
The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes

Abstract
:1. Introduction
- (1)
- The trapping approach takes advantage of observations in E. coli that inactivated ClpP stays associated with substrates that were selected by ClpX and translocated into the degradation chamber [20], but of course, very abundant soluble matrix proteins will also be observed in subsequent mass spectrometry as contaminants. Given that organisms are adapted to different environments and therefore have different expression profiles, the artifacts should vary upon analysis of many species, while true ClpP degradation substrates should be detected with high consistency.
- (2)
- Global proteome profiling represents a less laborious approach that can quantify steady-state accumulations also in complex tissues. However, it remains unclear if any protein accumulation is due to transcriptional or translational activation or to retarded protein turnover due to other reasons, so many effects may be indirect and may not represent true CLPP targets.
- (3)
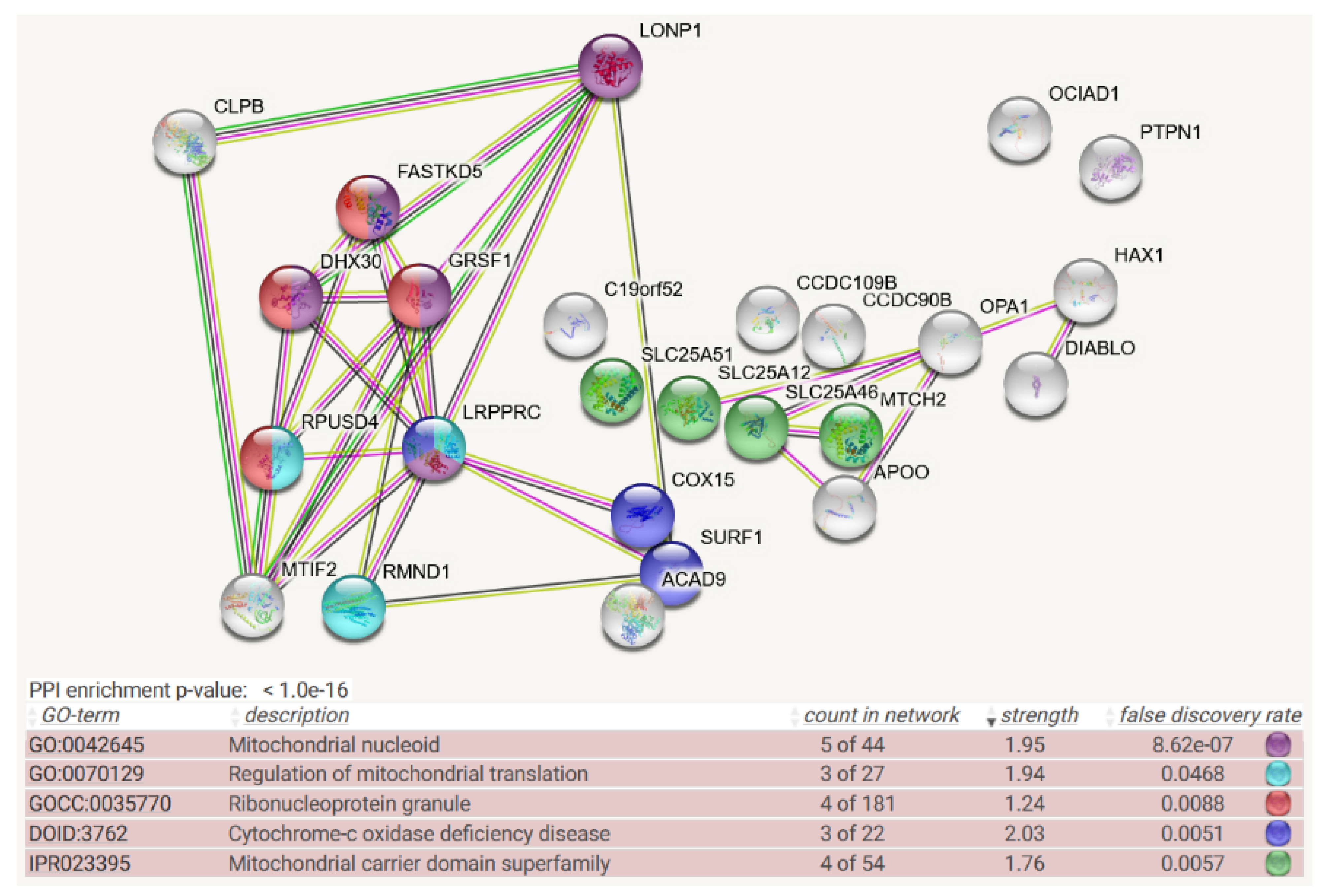
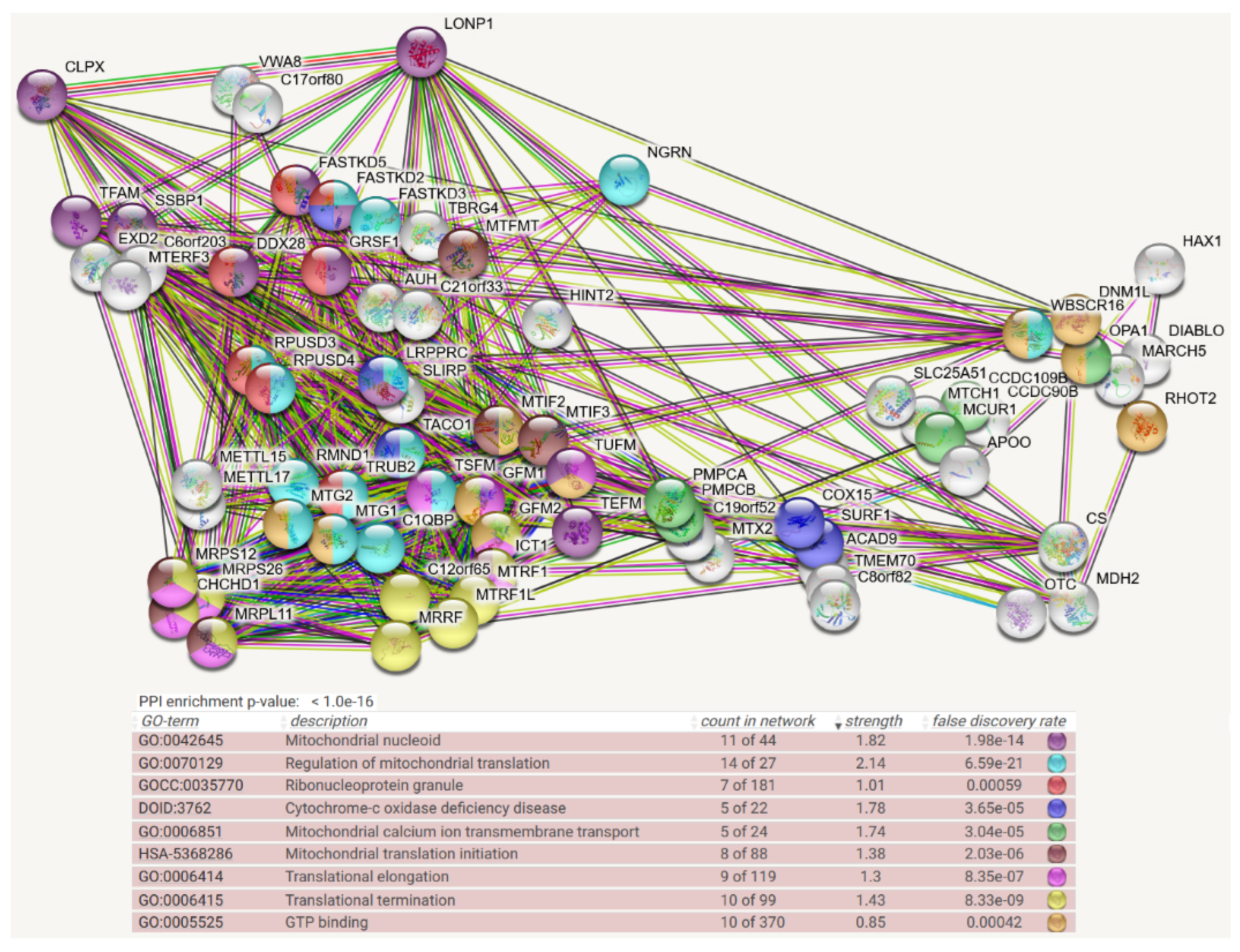
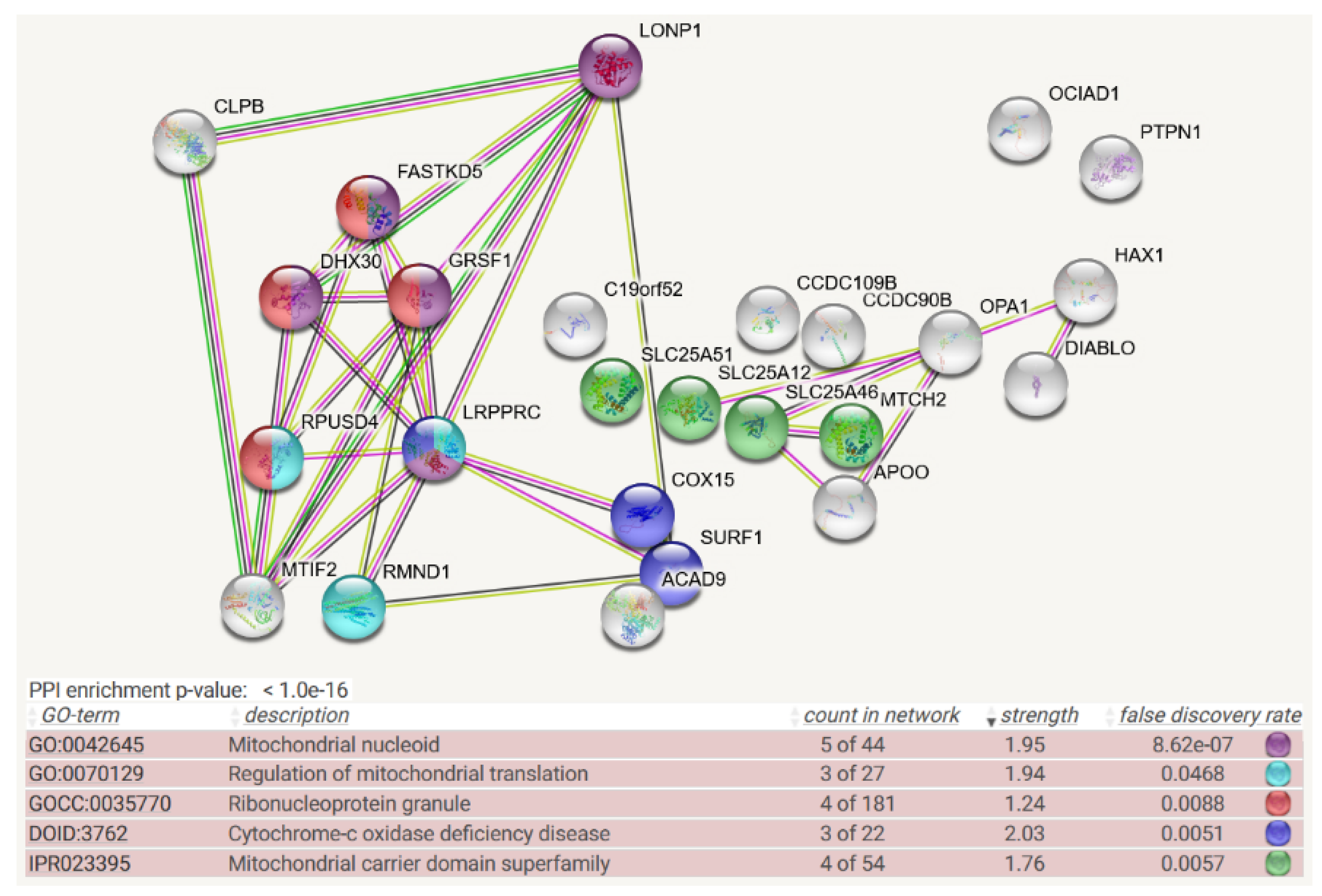
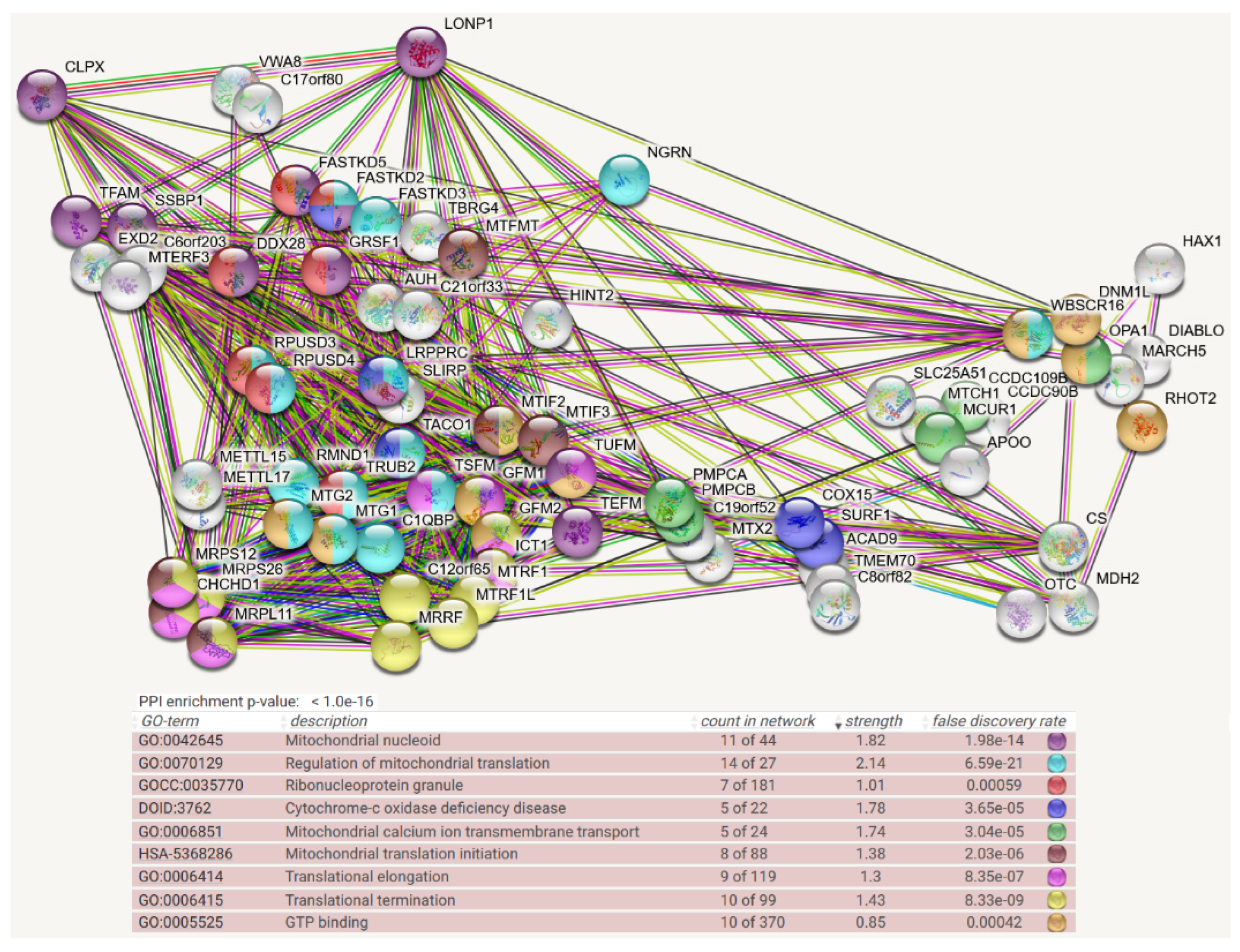
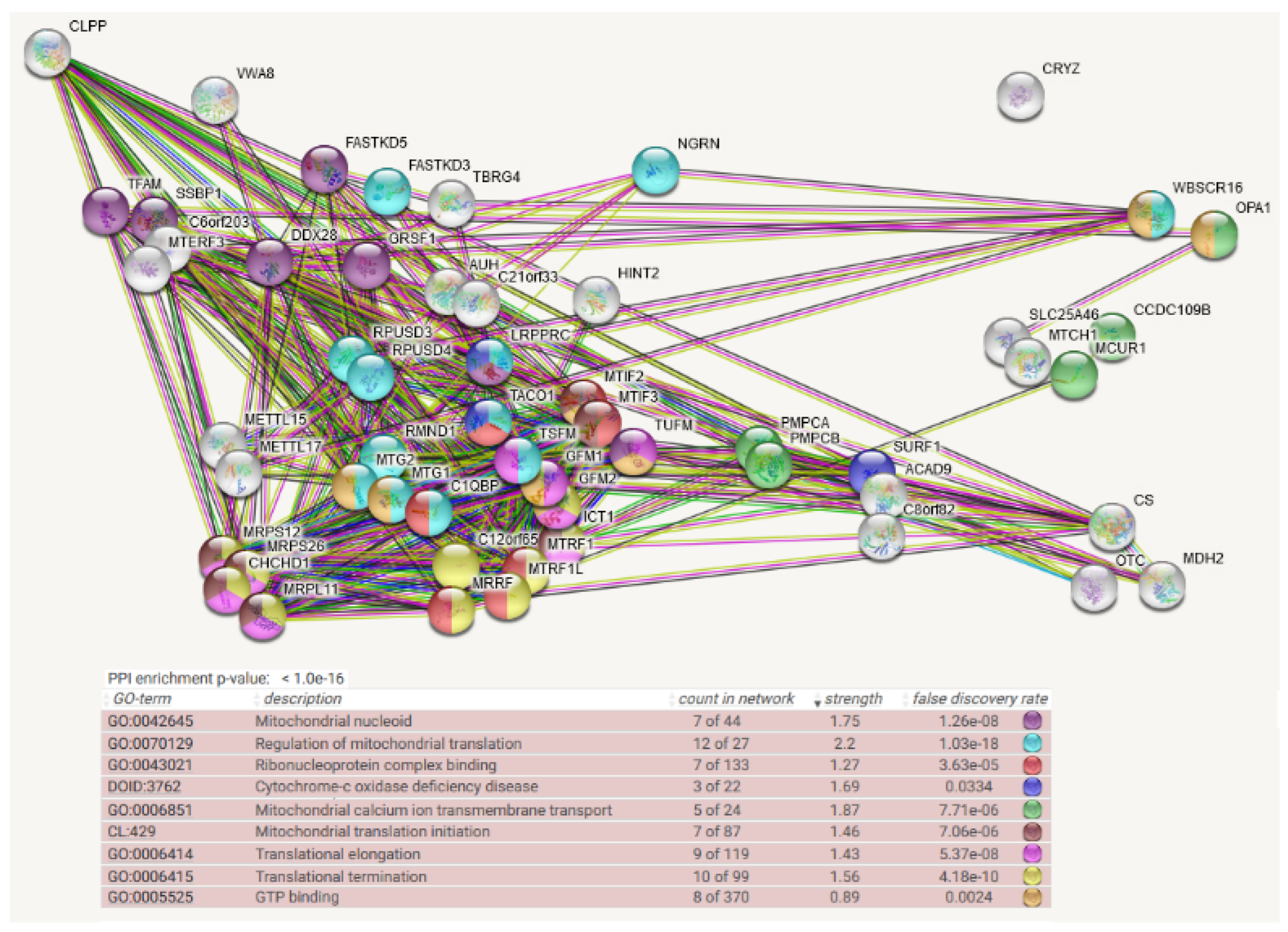
- Protein–protein interaction data may be useful, given that the association of a misfolded target protein with an unfoldase/disaggregase or with a peptidase during the degradation process would be more stable than the “kiss-and-run” interaction of targets with their protein kinases. Therefore, it may be possible to identify such associations by biochemical high-confidence interaction studies. As an abundant source of observations on CLPB/CLPX/CLPP