
Quantitative Acetylomics Uncover Acetylation-Mediated Pathway Changes Following Histone Deacetylase Inhibition in Anaplastic Large Cell Lymphoma
 ,
, 

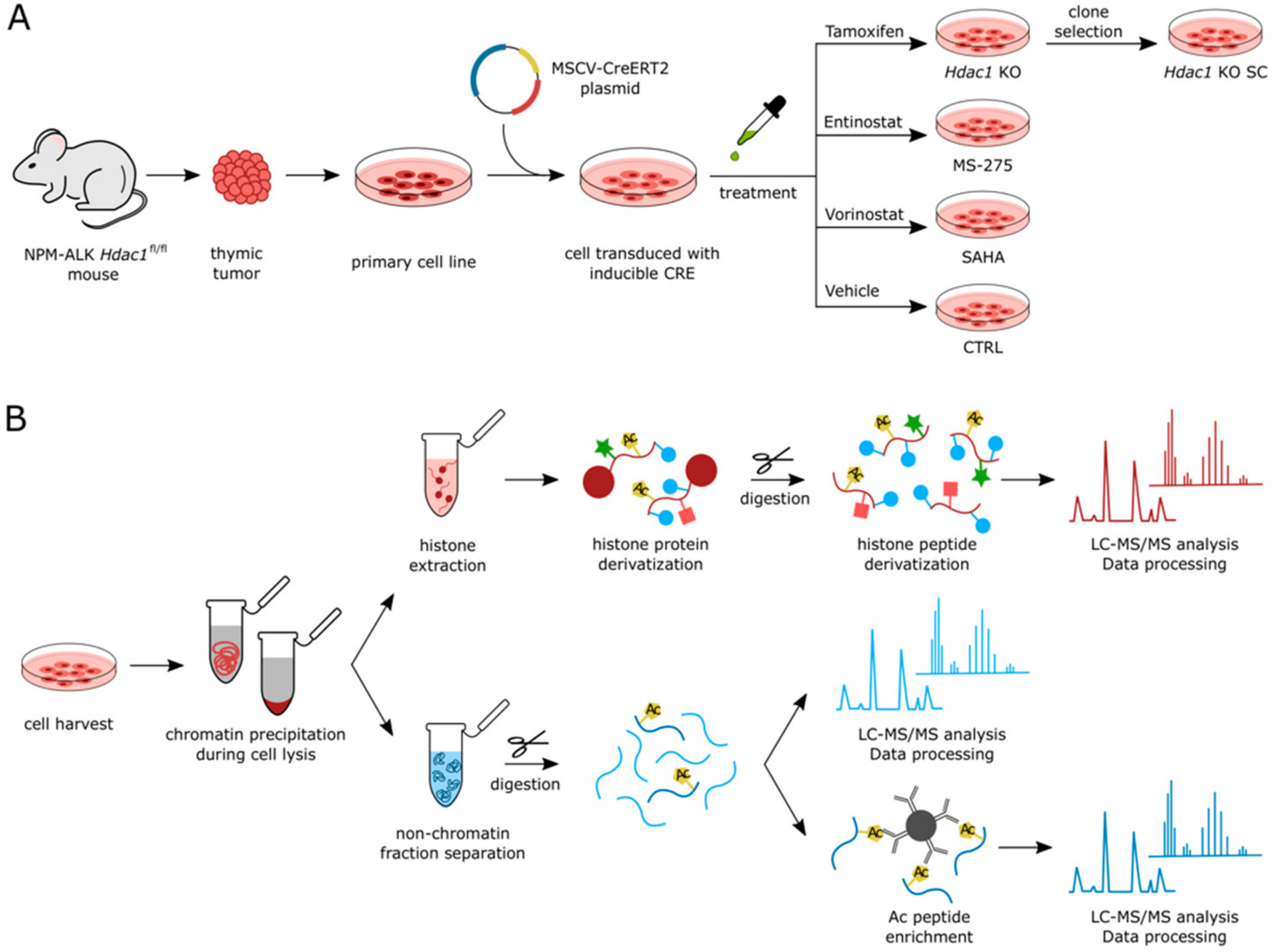{kind=link}
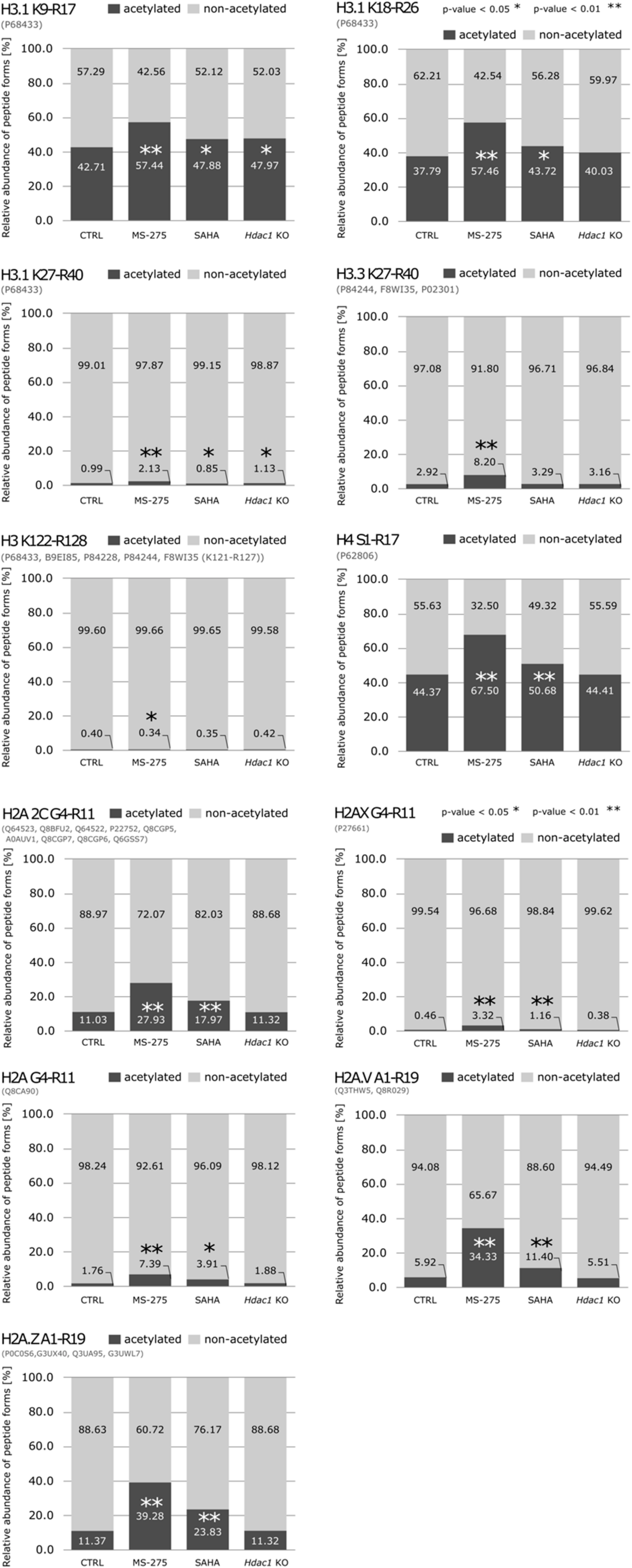{kind=link}
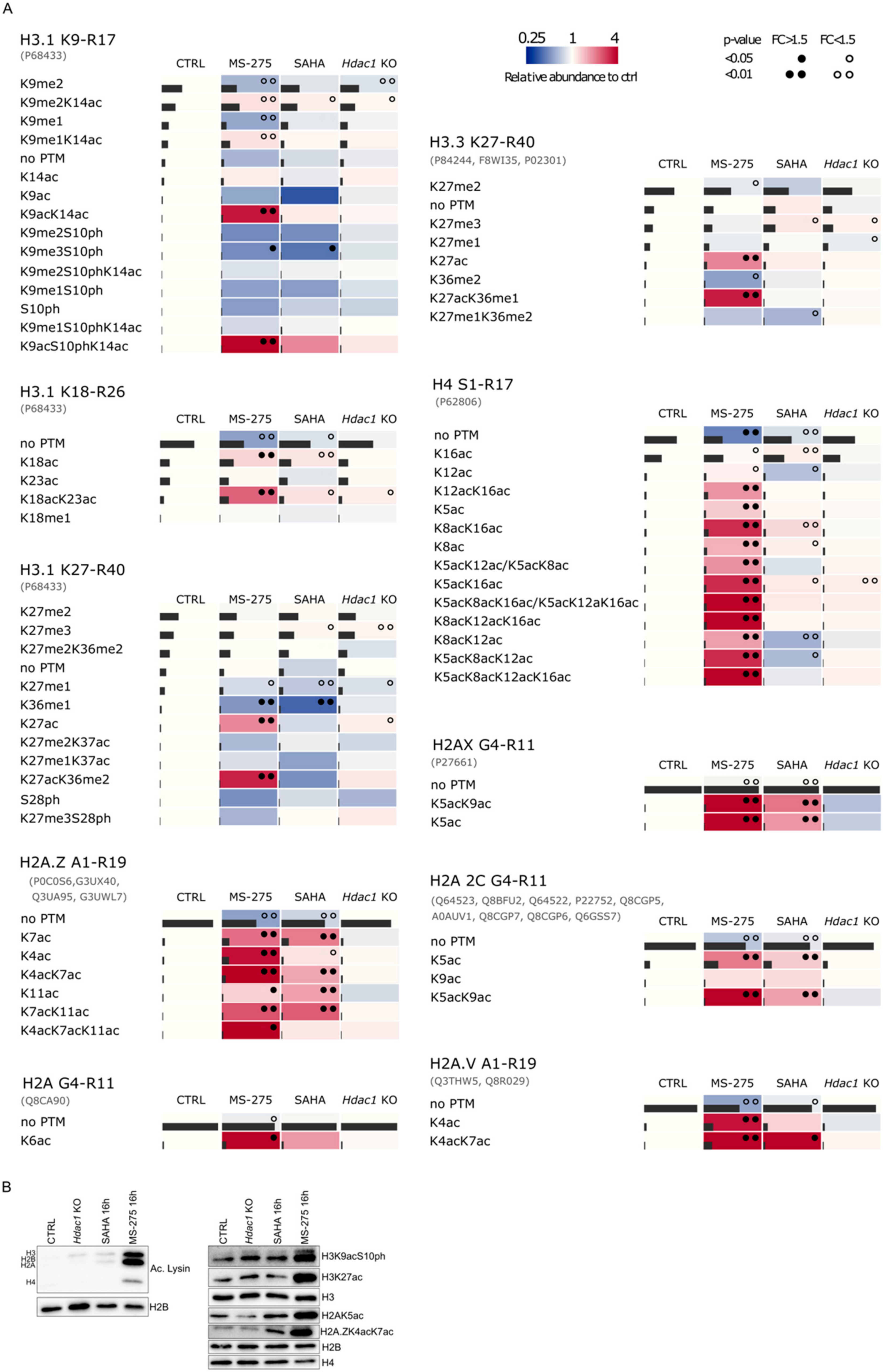{kind=link}
{kind=link}
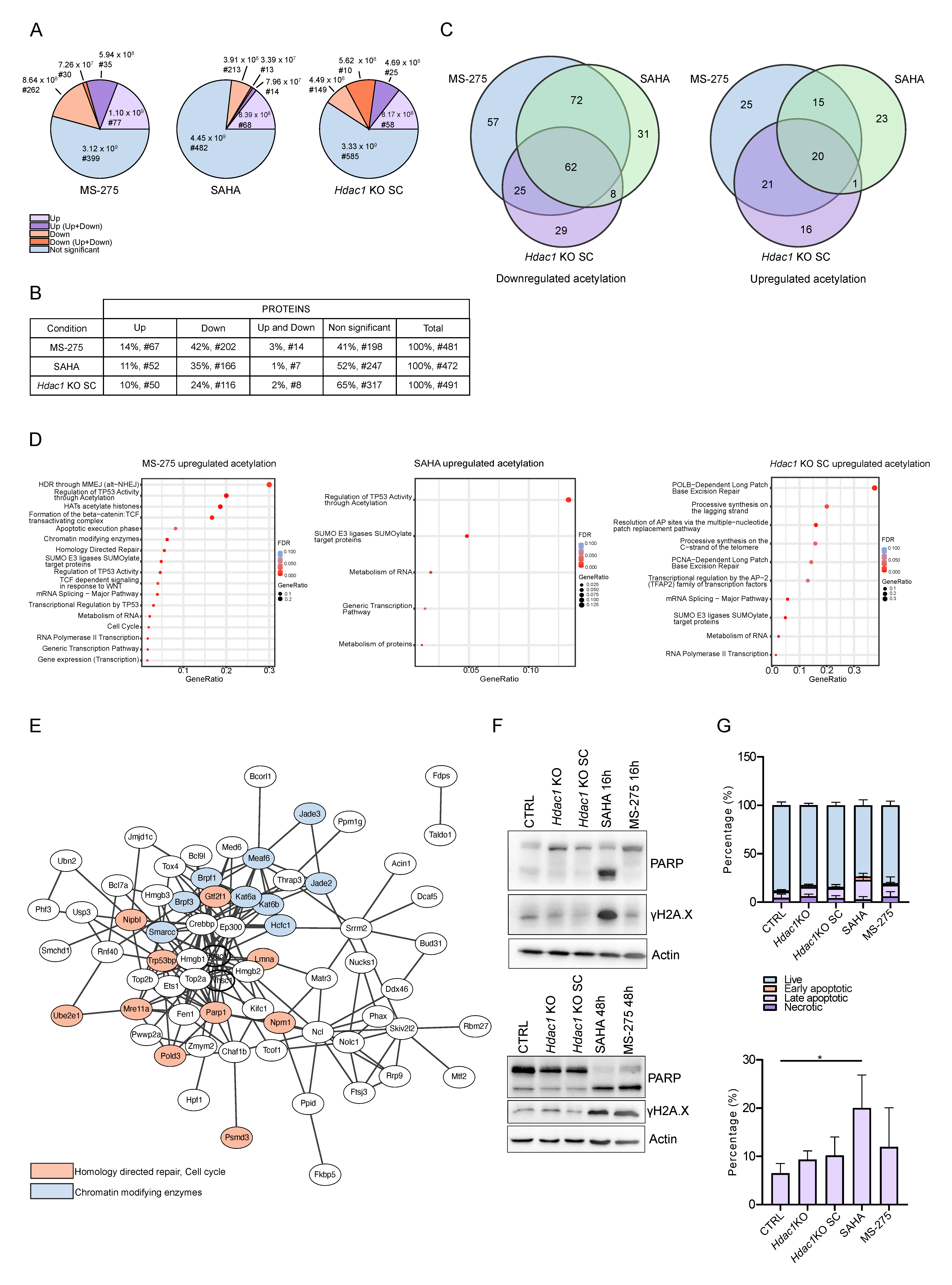{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Establishing, Passaging and Culturing of ALK+ Cell Lines
2.3. Transduction of Primary Cell Line with Inducible Cre
2.4. Virus Production and Lentiviral Transduction
2.5. Conditional Knockout System—4-OHT
2.6. Genotyping of Cell Lines
2.7. Inhibitor Treatments
2.8. Histone Extract Preparation for LC-MS/MS
2.9. Preparation of Non-Chromatin Peptides for Acetylomic Analysis
2.10. Purification of Samples Prior to LC-MS/MS Analysis
2.11. LC-MS/MS Analysis
2.12. Data Analysis
2.13. Isolation of Proteins, Isolation of Histones and Western Blotting
2.14. Characterization of Apoptotic Cells (Annexin V x DAPI Staining)
2.15. Irradiation of Cells
2.16. Immunofluorescence Staining
2.17. String Analysis
3. Results
3.1. Experimental Setup
3.2. Reduction of HDAC Activity Results in Global Changes in the Acetylation Status of Histones
3.3. Reduction in HDAC Activity Results in Deregulation of the Non-Chromatin Proteome
3.4. Reduction in HDAC Activity Causes Changes in Acetylation Levels of Specific non-Histone Proteins
