
Phenformin Down-Regulates c-Myc Expression to Suppress the Expression of Pro-Inflammatory Cytokines in Keratinocytes


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Primary Human Keratinocytes
2.2. Human Skin Ex Vivo Cultures (Skin Organ Cultures)
2.3. Mouse Model for Acute Skin Inflammation
2.4. Histology and IF Analysis
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR) Assay
2.6. ELISA
2.7. Western Blot Analysis
2.8. RNA-Seq Analysis
2.9. siRNA Transfection
2.10. Virus Infection
2.11. Statistical Analysis
2.12. Ethics Statement
3. Results
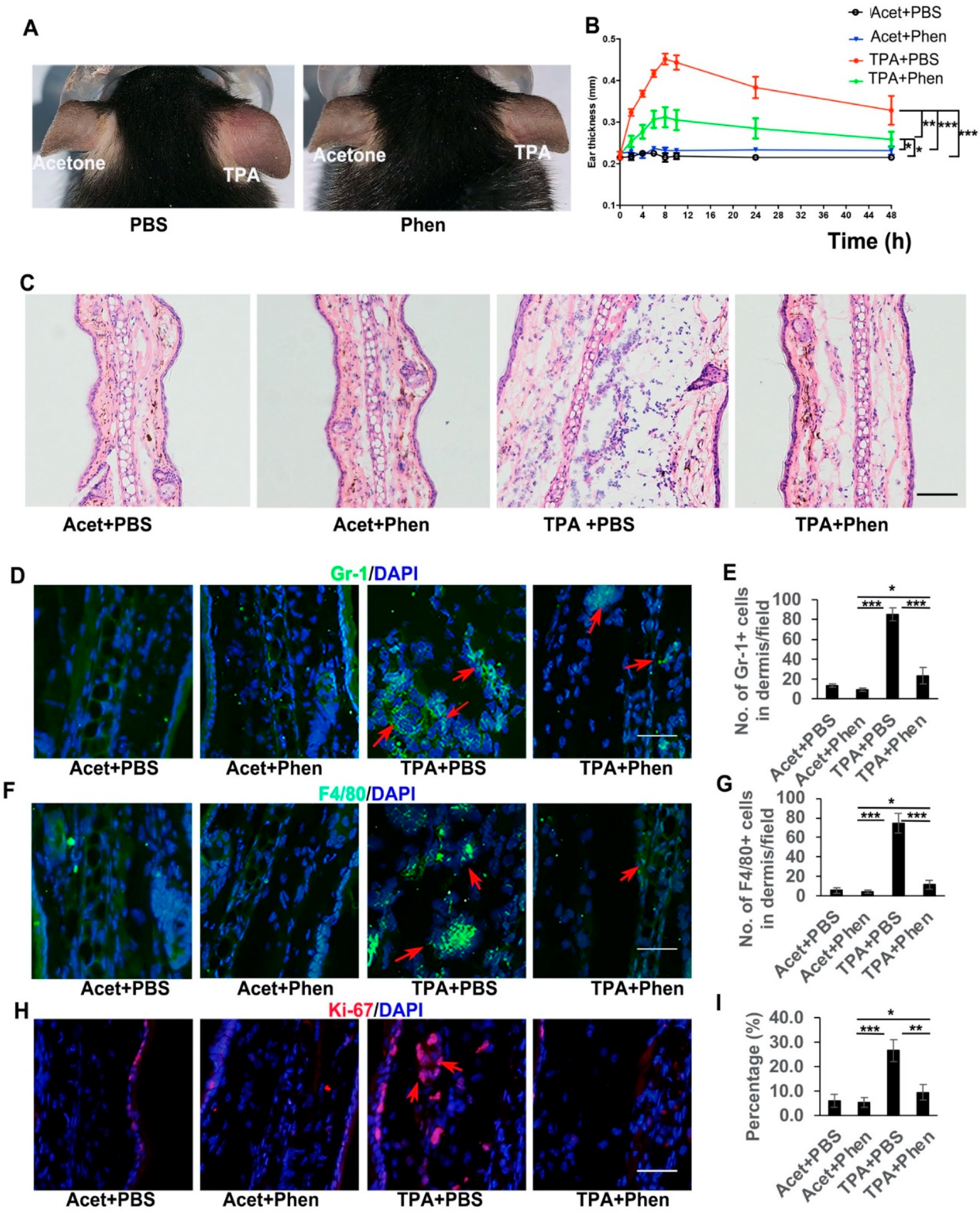
3.1. Phenformin Suppresses Acute Inflammatory Responses Induced by TPA in the Skin
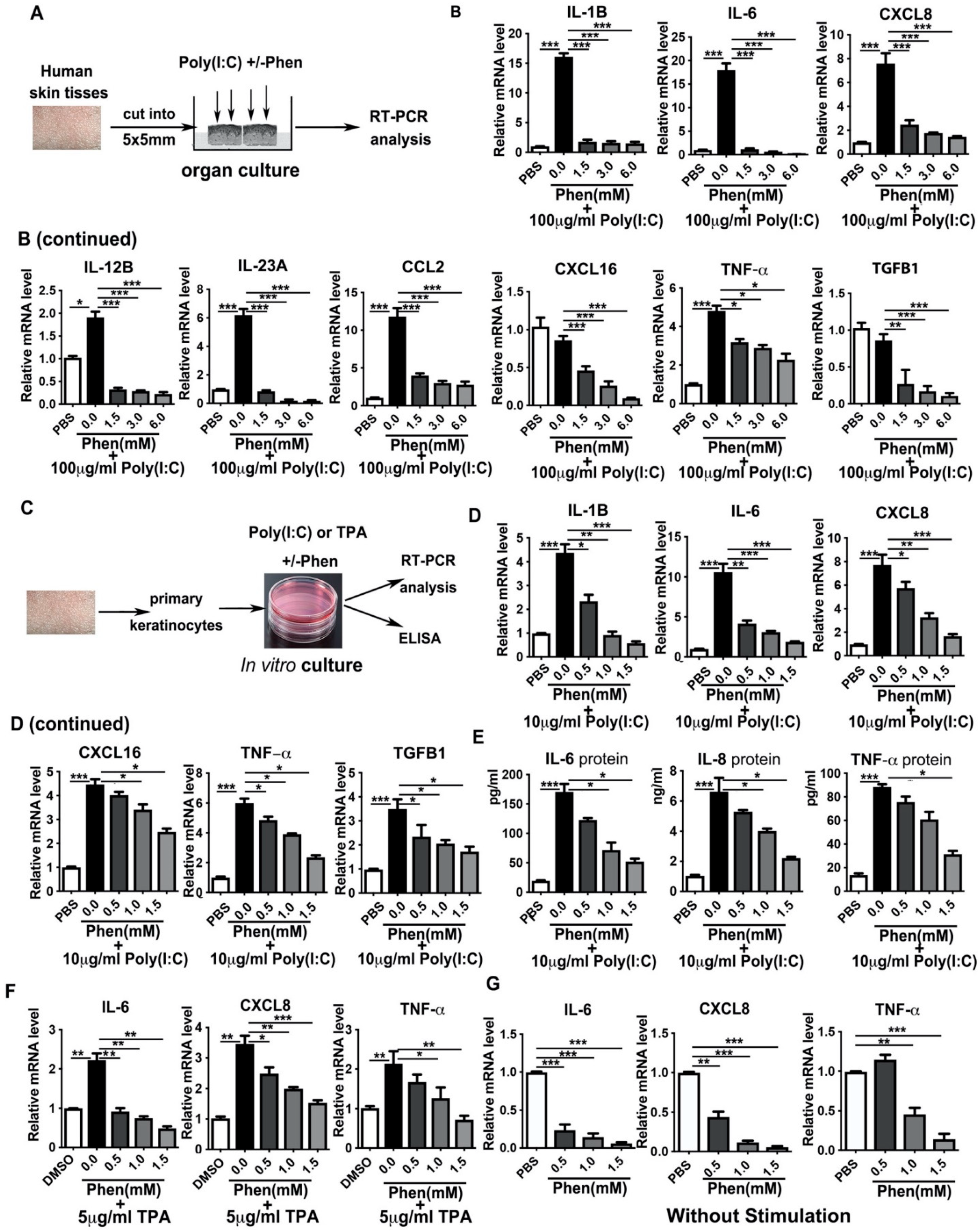
3.2. Phenformin Suppresses Pro-Inflammatory Cytokine Expression in Ex Vivo Cultured Skin and in In Vitro Cultured Keratinocytes Induced by Poly I:C or by TPA
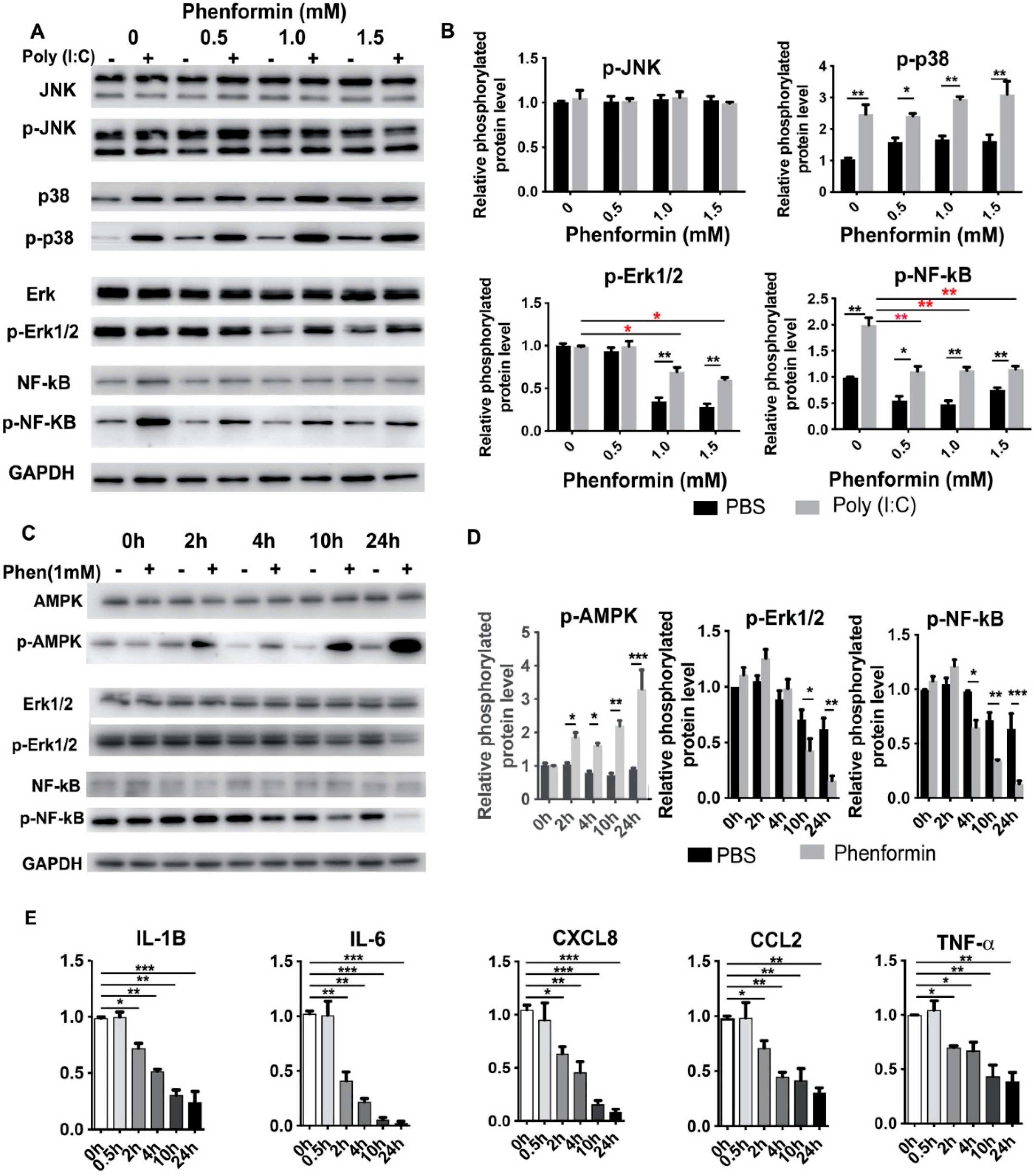
3.3. Phenformin Intially Suppresses the Expression of Pro-Inflammatory Cytokines but Not Directly by Blocking Activation of the NF-κB or MAPK Pathways in Human Keratinocytes
3.4. RNA-Seq Analysis Reveals That Phenformin Significantly Suppresses the c-Myc Pathway at Earlier Time Points
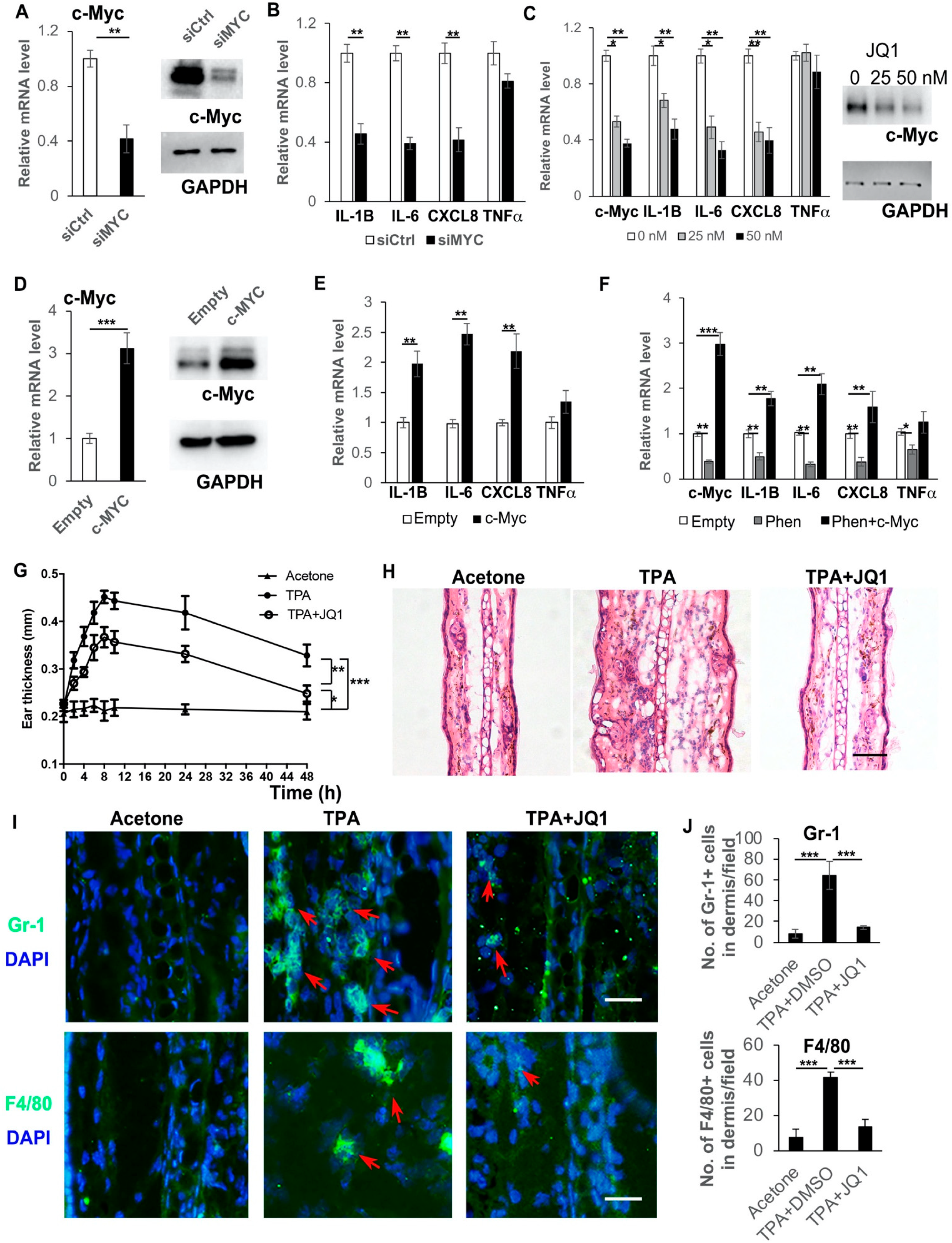
3.5. Inhibition of c-Myc Suppresses Cytokine Expression and the Over-Expression of c-Myc Rescues the Suppression of Cytokine Expression by Phenformin in Human Keratinocytes
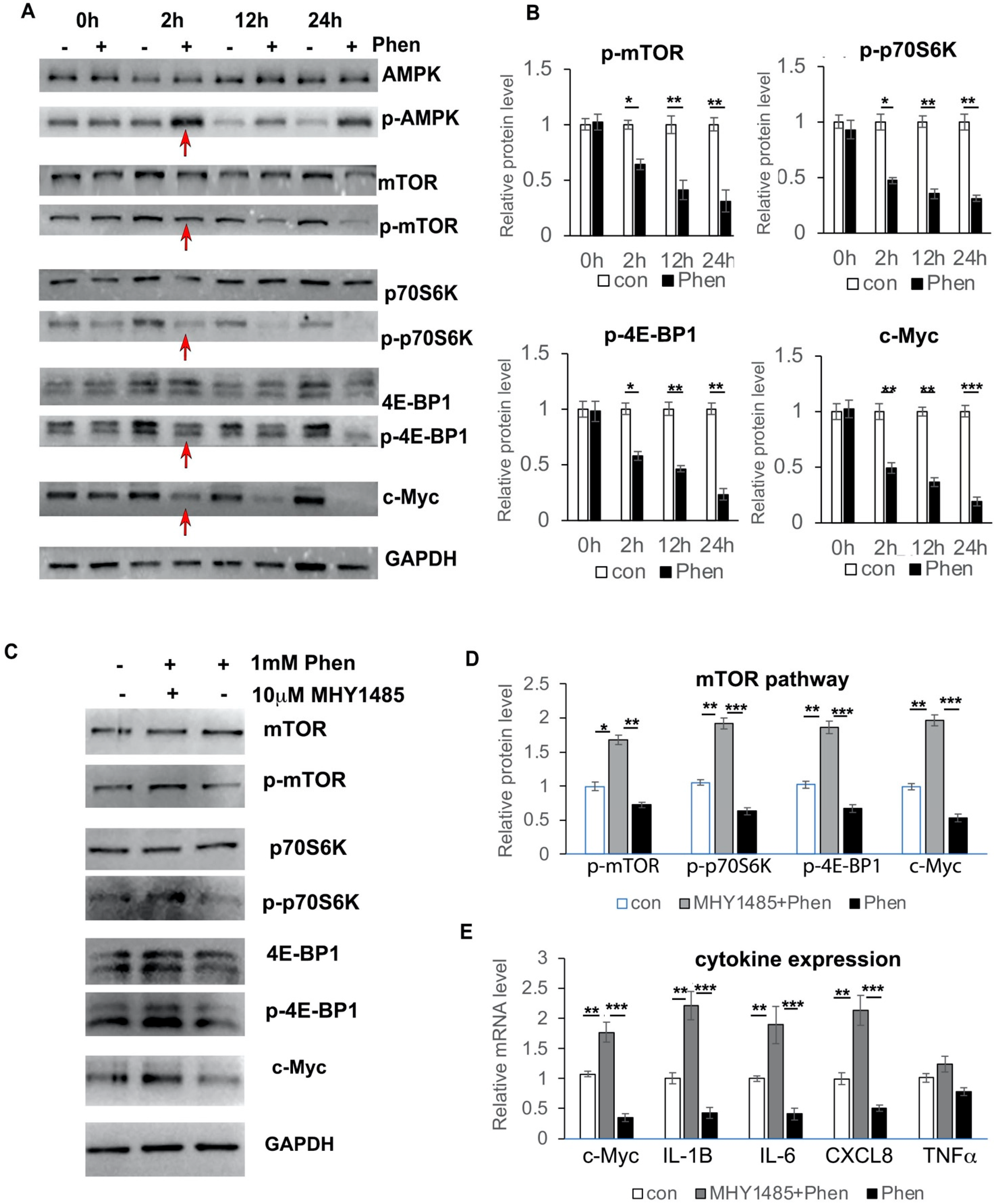
3.6. Phenformin Controls c-Myc Expression through Inhibition of the mTOR Pathway in Keratinocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.P.; et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Thibodaux, R.J.; Triche, M.W.; Espinoza, L.R. Ustekinumab for the treatment of psoriasis and psoriatic arthritis: A drug evaluation and literature review. Expert Opin. Biol. Ther. 2018, 18, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Pavel, A.B.; Diaz, A.; Werth, J.L.; Zang, C.; Vranic, I.; Purohit, V.S.; Zielinski, M.A.; Vlahos, B.; Estrada, Y.D.; et al. Crisaborole and atopic dermatitis skin biomarkers: An intrapatient randomized trial. J. Allergy Clin. Immunol. 