interactomes, we now decided to employ the Homo sapiens mitochondrial proximity interaction network detected by the BioID technique (Table S4 in [21]) and evaluate statistically/visually in a meta-analysis any enrichments, using the STRING webserver (https://string-db.org/ (accessed on 20 July 2022)) [22]. This effort complements our earlier work, where we had taken the complete BioGRID dataset into account [15]. However, the use of tagged and overexpressed recombinant proteins may trigger artificial interactions that occur in non-physiological sub-compartments of the cell.
- (4)
- The study of the assembly of endogenous mitochondrial proteins into complexes was optimized recently via blue-native (BN) or high-resolution clear native (CNE) gel electrophoresis in two dimensions or one dimension, followed by mass-spectrometry identification of the components [23]. The analysis of comigration patterns has greatly helped to define assembly intermediates of the respiratory chain [24], and mitochondrial data generated by such efforts may help to clarify the associations of CLPB and CLPXP. The advantage of this approach is the ability to observe endogenous proteins in their physiological association and their altered assembly/migration within protein complexes upon mutation impact. Their limitation consists of only quite abundant proteins and stable interactions being detectable.
- (5)
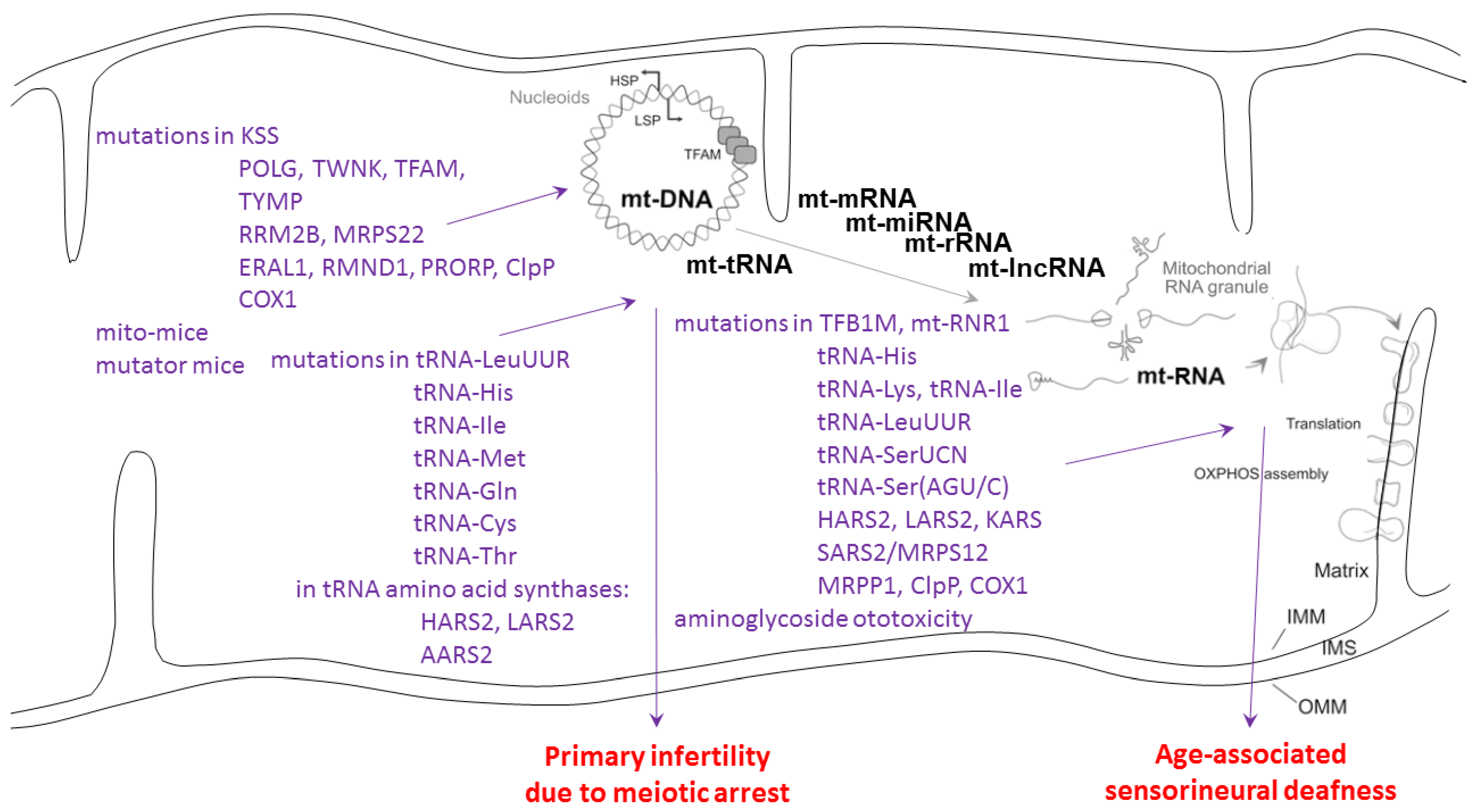
- The notion that CLPXP targets the mitochondrial RNA processing protein complexes is indeed supported by genotype–phenotype correlation data. However, any phenotype overlap is not necessarily triggered by protein–protein interactions that are in common.