3.5. Reduction in HDAC Activity Affects Acetylation of non-Histone Proteins Involved in Major Cellular Processes
3.6. Reduction in HDAC Activity Affects Acetylation of DDR Proteins
3.7. Acetylation of TP53BP1 Does Not Disrupt the Colocalization of TP53BP1 and γH2AX Foci during DNA Damage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Stengel, K.R.; Hiebert, S.W. Class I HDACs Affect DNA Replication, Repair, and Chromatin Structure: Implications for Cancer Therapy. Antioxid. Redox Signal 2015, 23, 51–65. [Google Scholar] [CrossRef]
- Lahue, R.S.; Frizzell, A. Histone deacetylase complexes as caretakers of genome stability. Epigenetics 2012, 7, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef]
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Martinez, E. Dual Regulation of c-Myc by p300 via Acetylation-Dependent Control of Myc Protein Turnover and Coactivation of Myc-Induced Transcription. Mol. Cell. Biol. 2005, 25, 10220–10234. [Google Scholar] [CrossRef]
- Yuan, Z.; Guan, Y.; Chatterjee, D.; Chin, Y.E. Stat3 Dimerization Regulated by Reversible Acetylation of a Single Lysine Residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Ikenoue, T.; Inoki, K.; Zhao, B.; Guan, K.-L. PTEN Acetylation Modulates Its Interaction with PDZ Domain. Cancer Res. 2008, 68, 6908–6912. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Guha, M. HDAC inhibitors still need a home run, despite recent approval. Nat. Rev. Drug Discov. 