2019, 144, 1274–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elenkov, I.J.; Iezzoni, D.G.; Daly, A.; Harris, A.G.; Chrousos, G.P. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation 2005, 12, 255–269. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Fulton, C.; Anderson, G.M.; Zasloff, M.; Bull, R.; Quinn, A.G. Expression of natural peptide antibiotics in human skin. Lancet 1997, 350, 1750–1751. [Google Scholar] [CrossRef]
- Martinez, A.; Elsasser, T.H.; Muro-Cacho, C.; Moody, T.W.; Miller, M.J.; Macri, C.J.; Cuttitta, F. Expression of adrenomedullin and its receptor in normal and malignant human skin: A potential pluripotent role in the integument. Endocrinology 1997, 138, 5597–5604. [Google Scholar] [CrossRef]
- Harder, J.; Schroder, J.M. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J. Biol. Chem. 2002, 277, 46779–46784. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.Y.; Destoumieux, D.; Wong, A.V.; Park, C.H.; Valore, E.V.; Liu, L.; Ganz, T. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J. Investig. Dermatol. 2002, 118, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, O.E.; Cowland, J.B.; Theilgaard-Monch, K.; Liu, L.; Ganz, T.; Borregaard, N. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J. Immunol. 2003, 170, 5583–5589. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.; Meyer-Hoffert, U.; Wehkamp, K.; Schwichtenberg, L.; Schroder, J.M. Differential gene induction of human beta-defensins (hBD-1, -2, -3, and -4) in keratinocytes is inhibited by retinoic acid. J. Investig. Dermatol. 2004, 123, 522–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braff, M.H.; Di Nardo, A.; Gallo, R.L. Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J. Investig. Dermatol. 2005, 124, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, A.P.; Ardern-Jones, M.R.; Kasprowicz, V.; Bowness, P.; Jones, L.; Bailey, A.S.; Ogg, G.S. Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol. 2007, 37, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Crispin, M.; Billick, E.; Mitsui, H.; Gulati, N.; Fujita, H.; Gilleaudeau, P.; Sullivan-Whalen, M.; Johnson-Huang, L.M.; Suarez-Farinas, M.; Krueger, J.G. Human keratinocytes’ response to injury upregulates CCL20 and other genes linking innate and adaptive immunity. J. Investig. Dermatol. 2012, 132, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Hanel, K.H.; Cornelissen, C.; Luscher, B.; Baron, J.M. Cytokines and the skin barrier. Int. J. Mol. Sci. 2013, 14, 6720–6745. [Google Scholar] [CrossRef] [Green Version]
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Dermatol. 2008, 17, 1063–1072. [Google Scholar] [CrossRef]
- Murata, T.; Honda, T.; Mostafa, A.; Kabashima, K. Stratum corneum as polymer sheet: Concept and cornification processes. Trends Mol. Med. 2022, 28, 350–359. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chen, S.C.; Hsu, S.Y.; Lin, Y.A.; Shih, C.M.; Huang, C.Y.; Wang, K.H.; Lee, A.W. Annoying Psoriasis and Atopic Dermatitis: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 4898. [Google Scholar] [CrossRef]
- Decensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Snima, K.S.; Pillai, P.; Cherian, A.M.; Nair, S.V.; Lakshmanan, V.K. Anti-diabetic drug metformin: Challenges and perspectives for cancer therapy. Curr. Cancer Drug Targets 2014, 14, 727–736. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kwak, H.J.; Cha, J.Y.; Jeong, Y.S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J. Biol. Chem. 2014, 289, 23246–23255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, F.; Ma, W.; Sun, Q. Metformin inhibits proliferation and proinflammatory cytokines of human keratinocytes in vitro via mTOR-signaling pathway. Pharm. Biol. 2016, 54, 1173–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petti, C.; Vegetti, C.; Molla, A.; Bersani, I.; Cleris, L.; Mustard, K.J.; Formelli, F.; Hardie, G.D.; Sensi, M.; Anichini, A. AMPK activators inhibit the proliferation of human melanomas bearing the activated MAPK pathway. Melanoma Res. 2012, 22, 341–350. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; Pasca di Magliano, M.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J. Investig. Dermatol. 2017, 137, 1740–1748. [Google Scholar] [CrossRef]
- Petrachi, T.; Romagnani, A.; Albini, A.; Longo, C.; Argenziano, G.; Grisendi, G.; Dominici, M.; Ciarrocchi, A.; Dallaglio, K. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget 2017, 8, 6914–6928. [Google Scholar] [CrossRef] [Green Version]
- Tzvetkov, M.V.; Vormfelde, S.V.; Balen, D.; Meineke, I.; Schmidt, T.; Sehrt, D.; Sabolic, I.; Koepsell, H.; Brockmoller, J. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin. Pharmacol. Ther. 2009, 86, 299–306. [Google Scholar] [CrossRef]
- Chen, L.; Pawlikowski, B.; Schlessinger, A.; More, S.S.; Stryke, D.; Johns, S.J.; Portman, M.A.; Chen, E.; Ferrin, T.E.; Sali, A.; et al. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharm. Genom. 2010, 20, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharm. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, R.; Takenaka, S.; Hashimoto, M.; Narawa, T.; Itoh, T. Expression of human solute carrier family transporters in skin: Possible contributor to drug-induced skin disorders. Sci. Rep. 2014, 4, 5251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Kim, S.H.; Perez-Lorenzo, R.; Liu, C.; Huang, M.; Dotto, G.P.; Zheng, B.; Wu, X. Phenformin Promotes Keratinocyte Differentiation via the Calcineurin/NFAT Pathway. J. Investig. Dermatol. 2021, 141, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, L., Jr.; Washenik, K.; Stenn, K. Full-thickness skin with mature hair follicles generated from tissue culture expanded human cells. Tissue Eng. Part A 2014, 20, 3314–3321. [Google Scholar] [CrossRef]
- Wen, J.; Zu, T.; Zhou, Q.; Leng, X.; Wu, X. Y-27632 simplifies the isolation procedure of human primary epidermal cells by selectively blocking focal adhesion of dermal cells. J. Tissue Eng. Regen. Med. 2018, 12, e1251–e1255. [Google Scholar] [CrossRef]
