [PubMed]
- Vazey, E.M.; Aston-Jones, G. The Emerging Role of Norepinephrine in Cognitive Dysfunctions of Parkinson’s Disease. Front. Behav. Neurosci. 2012, 6, 48. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Basile, M.J.; Mash, D.C. Catechols in Post-Mortem Brain of Patients with Parkinson Disease: Brain Catechols in Parkinson Disease. Eur. J. Neurol. 2011, 18, 703–710. [Google Scholar] [CrossRef]
- Nayyar, T.; Bubser, M.; Ferguson, M.C.; Neely, M.D.; Goodwin, J.S.; Montine, T.J.; Deutch, A.Y.; Ansah, T.A. Cortical Serotonin and Norepinephrine Denervation in Parkinsonism: Preferential Loss of the Beaded Serotonin Innervation. Eur. J. Neurosci. 2009, 30, 207–216. [Google Scholar] [CrossRef]
- Filevich, E.; Kühn, S.; Haggard, P. Intentional Inhibition in Human Action: The Power of ‘No’. Neurosci. Biobehav. Rev. 2012, 36, 1107–1118. [Google Scholar] [CrossRef]
- Brass, M.; Haggard, P. To Do or Not to Do: The Neural Signature of Self-Control. J. Neurosci. 2007, 27, 9141–9145. [Google Scholar] [CrossRef]
- Brass, M.; Haggard, P. The What, When, Whether Model of Intentional Action. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2008, 14, 319–325. [Google Scholar] [CrossRef]
- Cho, S.S.; Pellecchia, G.; Ko, J.H.; Ray, N.; Obeso, I.; Houle, S.; Strafella, A.P. Effect of Continuous Theta Burst Stimulation of the Right Dorsolateral Prefrontal Cortex on Cerebral Blood Flow Changes during Decision Making. Brain Stimulat. 2012, 5, 116–123. [Google Scholar] [CrossRef]
- Narayanan, N.S.; Horst, N.K.; Laubach, M. Reversible Inactivations of Rat Medial Prefrontal Cortex Impair the Ability to Wait for a Stimulus. Neuroscience 2006, 139, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, N.K. What We Can Do and What We Cannot Do with FMRI. Nature 2008, 453, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Herman, Z.S.; Brus, R.; Drybański, A.; Szkilnik, R.; Slomińska-Zurek, J. Influence of 6-Hydroxydopamine on the Behavioral Effects Induced by Apomorphine or Clonidine in Rats. Psychopharmacology 1976, 50, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Nassif, S.; Kempf, E.; Cardo, B.; Velley, L. Neurochemical Lesion of the Locus Coeruleus of the Rat Does Not Suppress the Sedative Effect of Clonidine. Eur. J. Pharmacol. 1983, 91, 69–76. [Google Scholar] [CrossRef]
- Spyraki, C.; Arbuthnott, G.W.; Fibiger, H.C. The Effect of DSP-4 on Some Positively Reinforced Operant Behaviors in the Rat. Pharmacol. Biochem. Behav. 1982, 16, 197–202. [Google Scholar] [CrossRef]
- Ma, C.-L.; Arnsten, A.F.T.; Li, B.-M. Locomotor Hyperactivity Induced by Blockade of Prefrontal Cortical A2-Adrenoceptors in Monkeys. Biol. Psychiatry 2005, 57, 192–195. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.J.; Liu, J.; Ali, U.; Gui, Z.H.; Hui, Y.P.; Wang, T.; Chen, L.; Li, Q. Noradrenergic Lesion of the Locus Coeruleus Increases the Firing Activity of the Medial Prefrontal Cortex Pyramidal Neurons and the Role of A2-Adrenoceptors in Normal and Medial Forebrain Bundle Lesioned Rats. Brain Res. 2010, 1324, 64–74. [Google Scholar] [CrossRef]