2015, 14, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.-P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. Available online: https://www.frontiersin.org/article/10.3389/fonc.2018.00092 (accessed on 14 January 2022). [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, Z. Mechanism of Action for HDAC Inhibitors—Insights from Omics Approaches. Int. J. Mol. Sci. 2019, 20, 1616. [Google Scholar] [CrossRef]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef]
- Heideman, M.R.; Wilting, R.H.; Yanover, E.; Velds, A.; de Jong, J.; Kerkhoven, R.M.; Dannenberg, J.H. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 2013, 121, 2038–2050. [Google Scholar] [CrossRef]
- Santoro, F.; Botrugno, O.A.; Dal Zuffo, R.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Minucci, S. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Ellmeier, W. Conditional Deletion of Histone Deacetylase 1 in T Cells Leads to Enhanced Airway Inflammation and Increased Th2 Cytokine Production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Boucheron, N.; Tschismarov, R.; Goeschl, L.; Moser, M.A.; Lagger, S.; Sakaguchi, S.; Ellmeier, W. CD4+ T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC. Nat. Immunol. 2014, 15, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Tschismarov, R.; Firner, S.; Gil-Cruz, C.; Göschl, L.; Boucheron, N.; Steiner, G.; Ellmeier, W. HDAC1 Controls CD8+ T Cell Homeostasis and Antiviral Response. PLoS ONE 2014, 9, e110576. