
Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gene Silencing with siRNA and Conditioned Medium Production
2.3. CCD-18Co Treatment with the Conditioned Medium
2.4. Protein Extraction and Western Blotting
2.5. mRNA Expression Analysis by qRT-PCR
2.6. Quantification of the Secreted Factors by ELISA
2.7. Wound-Healing Assay
2.8. Collagen Contraction Assay
2.9. Statistical Analysis
3. Results
3.1. CRC Cell-Secreted Factors Influence the Expression of α-SMA and the Secretion of TGFβ1 and HGF by CCD-18Co Fibroblasts
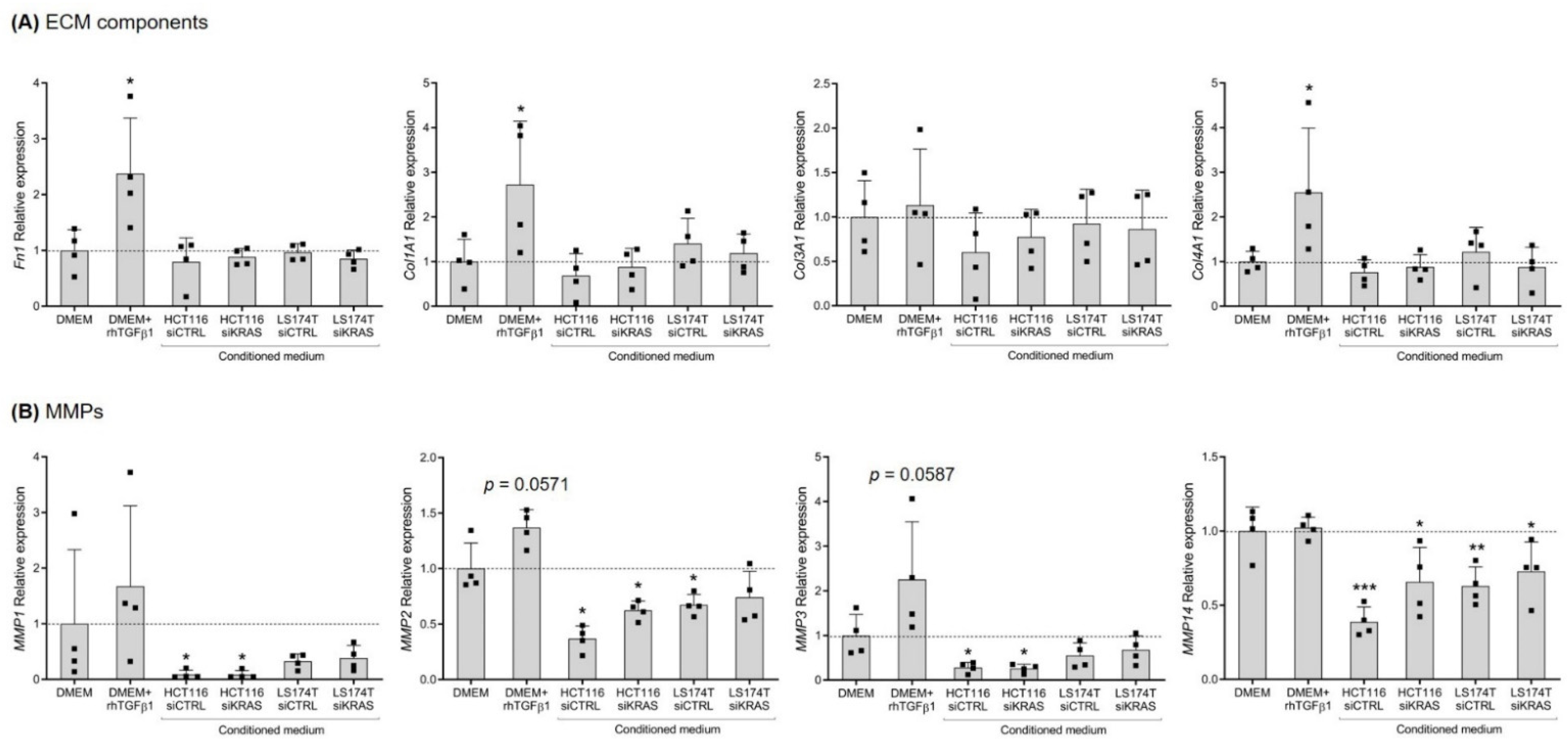
3.2. CRC Cell-Secretome Has No Influence on the mRNA Expression of Extracellular Matrix Components, but Alters the Expression of MMPs
3.3. HCT116 and LS174T Cell-Secreted Factors Differentially Affect Fibroblast Collagen Contraction and Migration Capacities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of ras mutations in cancer. Cancer Res. 2020, 80, 2669–2974. [Google Scholar] [CrossRef]
- Velho, S.; Haigis, K.M. Regulation of homeostasis and oncogenesis in the intestinal epithelium by Ras. Exp. Cell Res. 2011, 317, 2732–2739. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouché, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Clowers, M.J.; Velasco, W.V.; Ramos-Castaneda, M.; Moghaddam, S.J. Understanding the Complexity of the Tumor Microenvironment in K-ras Mutant Lung Cancer: Finding an Alternative Path to Prevention and Treatment. Front. Oncol. 2020, 9, 1556. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Gu, M.; Gao, Y.; Chang, P. KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas. Cancers 2021, 13, 2429. [Google Scholar] [CrossRef]
- Pereira, F.; Ferreira, A.; Reis, C.A.; Sousa, M.J.; Oliveira, M.J.; Preto, A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells 2022, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Tommelein, J.; Verset, L.; Boterberg, T.; Demetter, P.; Bracke, M.; De Wever, O. Cancer-Associated Fibroblasts Connect Metastasis-Promoting Communication in Colorectal Cancer. Front. Oncol. 2015, 5, 63. [Google Scholar] [CrossRef]
- Cullis, J.; Das, S.; Bar-Sagi, D. Kras and Tumor Immunity: Friend or Foe? Cold Spring Harb. Perspect. Med. 2018, 8, a031849. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Marco, M.R.; Choi, S.H.; Qu, X.; Chen, C.T.; Elkabets, M.; Fairchild, L.; Chow, O.; Barriga, F.M.; Dow, L.E.; et al. KRAS mutant rectal cancer cells interact with surrounding fibroblasts to deplete the extracellular matrix. Mol. Oncol. 2021, 15, 2766–2781. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; Denardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer- associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- De Jaeghere, E.A.; Denys, H.G.; De Wever, O. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer 2019, 5, 704–723. [Google Scholar] [CrossRef]