- Thierry, A.M.; Godbout, R.; Mantz, J.; Pirot, S.; Glowinski, J. Differential Influence of Dopaminergic and Noradrenergic Afferents on Their Target Cells in the Rat Prefrontal Cortex. Clin. Neuropharmacol. 1992, 15, 139A–140A. [Google Scholar] [CrossRef]
- Wilkinson, D.; Halligan, P. The Relevance of Behavioural Measures for Functional-Imaging Studies of Cognition. Nat. Rev. Neurosci. 2004, 5, 67–73. [Google Scholar] [CrossRef]
- Nair, V.; Mahadevan, S. Randomised Controlled Study-Efficacy of Clonidine versus Carbamazepine in Children with ADHD. J. Trop. Pediatr. 2009, 55, 116–121. [Google Scholar] [CrossRef]
- Narabayashi, H. The Neural Mechanisms and Progressive Nature of Symptoms of Parkinson’s Disease—Based on Clinical, Neurophysiological and Morphological Studies. J. Neural Transm. Park. Dis. Dement. Sect. 1995, 10, 63–75. [Google Scholar] [CrossRef]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal Loss Is Greater in the Locus Coeruleus Than Nucleus Basalis and Substantia Nigra in Alzheimer and Parkinson Diseases. Arch. Neurol. 2003, 60, 337. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Moreau, C.; Delval, A.; Defebvre, L.; Dujardin, K.; Duhamel, A.; Petyt, G.; Vuillaume, I.; Corvol, J.-C.; Brefel-Courbon, C.; Ory-Magne, F.; et al. Methylphenidate for Gait Hypokinesia and Freezing in Patients with Parkinson’s Disease Undergoing Subthalamic Stimulation: A Multicentre, Parallel, Randomised, Placebo-Controlled Trial. Lancet Neurol. 2012, 11, 589–596. [Google Scholar] [CrossRef]
- Taylor, N.L.; Wainstein, G.; Quek, D.; Lewis, S.J.G.; Shine, J.M.; Ehgoetz Martens, K.A. The Contribution of Noradrenergic Activity to Anxiety-Induced Freezing of Gait. Mov. Disord. 2022, 37, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.A.; Sato, T.; Muramatsu, S. Freezing of Gait in Parkinson’s Disease Is Associated with Reduced 6-[18F]Fluoro-l-m-Tyrosine Uptake in the Locus Coeruleus. Park. Dis. 2016, 2016, 5430920. [Google Scholar] [CrossRef] [PubMed]
- Marsh, L.; Biglan, K.; Gerstenhaber, M.; Williams, J.R. Atomoxetine for the Treatment of Executive Dysfunction in Parkinson’s Disease: A Pilot Open-Label Study. Mov. Disord. 2009, 24, 277–282. [Google Scholar] [CrossRef]
- Ye, Z.; Altena, E.; Nombela, C.; Housden, C.R.; Maxwell, H.; Rittman, T.; Huddleston, C.; Rae, C.L.; Regenthal, R.; Sahakian, B.J.; et al. Improving Response Inhibition in Parkinson’s Disease with Atomoxetine. Biol. Psychiatry 2015, 77, 740–748. [Google Scholar] [CrossRef]
- Yssel, J.D.; O’Neill, E.; Nolan, Y.M.; Connor, T.J.; Harkin, A. Treatment with the Noradrenaline Re-Uptake Inhibitor Atomoxetine Alone and in Combination with the A2-Adrenoceptor Antagonist Idazoxan Attenuates Loss of Dopamine and Associated Motor Deficits in the LPS Inflammatory Rat Model of Parkinson’s Disease. Brain. Behav. Immun. 2018, 69, 456–469. [Google Scholar] [CrossRef]
- Ye, Z.; Rae, C.L.; Nombela, C.; Ham, T.; Rittman, T.; Jones, P.S.; Rodríguez, P.V.; Coyle-Gilchrist, I.; Regenthal, R.; Altena, E.; et al. Predicting Beneficial Effects of Atomoxetine and Citalopram on Response Inhibition in Parkinson’s Disease with Clinical and Neuroimaging Measures: Predicting Treatment Response in PD. Hum. Brain Mapp. 2016, 37, 1026–1037. [Google Scholar] [CrossRef]