2. Substrate-Trapping Assays
3. Proteome Profiles
4. Protein Interaction Data
5. Complexomics Data
6. Genotype–Phenotype Correlation
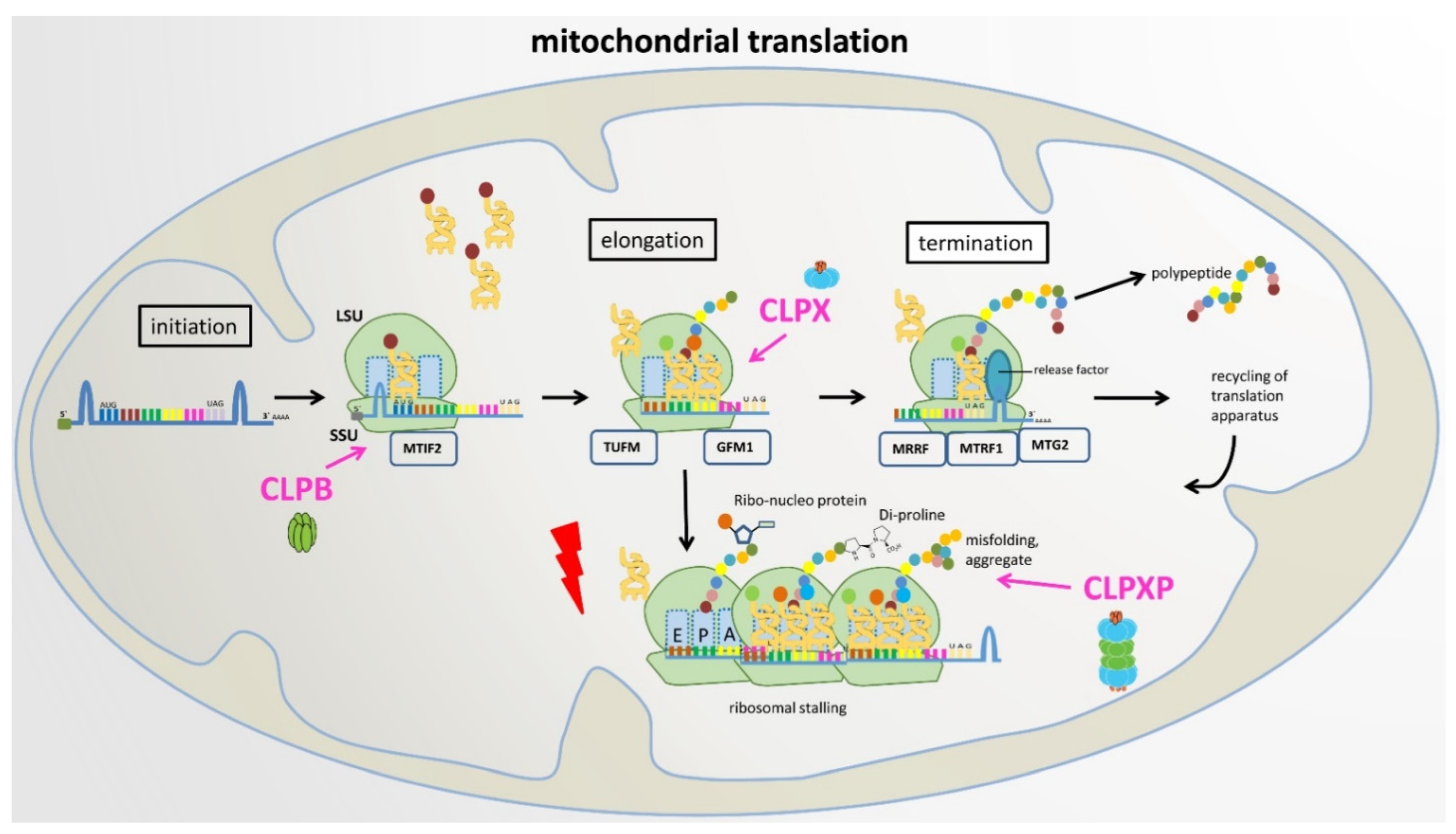
7. Discussion
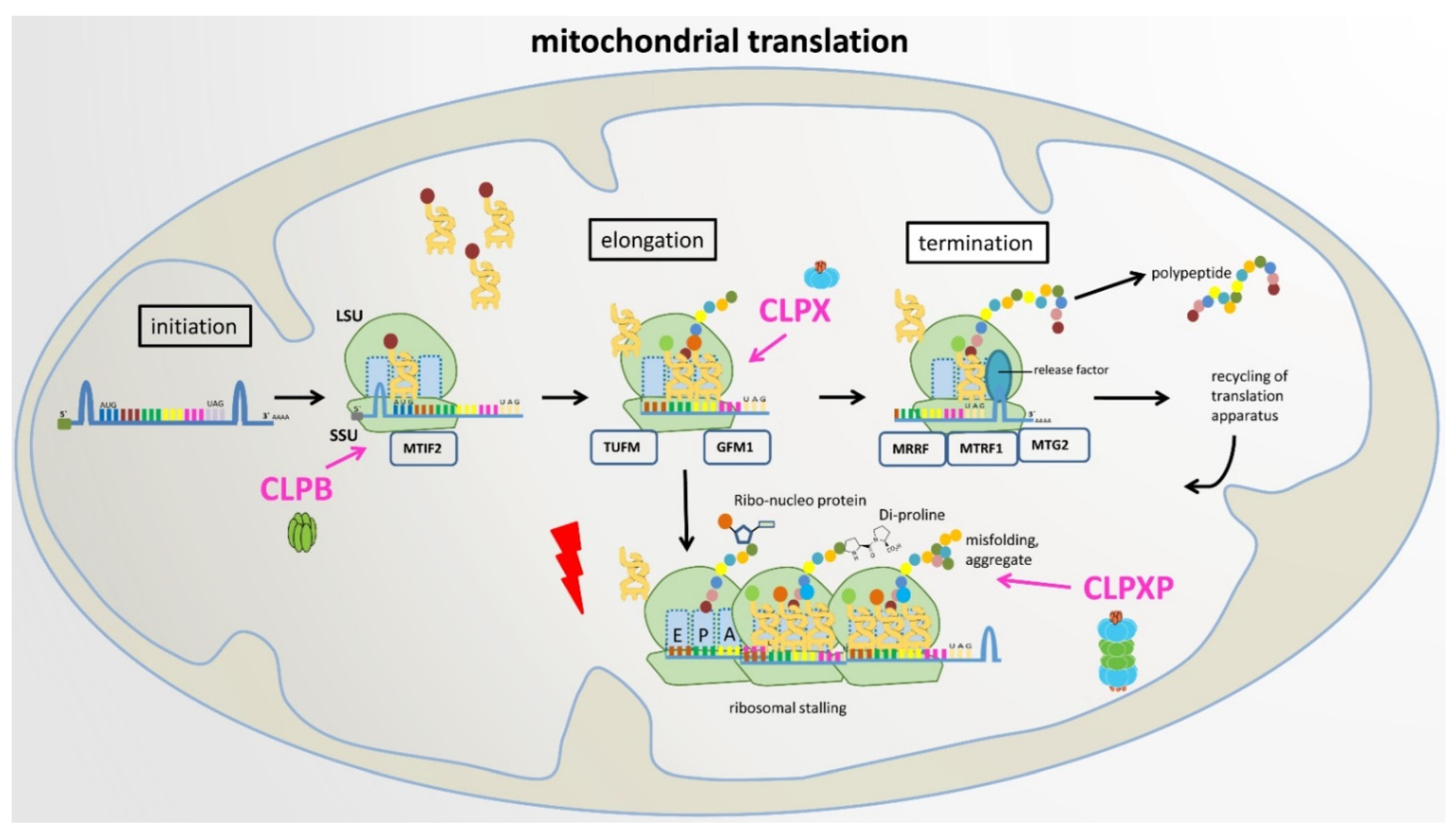
8. Proposal of a Plausible and Testable Scenario
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S | the RNA component in the 30S subunit of prokaryotic ribosomes |
| 30S | prokaryotic small ribosomal subunit |
| 2D-DIGE | two-dimensional difference gel electrophoresis |
| 4Fe-4S | iron-sulfur cluster with [4Fe–4S] stoichiometry |
| ΔclpP | organism that lacks ClpP |
| ΔclpX | organism that lacks ClpX |
| A. thaliana | Arabidopsis thaliana (thale cress) is a small flowering plant |
| AAA+ | protein superfamily of ring-shaped P-loop NTPases |
| AARS2 | protein, alanyl—tRNA synthetase, mitochondrial |
| ACAD9 | protein, acyl-CoA dehydrogenase family member 9 mitochondrial |
| ACADSB | protein, short/branched chain specific acyl-CoA dehydrogenase |
| AcCoA | metabolite, acetyl-coenzyme-A |
| ACO2 | protein, aconitase 2, mitochondrial |
| ACSS1 | protein, acyl-CoA synthetase short chain family member 1 |
| ADP | nucleotide, adenosine di-phosphate |
| ALAS | protein, aminolevulinic acid synthase |
| ALAS1 | protein, Delta-aminolevulinate synthase 1 |
| ALDH6A1 | protein, aldehyde dehydrogenase 6 family, member A1 |
| AML | acute myeloid leukemia |
| APEX | ascorbic acid peroxidase |
| ATP | nucleotide, adenosine tri-phosphate |
| ATPase | enzymes that catalyze the decomposition of ATP into ADP |
| AUH | protein, AU RNA binding methylglutaconyl-CoA hydratase |
| B. subtilis | Bacillus subtilis is a Gram-positive, catalase-positive bacterium |
| BioID | biotinylation identification, a method to detect protein interactions |
| BN | blue native |
| C12orf65 | chromosome 12 open reading frame 65 = MTRFR |
| C1QBP | complement C1q binding protein |
| C. crescentus | Caulobacter crescentus is a Gram-negative, oligotrophic bacterium |
| CARS2 | protein, cysteinyl-tRNA synthetase 2, mitochondrial |
| CBR4 | protein, carbonyl reductase 4 |
| CCDC109B | mitochondrial calcium uniporter dominant negative beta subunit |
| CCDC90B | coiled-coil domain-containing protein 90B, mitochondrial |
| CDP | nucleotide, cytidine di-phosphate |
| CHCHD1 | coiled-coil-helix-coiled-coil-helix domain containing 1 protein |
| ClpA | caseinolytic mitochondrial matrix peptidase chaperone subunit A |
| CLPB | caseinolytic mitochondrial matrix peptidase chaperone subunit B |
| CLPP | caseinolytic mitochondrial matrix peptidase chaperone subunit P |
| CLPX | caseinolytic mitochondrial matrix peptidase chaperone subunit X |
| CNE | clear native |
| CODAS | syndrome, cerebral, ocular, dental, auricular, skeletal anomalies |
| COX1 | protein, mitochondrially encoded cytochrome C oxidase I |
| COX15 | COX15 (yeast) homolog, cytochrome C oxidase assembly protein |
| COX5A | cytochrome C oxidase subunit 5A, mitochondrial |