- Sandberg, T.P.; Stuart, M.P.M.E.; Oosting, J.; Tollenaar, R.A.E.M.; Sier, C.F.M.; Mesker, W.E. Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer 2019, 19, 284. [Google Scholar] [CrossRef]
- Gao, L.-F.; Zhong, Y.; Long, T.; Wang, X.; Zhu, J.-X.; Wang, X.-Y.; Hu, Z.-Y.; Li, Z.-G. Tumor bud-derived CCL5 recruits fibroblasts and promotes colorectal cancer progression via CCR5-SLC25A24 signaling. J. Exp. Clin. Cancer Res. 2022, 41, 81. [Google Scholar] [CrossRef]
- Tsujino, T.; Seshimo, I.; Yamamoto, H.; Chew, Y.N.; Ezumi, K.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; Matsuura, N.; Monden, M. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin. Cancer Res. 2007, 13, 2082–2090. [Google Scholar] [CrossRef]
- Dias Carvalho, P.; Martins, F.; Carvalho, J.; Oliveira, M.J.; Velho, S. Mutant KRAS-Associated Proteome Is Mainly Controlled by Exogenous Factors. Cells 2022, 11, 1988. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Martins, F.; Mendonça, S.; Ribeiro, A.; Machado, A.L.; Carvalho, J.; Oliveira, M.J.; Velho, S. Mutant KRAS modulates colorectal cancer cells invasive response to fibroblast-secreted factors through the HGF/C-MET axis. Int. J. Cancer 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Nguyen, Q.-D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, X.; Zhou, Q.; Li, P.; Yu, B.; Li, J.; Qu, Y.; Yan, J.; Yu, Y.; Yan, M.; et al. Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer Lett. 2013, 335, 128–135. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef]
- Denys, H.; Derycke, L.; Hendrix, A.; Westbroek, W.; Gheldof, A.; Narine, K.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential impact of TGF-B1 and EGF on fibroblast differentiation and invasion reciprocally promotes colon cancer cell invasion. Cancer Lett. 2008, 266, 263–274. [Google Scholar] [CrossRef]
- Hawinkels, L.J.A.C.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; Van Der Zon, J.M.; Van Der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.N.; Mesker, W.; Ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; Byrom, D.; et al. Dependency of Colorectal Cancer on a TGF- b -Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef]