- Delaville, C.; Deurwaerdère, P.D.; Benazzouz, A. Noradrenaline and Parkinson’s Disease. Front. Syst. Neurosci. 2011, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; LeWitt, P.A.; Kaufmann, H. Norepinephrine Deficiency in Parkinson’s Disease: The Case for Noradrenergic Enhancement: Norepinephrine Deficiency in PD. Mov. Disord. 2014, 29, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Colpaert, F.C. Pharmacological Characteristics of Tremor, Rigidity and Hypokinesia Induced by Reserpine in Rat. Neuropharmacology 1987, 26, 1431–1440. [Google Scholar] [CrossRef]
- Bezard, E.; Brefel, C.; Tison, F.; Peyro-SaintPaul, H.; Ladure, P.; Rascol, O.; Gross, C.E. Effect of the A2 Adrenoreceptor Antagonist, Idazoxan, on Motor Disabilities in MPTP-Treated Monkey. Prog. Neuropsychopharmacol. Biol. Psychiatry 1999, 23, 1237–1246. [Google Scholar] [CrossRef]
- Savola, J.-M.; Hill, M.; Engstrom, M.; Merivuori, H.; Wurster, S.; McGuire, S.G.; Fox, S.H.; Crossman, A.R.; Brotchie, J.M. Fipamezole (JP-1730) Is a Potent A2 Adrenergic Receptor Antagonist That Reduces Levodopa-Induced Dyskinesia in the MPTP-Lesioned Primate Model of Parkinson’s Disease. Mov. Disord. 2003, 18, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Grondin, R.; Hadj Tahar, A.; Doan, V.D.; Ladure, P.; Bédard, P.J. Noradrenoceptor Antagonism with Idazoxan Improves l-Dopa-Induced Dyskinesias in MPTP Monkeys. Naunyn. Schmiedebergs Arch. Pharmacol. 2000, 361, 181–186. [Google Scholar] [CrossRef]
- Domino, E.F.; Ni, L.; Colpaert, F.; Marien, M. Effects of (+/−)-Idazoxan Alone and in Combination with L-DOPA Methyl Ester in MPTP-Induced Hemiparkinsonian Monkeys. Receptors Channels 2003, 9, 335–338. [Google Scholar] [CrossRef]
- Henry, B.; Fox, S.H.; Peggs, D.; Crossman, A.R.; Brotchie, J.M. The A2-Adrenergic Receptor Antagonist Idazoxan Reduces Dyskinesia and Enhances Anti-Parkinsonian Actions of L-Dopa in the MPTP-Lesioned Primate Model of Parkinson’s Disease. Mov. Disord. 1999, 14, 744–753. [Google Scholar] [CrossRef]
- Rascol, O.; Arnulf, I.; Peyro-Saint Paul, H.; Brefel-Courbon, C.; Vidailhet, M.; Thalamas, C.; Bonnet, A.m.; Descombes, S.; Bejjani, B.; Fabre, N.; et al. Idazoxan, an Alpha-2 Antagonist, and L-DOPA-Induced Dyskinesias in Patients with Parkinson’s Disease. Mov. Disord. 2001, 16, 708–713. [Google Scholar] [CrossRef]
- Colosimo, C.; Craus, A. Noradrenergic Drugs for Levodopa-Induced Dyskinesia. Clin. Neuropharmacol. 2003, 26, 299–305. [Google Scholar] [CrossRef]
- Bylund, D.B. Heterogeneity of Alpha-2 Adrenergic Receptors. Pharmacol. Biochem. Behav. 1985, 22, 835–843. [Google Scholar] [CrossRef]
- Bylund, D.B. Pharmacological Characteristics of Alpha-2 Adrenergic Receptor Subtypes. Ann. N. Y. Acad. Sci. 1995, 763, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scheinin, M.; Lomasney, J.W.; Hayden-Hixson, D.M.; Schambra, U.B.; Caron, M.G.; Lefkowitz, R.J.; Fremeau, R.T. Distribution of Alpha 2-Adrenergic Receptor Subtype Gene Expression in Rat Brain. Brain Res. Mol. Brain Res. 1994, 21, 133–149. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Hauser, R.A.; Lu, M.; Nicholas, A.P.; Weiner, W.; Coppard, N.; Leinonen, M.; Savola, J.-M. Randomized Clinical Trial of Fipamezole for Dyskinesia in Parkinson Disease (FJORD Study). Neurology 2012, 79, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.G.; Podell, D.M.; Arnsten, A.F. Noradrenergic Alpha-2 Receptor Agonists Reverse Working Memory Deficits Induced by the Anxiogenic Drug, FG7142, in Rats. Pharmacol. Biochem. Behav. 2000, 67, 397–403. [Google Scholar] [CrossRef]