- Qian, H.; Leng, X.; Wen, J.; Zhou, Q.; Xu, X.; Wu, X. One-Step Simple Isolation Method to Obtain Both Epidermal and Dermal Stem Cells from Human Skin Specimen. Methods Mol. Biol. 2019, 1879, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qin, J.; Xie, Z.; Liu, C.; Su, Y.; Chen, Z.; Zhou, Q.; Ma, C.; Liu, G.; Paus, R.; et al. Y-27632 preserves epidermal integrity in a human skin organ-culture (hSOC) system by regulating AKT and ERK signaling pathways. J. Dermatol. Sci. 2019, 96, 99–109. [Google Scholar] [CrossRef]
- Zeng, W.J.; Tan, Z.; Lai, X.F.; Xu, Y.N.; Mai, C.L.; Zhang, J.; Lin, Z.J.; Liu, X.G.; Sun, S.L.; Zhou, L.J. Topical delivery of l-theanine ameliorates TPA-induced acute skin inflammation via downregulating endothelial PECAM-1 and neutrophil infiltration and activation. Chem. Biol. Interact 2018, 284, 69–79. [Google Scholar] [CrossRef]
- Wen, J.; Li, X.; Leng, X.; Xu, X.; Wu, X. An advanced mouse model for human skin wound healing. Exp. Dermatol. 2017, 26, 433–435. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Tommasi di Vignano, A.; Zhou, Q.; Michel-Dziunycz, P.J.; Bai, F.; Mi, J.; Qin, J.; Zu, T.; Hofbauer, G.F.L. The ARE-binding protein Tristetraprolin (TTP) is a novel target and mediator of calcineurin tumor suppressing function in the skin. PLoS Genet. 2018, 14, e1007366. [Google Scholar] [CrossRef]
- Piipponen, M.; Li, D.; Landen, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. [Google Scholar] [CrossRef]
- Hanada, T.; Yoshimura, A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002, 13, 413–421. [Google Scholar] [CrossRef]
- Moens, U.; Kostenko, S.; Sveinbjornsson, B. The Role of Mitogen-Activated Protein Kinase-Activated Protein Kinases (MAPKAPKs) in Inflammation. Genes 2013, 4, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Qiu, S.; Zhu, X.; Lin, H.; Li, L. OCT1-Mediated Metformin Uptake Regulates Pancreatic Stellate Cell Activity. Cell Physiol. Biochem. 