- Gusenbauer, S.; Vlaicu, P.; Ullrich, A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene 2013, 32, 3846–3856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, F.; Shao, S.; Li, G.; Feng, Y. Human colorectal cancer-derived carcinoma associated fibroblasts promote CD44-mediated adhesion of colorectal cancer cells to endothelial cells by secretion of HGF. Cancer Cell Int. 2019, 19, 192. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Ji, J.; Jiang, J.; Cai, Q.; Wang, C.; Shi, M.; Yu, Y.; Zhu, Z.; Zhang, J. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018, 9, 867. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef]
- Singer, C.F.; Kronsteiner, N.; Marton, E.; Kubista, M.; Cullen, K.J.; Hirtenlehner, K.; Seifert, M.; Kubista, E. MMP-2 and MMP-9 expression in breast cancer-derived human fibroblasts is differentially regulated by stromal-epithelial interactions. Breast Cancer Res. Treat. 2002, 72, 69–77. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kim, H.J.; Liu, X.; Sugiura, H.; Kohyama, T.; Fang, Q.; Wen, F.Q.; Abe, S.; Wang, X.; Atkinson, J.J.; et al. Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 306, L1006–L1015. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Kagami, S.; Urushihara, M.; Kitamura, A.; Shimizu, M.; Strutz, F.; Müller, G.A.; Kuroda, Y. Transforming growth factor-β1 stimulates collagen matrix remodeling through increased adhesive and contractive potential by human renal fibroblasts. Biochim. Biophys. Acta 2004, 1693, 91–100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Saotome, T.; Usui, T.; Ohama, T.; Sato, K. Regulation of intestinal myofibroblasts by KRas-mutated colorectal cancer cells through heparin-binding epidermal growth factor-like growth factor. Oncol. Rep. 2017, 37, 3128–3136. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias Carvalho, P.; Mendonça, S.; Martins, F.; Oliveira, M.J.; Velho, S. Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS. Cells 2022, 11, 2490. https://doi.org/10.3390/cells11162490
Dias Carvalho P, Mendonça S, Martins F, Oliveira MJ, Velho S. Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS. Cells. 2022; 11(16):2490. https://doi.org/10.3390/cells11162490
Chicago/Turabian StyleDias Carvalho, Patrícia, Susana Mendonça, Flávia Martins, Maria José Oliveira, and Sérgia Velho. 2022. "Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS" Cells 11, no. 16: 2490. https://doi.org/10.3390/cells11162490
APA StyleDias Carvalho, P., Mendonça, S., Martins, F., Oliveira, M. J., & Velho, S. (2022). Modulation of Fibroblast Phenotype by Colorectal Cancer Cell-Secreted Factors Is Mostly Independent of Oncogenic KRAS. Cells, 11(16), 2490. https://doi.org/10.3390/cells11162490