| COX5B | cytochrome C oxidase subunit 5B, mitochondrial |
| COX6A1 | cytochrome C oxidase subunit 6A1 |
| COXPD1 | combined oxidative phosphorylation defect type 1 |
| COXPD3 | combined oxidative phosphorylation defect type 3 |
| COXPD39 | combined oxidative phosphorylation defect type 39 |
| COXPD4 | combined oxidative phosphorylation defect type 4 |
| CP29 | chloroplast RNA-binding protein 29, in A. thaliana |
| CS | protein, citrate synthase, mitochondrial |
| DEAD-box | proteins with a DEAD (Asp-Glu-Ala-Asp) motif are RNA helicases |
| DFNA4 | deafness, autosomal dominant, type 4 |
| DFNA67 | autosomal dominant nonsyndromic deafness 67 |
| dGTP | nucleotide precursor, deoxy-guanosine triphosphate |
| DHX30 | protein, ATP-dependent DExH-box helicase 30 |
| DNA | deoxyribo-nucleic acid, polymer of polynucleotide chains |
| DNM1L | dynamin-1-like protein, a GTPase |
| dTTP | nucleotide precursor, deoxy-thymidine triphosphate |
| E. coli | Escherichia coli, a Gram-negative, facultative anaerobic bacterium |
| EFG2 | elongation factor G 2, mitochondrial = GFM2 |
| ERAL1 | protein, era-like 12S mitochondrial rRNA chaperone 1 |
| EXD2 | protein, exonuclease 3′-5′ domain containing 2 |
| FAD | coenzyme, flavin adenine dinucleotide |
| FASTKD2 | protein, FAST kinase domain-containing protein 2, mitochondrial |
| FASTKD3 | protein, FAST kinase domain-containing protein 3, mitochondrial |
| FASTKD4 | protein, FAST kinase domain-containing protein 4, mitochondrial |
| FeS | iron-sulfur cluster |
| GATB | protein, glutamyl-tRNA amidotransferase subunit B |
| GDP | nucleotide, guanosine diphosphate |
| GFM1 | protein, mitochondrial elongation factor G1 |
| GFM2 | protein, mitochondrial elongation factor G2 |
| GO-term | Gene Ontology term |
| GRSF1 | G-rich RNA sequence binding factor 1 |
| GTP | nucleotide, guanosine-5′-triphosphate |
| GWAS | genome-wide association study |
| H. sapiens | Homo sapiens (human), the most abundant primate species |
| HARS2 | protein, histidyl-tRNA synthetase 2, mitochondrial |
| HINT2 | histidine triad nucleotide-binding protein 2, mitochondrial |
| HNRNPA2B1 | heterogeneous nuclear ribonucleoprotein A2/B1 |
| Hsp100 | heat shock proteins with size around 100 kDa |
| HSPA9 | heat shock protein family A (Hsp70) member 9 |
| IARS2 | isoleucyl-tRNA synthetase 2, mitochondrial |
| ICT1 | peptidyl-tRNA hydrolase ICT1, mitochondrial = MRPL58 |
| IDH3B | isocitrate dehydrogenase (NAD(+)) 3 non-catalytic subunit beta |
| IMM | inner mitochondrial membrane |
| IMS | inter-membrane space, mitochondrial |
| KARS | lysyl-tRNA synthetase 1 |
| kDa | kiloDalton |
| KSS | Kearns–Sayre syndrome |
| LARS2 | Leucyl-tRNA synthetase 2, mitochondrial |
| LONP1 | mitochondrial Lon protease-like protein 1 |
| LRPPRC | leucine-rich pentatricopeptide-repeat containing protein |
| M. musculus | Mus musculus (mouse) is a small mammal of the order Rodentia |