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NPM, in Non-Hodgkin’s Lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Werner, M.T.; Zhang, Q.; Wasik, M.A. From Pathology to Precision Medicine in Anaplastic Large Cell Lymphoma Expressing Anaplastic Lymphoma Kinase (ALK+ ALCL). Cancers 2017, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer. 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.M.; Lai, R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood 2007, 110, 2259–2267. [Google Scholar] [CrossRef]
- Prokoph, N.; Larose, H.; Lim, M.S.; Burke, G.A.A.; Turner, S.D. Treatment Options for Paediatric Anaplastic Large Cell Lymphoma (ALCL): Current Standard and beyond. Cancers 2018, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Kuchaříková, H.; Dobrovolná, P.; Lochmanová, G.; Zdráhal, Z. Trimethylacetic Anhydride–Based Derivatization Facilitates Quantification of Histone Marks at the MS1 Level. Mol. Cell Proteom. 2021, 20, 100114. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Gong, J.Z.; Guasparri, I.; Pesci, A.; Cai, J.; Liu, J.; Inghirami, G. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003, 101, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Činčárová, L.; Lochmanová, G.; Nováková, K.; Šultesová, P.; Konečná, H.; Fajkusová, L.; Zdráhal, Z. A combined approach for the study of histone deacetylase inhibitors. Mol. Biosyst. 