- Li, B.M.; Mao, Z.M.; Wang, M.; Mei, Z.T. Alpha-2 Adrenergic Modulation of Prefrontal Cortical Neuronal Activity Related to Spatial Working Memory in Monkeys. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 21, 601–610. [Google Scholar] [CrossRef]
- Jäkälä, P.; Riekkinen, M.; Sirviö, J.; Koivisto, E.; Kejonen, K.; Vanhanen, M.; Riekkinen, P. Guanfacine, but Not Clonidine, Improves Planning and Working Memory Performance in Humans. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 20, 460–470. [Google Scholar] [CrossRef]
- Björklund, M.; Sirviö, J.; Sallinen, J.; Scheinin, M.; Kobilka, B.K.; Riekkinen, P. Alpha2C-Adrenoceptor Overexpression Disrupts Execution of Spatial and Non-Spatial Search Patterns. Neuroscience 1999, 88, 1187–1198. [Google Scholar] [CrossRef]
- Khokhar, J.Y.; Henricks, A.M.; Kirk, E.; Green, A.I. Unique Effects of Clozapine: A Pharmacological Perspective. Adv. Pharmacol. 2018, 82, 137–162. [Google Scholar] [CrossRef]
- Durif, F.; Debilly, B.; Galitzky, M.; Morand, D.; Viallet, F.; Borg, M.; Thobois, S.; Broussolle, E.; Rascol, O. Clozapine Improves Dyskinesias in Parkinson Disease: A Double-Blind, Placebo-Controlled Study. Neurology 2004, 62, 381–388. [Google Scholar] [CrossRef]
- Nahimi, A.; Kinnerup, M.B.; Sommerauer, M.; Gjedde, A.; Borghammer, P. Molecular Imaging of the Noradrenergic System in Idiopathic Parkinson’s Disease. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 141, pp. 251–274. [Google Scholar]
- Nahimi, A.; Sommerauer, M.; Kinnerup, M.B.; Østergaard, K.; Wintherdahl, M.; Jacobsen, J.; Schacht, A.; Johnsen, B.; Damholdt, M.F.; Borghammer, P.; et al. Noradrenergic Deficits in Parkinson Disease Imaged with 11 C-MeNER. J. Nucl. Med. 2018, 59, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Doppler, C.E.J.; Kinnerup, M.B.; Brune, C.; Farrher, E.; Betts, M.; Fedorova, T.D.; Schaldemose, J.L.; Knudsen, K.; Ismail, R.; Seger, A.D.; et al. Regional Locus Coeruleus Degeneration Is Uncoupled from Noradrenergic Terminal Loss in Parkinson’s Disease. Brain 2021, 144, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Kinnerup, M.B.; Sommerauer, M.; Damholdt, M.F.; Schaldemose, J.L.; Ismail, R.; Terkelsen, A.J.; Stær, K.; Hansen, A.; Fedorova, T.D.; Knudsen, K.; et al. Preserved Noradrenergic Function in Parkinson’s Disease Patients with Rest Tremor. Neurobiol. Dis. 2021, 152, 105295. [Google Scholar] [CrossRef]
- Andersen, K.B.; Hansen, A.K.; Sommerauer, M.; Fedorova, T.D.; Knudsen, K.; Vang, K.; Van Den Berge, N.; Kinnerup, M.; Nahimi, A.; Pavese, N.; et al. Altered Sensorimotor Cortex Noradrenergic Function in Idiopathic REM Sleep Behaviour Disorder – A PET Study. Parkinsonism Relat. Disord. 2020, 75, 63–69. [Google Scholar] [CrossRef]
- Sommerauer, M.; Fedorova, T.D.; Hansen, A.K.; Knudsen, K.; Otto, M.; Jeppesen, J.; Frederiksen, Y.; Blicher, J.U.; Geday, J.; Nahimi, A.; et al. Evaluation of the Noradrenergic System in Parkinson’s Disease: An 11C-MeNER PET and Neuromelanin MRI Study. Brain 2018, 141, 496–504. [Google Scholar] [CrossRef]