2018, 47, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Veiga, S.R.; Ge, X.; Mercer, C.A.; Hernandez-Alvarez, M.I.; Thomas, H.E.; Hernandez-Losa, J.; Ramon, Y.C.S.; Zorzano, A.; Thomas, G.; Kozma, S.C. Phenformin-Induced Mitochondrial Dysfunction Sensitizes Hepatocellular Carcinoma for Dual Inhibition of mTOR. Clin. Cancer Res. 2018, 24, 3767–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [Green Version]
- Tokajuk, A.; Krzyzanowska-Grycel, E.; Tokajuk, A.; Grycel, S.; Sadowska, A.; Car, H. Antidiabetic drugs and risk of cancer. Pharmacol. Rep. 2015, 67, 1240–1250. [Google Scholar] [CrossRef]
- Barker, J.N.; Mitra, R.S.; Griffiths, C.E.; Dixit, V.M.; Nickoloff, B.J. Keratinocytes as initiators of inflammation. Lancet 1991, 337, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–d26. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M.; DePalo, V.A. Anti-inflammatory cytokines. Chest 2000, 117, 1162–1172. [Google Scholar] [CrossRef] [Green Version]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachstetter, A.D.; Van Eldik, L.J. The p38 MAP Kinase Family as Regulators of Proinflammatory Cytokine Production in Degenerative Diseases of the CNS. Aging Dis. 2010, 1, 199–211. [Google Scholar] [PubMed]
- Mori, H.; Murakami, M.; Tsuda, T.; Kameda, K.; Utsunomiya, R.; Masuda, K.; Shiraishi, K.; Dai, X.; Tohyama, M.; Nakaoka, H.; et al. Reduced-HMGB1 suppresses poly(I:C)-induced inflammation in keratinocytes. J. Dermatol. Sci. 2018, 90, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sayama, K.; Tohyama, M.; Shirakata, Y.; Yang, L.; Hirakawa, S.; Tokumaru, S.; Hashimoto, K. The NF-κB, p38 MAPK and STAT1 pathways differentially regulate the dsRNA-mediated innate immune responses of epidermal keratinocytes. Int. Immunol. 2008, 20, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penn, L.J.; Laufer, E.M.; Land, H. C-MYC: Evidence for multiple regulatory functions. Semin. Cancer Biol. 1990, 1, 69–80. [Google Scholar] [PubMed]