| MALDI-TOF | matrix-assisted laser-desorption-Ionization, with time-of-flight |
| MARCH5 | membrane associated ring-CH-type finger 5 protein |
| MC4DN5 | mitochondrial complex IV deficiency, nuclear type 5 |
| MC4DN6 | mitochondrial complex IV deficiency, nuclear type 6 |
| MDH2 | protein, malate dehydrogenase 2, mitochondrial |
| MEF | mouse embryonic fibroblasts |
| METTL15 | 12S rRNA M4C methyltransferase protein |
| METTL17 | methyltransferase-like protein 17, mitochondrial |
| mRNA | messenger RNA |
| MRPP1 | mitochondrial ribonuclease P protein 1 = tRNA (adenine-guanine(9)-N(1)) methyltransferase TRMT10C |
| MRPS12 | mitochondrial small ribosomal subunit protein US12m |
| MRPS22 | mitochondrial small ribosomal subunit protein MS22 |
| MRPS26 | mitochondrial small ribosomal subunit protein MS26 |
| MRPS7 | mitochondrial small ribosomal subunit protein US7m |
| MRPL11 | mitochondrial large ribosomal subunit protein UL11m |
| MRPL13 | mitochondrial large ribosomal subunit protein UL13m |
| MRPL18 | mitochondrial large ribosomal subunit protein UL18m |
| MRRF | ribosome-recycling factor, mitochondrial |
| mtDNA | mitochondrial DNA |
| MTERF3 | mitochondrial transcription termination factor 3 |
| MTFMT | mitochondrial methionyl-tRNA formyl-transferase protein |
| MTG1 | mitochondrial ribosome associated GTPase 1 |
| MTG2 | mitochondrial ribosome associated GTPase 2 |
| mt-HSP70 | mitochondrial heat shock protein 70 kDa = HSPA9 |
| MTIF2 | mitochondrial translational initiation factor 2 |
| MTIF3 | mitochondrial translational initiation factor 3 |
| mt-lncRNA | mitochondrial long non-coding RNA |
| mt-mRNA | mitochondrial messenger RNA |
| mt-rRNA | mitochondrial ribosomal RNA |
| mt-tRNA | mitochondrial transfer RNA |
| MTRF1 | mitochondrial translation release factor 1 |
| MTRF1L | mitochondrial translation release factor 1 like |
| MTRFR | mitochondrial translation release factor in rescue |
| mtRNA | mitochondrial RNA |
| MTX2 | mitochondrial outer membrane import complex protein 2 |
| MUT | methylmalonyl-CoA mutase protein, mitochondrial |
| NAD+ | nicotinamide adenine dinucleotide, oxidized |
| NADH | nicotinamide adenine dinucleotide, reduced |
| NADP+ | nicotinamide adenine dinucleotide phosphate, oxidized |
| NADPH | nicotinamide adenine dinucleotide phosphate, reduced |
| NARS2 | asparaginyl-tRNA synthetase 2, mitochondrial |
| NDUFA4 | cytochrome C oxidase subunit FA4 |
| NDUFAF1 | NADH:ubiquinone oxidoreductase complex assembly factor 1 |
| NEDMIAL | Neurodevelopmental disorder with severe motor impairment and absent language |
| NFS1 | nitrogen-fixing bacteria S-like protein |
| NGRN | neugrin, neurite outgrowth associated |
| OAT | ornithine Aminotransferase |
| OMM | outer mitochondrial membrane |
| OPA1 | optic atrophy 1 protein, mitochondrial dynamin-like GTPase |
| OTC | ornithine transcarbamylase protein |
| OXPHOS | oxidative phosphorylation (respiratory) chain complexes |