- Sommerauer, M.; Hansen, A.K.; Parbo, P.; Fedorova, T.D.; Knudsen, K.; Frederiksen, Y.; Nahimi, A.; Barbe, M.T.; Brooks, D.J.; Borghammer, P. Decreased Noradrenaline Transporter Density in the Motor Cortex of Parkinson’s Disease Patients: Cortical Noradrenaline Transporter. Mov. Disord. 2018, 33, 1006–1010. [Google Scholar] [CrossRef]
- Meyer, G.M.; Spay, C.; Laurencin, C.; Ballanger, B.; Sescousse, G.; Boulinguez, P. Functional Imaging Studies of Impulse Control Disorders in Parkinson’s Disease Need a Stronger Neurocognitive Footing. Neurosci. Biobehav. Rev. 2019, 98, 164–176. [Google Scholar] [CrossRef]
- Laurencin, C.; Lancelot, S.; Gobert, F.; Redouté, J.; Mérida, I.; Iecker, T.; Liger, F.; Irace, Z.; Greusard, E.; Lamberet, L.; et al. Modeling [11C]Yohimbine PET Human Brain Kinetics with Test-Retest Reliability, Competition Sensitivity Studies and Search for a Suitable Reference Region. NeuroImage 2021, 240, 118328. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Controls (n = 15) | PD Patients (n = 12) | Group Difference | |
|---|---|---|---|
| Age in years (SD) | 52.5 (11.2) | 56.2 (8.9) | ns |
| Male/female | 6M/9F | 8M/4F | - |
| UPDRS-III (ON medication) | 12.7 (4.8) | - | |
| Disease duration in years (SD) | 6.1 (2.3) | - | |
| LED (mg/day) | 948 (320) | - | |
| Mattis | 141 (3) | 137 (3) | p < 0.006 |
| Beck Depression Inventory | 3.9 (3.3) | 11.1 (6.2) | p < 0.0005 |
| MNI Coordinates | P Corr | Cluster | ||||||
|---|---|---|---|---|---|---|---|---|
| Areas | BA | Side | x | y | z | Z-Score | Cluster | Size |
| Anterior Cingulate Cortex | 32 | R | 8 | 44 | 16 | 4.21 | 0.037 | 311 |
| Superior Frontal Gyrus | 32 | R | 16 | 50 | 18 | 4.03 | ||
| Superior Medial Frontal Gyrus | 9/32 | R | 10 | 44 | 34 | 3.72 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criaud, M.; Laurencin, C.; Poisson, A.; Metereau, E.; Redouté, J.; Thobois, S.; Boulinguez, P.; Ballanger, B. Noradrenaline and Movement Initiation Disorders in Parkinson’s Disease: A Pharmacological Functional MRI Study with Clonidine. Cells 2022, 11, 2640. https://doi.org/10.3390/cells11172640
Criaud M, Laurencin C, Poisson A, Metereau E, Redouté J, Thobois S, Boulinguez P, Ballanger B. Noradrenaline and Movement Initiation Disorders in Parkinson’s Disease: A Pharmacological Functional MRI Study with Clonidine. Cells. 2022; 11(17):2640. https://doi.org/10.3390/cells11172640
Chicago/Turabian StyleCriaud, Marion, Chloé Laurencin, Alice Poisson, Elise Metereau, Jérôme Redouté, Stéphane Thobois, Philippe Boulinguez, and Bénédicte Ballanger. 2022. "Noradrenaline and Movement Initiation Disorders in Parkinson’s Disease: A Pharmacological Functional MRI Study with Clonidine" Cells 11, no. 17: 2640. https://doi.org/10.3390/cells11172640
APA StyleCriaud, M., Laurencin, C., Poisson, A., Metereau, E., Redouté, J., Thobois, S., Boulinguez, P., & Ballanger, B. (2022). Noradrenaline and Movement Initiation Disorders in Parkinson’s Disease: A Pharmacological Functional MRI Study with Clonidine. Cells, 11(17), 2640. https://doi.org/10.3390/cells11172640