- Felsher, D.W.; Bishop, J.M. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.; Liebermann, D.; Hoffman, B. The c-myc apoptotic response is not intrinsic to blocking terminal myeloid differentiation. J. Cell Physiol. 2008, 216, 120–127. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-MYC. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Sipos, F.; Firneisz, G.; Muzes, G. Therapeutic aspects of c-MYC signaling in inflammatory and cancerous colonic diseases. World J. Gastroenterol. 2016, 22, 7938–7950. [Google Scholar] [CrossRef]
- Borrello, M.G.; Degl’Innocenti, D.; Pierotti, M.A. Inflammation and cancer: The oncogene-driven connection. Cancer Lett. 2008, 267, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, Y.; Ko, K.S.; Yang, H. Interactions between Myc and Mediators of Inflammation in Chronic Liver Diseases. Mediat. Inflamm. 2015, 2015, 276850. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchors, K.; Shchors, E.; Rostker, F.; Lawlor, E.R.; Brown-Swigart, L.; Evan, G.I. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006, 20, 2527–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, H.C.; Li, J.C.; Pong, J.C.; Lau, A.S. A role for c-Myc in regulating anti-mycobacterial responses. Proc. Natl. Acad. Sci. USA 2011, 108, 17749–17754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Vlotides, G.; Tanyeri, A.; Spampatti, M.; Zitzmann, K.; Chourdakis, M.; Spttl, C.; Maurer, J.; Nolting, S.; Goke, B.; Auernhammer, C.J. Anticancer effects of metformin on neuroendocrine tumor cells in vitro. Hormones 2014, 13, 498–508. [Google Scholar] [CrossRef]
- Shin, J.M.; Jeong, Y.J.; Cho, H.J.; Magae, J.; Bae, Y.S.; Chang, Y.C. Suppression of c-Myc induces apoptosis via an AMPK/mTOR-dependent pathway by 4-O-methyl-ascochlorin in leukemia cells. Apoptosis 2016, 21, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.M.; Shan, Y.S.; Chu, P.Y.; Jiang, S.S.; Hung, W.C.; Chen, Y.L.; Tu, H.C.; Lin, H.Y.; Tsai, H.J.; Chen, L.T. The regulatory role of aberrant Phosphatase and Tensin Homologue and Liver Kinase B1 on AKT/mTOR/c-Myc axis in pancreatic neuroendocrine tumors. Oncotarget 2017, 8, 98068–98083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.K.; Han, S.Y.; Jeong, Y.J.; Magae, J.; Bae, Y.S.; Chang, Y.C. 4-O-methylascochlorin activates autophagy by activating AMPK and suppressing c-Myc in glioblastoma. J. Biochem. Mol. Toxicol. 