2012, 8, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Lochmanová, G.; Ihnatová, I.; Kuchaříková, H.; Brabencová, S.; Zachová, D.; Fajkus, J.; Fojtová, M. Different Modes of Action of Genetic and Chemical Downregulation of Histone Deacetylases with Respect to Plant Development and Histone Modifications. Int. J. Mol. Sci. 2019, 20, 5093. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Vizcaíno, J.A. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Sehested, M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2007, 409, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Moser, M.A.; Meunier, D.; Fischer, C.; Machat, G.; Mattes, K.; Seiser, C. Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. EMBO J. 2013, 32, 3176–3191. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; von Mering, C. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose Metabolism as a Target of Histone Deacetylase Inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Meneceur, S.; Hommel, K.; Schulz, W.A.; Niegisch, G. Downregulation of Cell Cycle and Checkpoint Genes by Class I HDAC Inhibitors Limits Synergism with G2/M Checkpoint Inhibitor MK-1775 in Bladder Cancer Cells. Genes 2021, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, Y.; Liu, S.; Lu, J.; Huang, B.; Zhang, Y. HDAC inhibitor PAC-320 induces G2/M cell cycle arrest and apoptosis in human prostate cancer. Oncotarget 2017, 9, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Luchenko, V.L.; Litman, T.; Chakraborty, A.R.; Heffner, A.; Devor, C.; Wilkerson, J.; Bates, S.E. Histone deacetylase inhibitor-mediated cell death is distinct from its global effect on chromatin. Mol. Oncol. 2014, 8, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Gomes, I.D.; Wambua, M.K.; Pflum, M.K.H. HDAC Inhibitor-Induced Mitotic Arrest Is Mediated by Eg5/KIF11 Acetylation. Cell Chem. Biol. 2017, 24, 481–492. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Vega, F.; Medeiros, L.J.; Leventaki, V.; Atwell, C.; Cho-Vega, J.H.; Tian, L.; Rassidakis, G.Z. Activation of Mammalian Target of Rapamycin Signaling Pathway Contributes to Tumor Cell Survival in Anaplastic Lymphoma Kinase–Positive Anaplastic Large Cell Lymphoma. Cancer Res. 2006, 66, 6589–6597. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Yeung, D.; Hadfield, K.; Cook, S.J.; Alexander, D.R. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell Signal 2007, 19, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Rassool, F.V. Chapter Three-HDAC Inhibitors: Roles of DNA Damage and Repair. In Advances in Cancer Research; Grant, S., Ed.; Academic Press: Cambridge, MA, USA, 2012; Histone Deacetylase Inhibitors as Cancer Therapeutics; Volume 116, pp. 87–129. Available online: https://www.sciencedirect.com/science/article/pii/B9780123943873000033 (accessed on 14 January 2022).