2020, 34, e22552. [Google Scholar] [CrossRef] [PubMed]
- Buckle, R.S.; Mechali, M. Analysis of c-Myc and Max binding to the c-myc promoter: Evidence that autosuppression occurs via an indirect mechanism. Oncogene 1995, 10, 1249–1255. [Google Scholar] [PubMed]
- Iguchi-Ariga, S.M.; Okazaki, T.; Itani, T.; Ogata, M.; Sato, Y.; Ariga, H. An initiation site of DNA replication with transcriptional enhancer activity present upstream of the c-myc gene. EMBO J. 1988, 7, 3135–3142. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Li, D.; Zhang, L.; Xu, Q.; Zhuang, D.; Liu, P.; Hu, L.; Deng, H.; Sun, J.; Wang, S.; et al. Phenformin Down-Regulates c-Myc Expression to Suppress the Expression of Pro-Inflammatory Cytokines in Keratinocytes. Cells 2022, 11, 2429. https://doi.org/10.3390/cells11152429
Liu G, Li D, Zhang L, Xu Q, Zhuang D, Liu P, Hu L, Deng H, Sun J, Wang S, et al. Phenformin Down-Regulates c-Myc Expression to Suppress the Expression of Pro-Inflammatory Cytokines in Keratinocytes. Cells. 2022; 11(15):2429. https://doi.org/10.3390/cells11152429
Chicago/Turabian StyleLiu, Guanyi, Dingyang Li, Liwei Zhang, Qiuping Xu, Dexuan Zhuang, Panpan Liu, Ling Hu, Huiting Deng, Jianfeng Sun, Shuangshuang Wang, and et al. 2022. "Phenformin Down-Regulates c-Myc Expression to Suppress the Expression of Pro-Inflammatory Cytokines in Keratinocytes" Cells 11, no. 15: 2429. https://doi.org/10.3390/cells11152429
APA StyleLiu, G., Li, D., Zhang, L., Xu, Q., Zhuang, D., Liu, P., Hu, L., Deng, H., Sun, J., Wang, S., Zheng, B., Guo, J., & Wu, X. (2022). Phenformin Down-Regulates c-Myc Expression to Suppress the Expression of Pro-Inflammatory Cytokines in Keratinocytes. Cells, 11(15), 2429. https://doi.org/10.3390/cells11152429