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Iesmantavicius, V.; Moustafa, T.; Schölz, C.; Wagner, S.A.; Magnes, C.; Choudhary, C. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol. Syst. Biol. 2014, 10, 716. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Steiner, P. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef]
- Becher, I.; Dittmann, A.; Savitski, M.M.; Hopf, C.; Drewes, G.; Bantscheff, M. Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes. ACS Chem. Biol. 2014, 9, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat Res. Mol. Mech Mutagen. 2013, 750, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Seto, E. A Functional Link Between SIRT1 Deacetylase and NBS1 in DNA Damage Response. Cell Cycle 2007, 6, 2869–2871. [Google Scholar] [CrossRef]
- Eliezer, Y.; Argaman, L.; Rhie, A.; Doherty, A.J.; Goldberg, M. The Direct Interaction between 53BP1 and MDC1 Is Required for the Recruitment of 53BP1 to Sites of Damage. J. Biol. Chem. 2009, 284, 426–435. [Google Scholar] [CrossRef]
- Nelson, G.; Buhmann, M.; von Zglinicki, T. DNA damage foci in mitosis are devoid of 53BP. Cell Cycle 2009, 8, 3379–3383. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskaya, R.; Jackson, S.P. Keeping 53BP1 out of focus in mitosis. Cell Res. 2014, 24, 781–782. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kelly, R.D.W.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Mol. Cell. 2014, 54, 5–16. [Google Scholar] [CrossRef]
- Guo, X.; Bai, Y.; Zhao, M.; Zhou, M.; Shen, Q.; Yun, C.H.; Wang, J. Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Res. 2018, 46, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, O.; Wu, X.; Hwang, S.T.; Akilov, O.E. The synergistic proapoptotic effect of PARP-1 and HDAC inhibition in cutaneous T-cell lymphoma is mediated via Blimp-1. Blood Adv. 2020, 4, 4788–4797. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2017, 9, 3908–3921. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zrimšek, M.; Kuchaříková, H.; Draganić, K.; Dobrovolná, P.; Heiss Spornberger, V.; Winkelmayer, L.; Hassler, M.R.; Lochmanová, G.; Zdráhal, Z.; Egger, G. Quantitative Acetylomics Uncover Acetylation-Mediated Pathway Changes Following Histone Deacetylase Inhibition in Anaplastic Large Cell Lymphoma. Cells 2022, 11, 2380. https://doi.org/10.3390/cells11152380
Zrimšek M, Kuchaříková H, Draganić K, Dobrovolná P, Heiss Spornberger V, Winkelmayer L, Hassler MR, Lochmanová G, Zdráhal Z, Egger G. Quantitative Acetylomics Uncover Acetylation-Mediated Pathway Changes Following Histone Deacetylase Inhibition in Anaplastic Large Cell Lymphoma. Cells. 2022; 11(15):2380. https://doi.org/10.3390/cells11152380
Chicago/Turabian StyleZrimšek, Maša, Hana Kuchaříková, Kristina Draganić, Pavlína Dobrovolná, Verena Heiss Spornberger, Lisa Winkelmayer, Melanie R. Hassler, Gabriela Lochmanová, Zbyněk Zdráhal, and Gerda Egger. 2022. "Quantitative Acetylomics Uncover Acetylation-Mediated Pathway Changes Following Histone Deacetylase Inhibition in Anaplastic Large Cell Lymphoma" Cells 11, no. 15: 2380. https://doi.org/10.3390/cells11152380
APA StyleZrimšek, M., Kuchaříková, H., Draganić, K., Dobrovolná, P., Heiss Spornberger, V., Winkelmayer, L., Hassler, M. R., Lochmanová, G., Zdráhal, Z., & Egger, G. (2022). Quantitative Acetylomics Uncover Acetylation-Mediated Pathway Changes Following Histone Deacetylase Inhibition in Anaplastic Large Cell Lymphoma. Cells, 11(15), 2380. https://doi.org/10.3390/cells11152380