| P. anserina | Podospora anserine, a filamentous ascomycete fungus |
| P5P | cofactor, pyridoxal-5′-phosphate |
| PEO1 | progressive external ophthalmoplegia 1 protein = Twinkle |
| PET112 | glutamyl-tRNA amidotransferase subunit B = GATB |
| PMID | Public Library of Medicine (PubMed) identification number |
| PMPCA | peptidase, mitochondrial processing subunit alpha |
| PMPCB | peptidase, mitochondrial processing subunit beta |
| POLDIP2 | DNA polymerase delta interacting protein 2 |
| POLG | polymerase gamma catalytic subunit, mitochondrial |
| POLG2 | mitochondrial DNA polymerase, accessory subunit |
| PPI | peptidyl-prolyl-cis-trans-isomerase, rotates proline peptide bond |
| PPI (in STRING) | Protein–protein-interaction |
| PRLTS | Perrault syndrome (autosomal recessive ovarian failure + deafness) |
| PRLTS2 | Perrault syndrome type 2 |
| PRLTS3 | Perrault syndrome type 3 |
| PRLTS4 | Perrault syndrome type 4 |
| PRLTS5 | Perrault syndrome type 5 |
| PRLTS6 | Perrault syndrome type6 |
| QRSL1 | glutaminyl-tRNA amidotransferase subunit QRSL1 |
| RMND1 | protein, required for meiotic nuclear division 1 homolog |
| RNA | ribonucleic acid |
| RNAse P | RNA cleaving enzyme, for mitochondrial tRNA 5′-end processing |
| RNP | ribonucleoprotein |
| RPS12 | small ribosomal subunit protein ES12 |
| RPSA | small ribosomal subunit protein US2 = 67 KDa laminin receptor |
| RPUSD3 | mitochondrial mRNA pseudouridine synthase D3 |
| RPUSD4 | mitochondrial RNA pseudouridine Synthase D4 |
| RRM2B | ribonucleotide reductase regulatory TP53 inducible subunit M2B |
| rRNA | ribosomal RNA |
| S. aureus | Staphylococcus aureus, Gram-positive aerobic/anaerobic bacterium |
| SAM | S-adenosyl methionine, cosubstrate for methyl-group transfers |
| SARS2 | protein, seryl-tRNA synthetase 2, mitochondrial |
| SDHA | succinate dehydrogenase complex flavoprotein subunit A |
| SDHC | succinate dehydrogenase complex subunit C |
| Ser-UCN | serine encoded by UCU, UCC, UCA, UCG (UCN) triplet |
| SHTM2 | protein, serine hydroxymethyltransferase 2 |
| SLC25A46 | OMM protein, solute carrier family 25, member 46 |
| SLC25A51 | mitochondrial NAD+ transporter, solute carrier family 25 member 51 |
| SLIRP | SRA stem-loop-interacting RNA-binding protein, mitochondrial |
| SSBP1 | single-stranded DNA binding protein 1 |
| ssDNA | single-stranded DNA |
| STRING | search tool for the retrieval of interacting genes/proteins |
| SURF1 | Surfeit locus protein 1, cytochrome C oxidase assembly factor |
| TACO1 | translational activator of cytochrome C oxidase I |
| TBRG4 | transforming growth factor beta regulator 4 = FASTKD4 |
| TCA | tricarboxylic acid cycle |
| TEFM | transcription elongation factor of mitochondria |
| TFAM | mitochondrial transcription factor A |
| THF | cofactor, tetrahydrofolate |
| TMEM70 | transmembrane protein 70, mitochondrial; ATP synthase scaffold |
| tmRNA | transfer and messenger RNA, e.g., in E. coli |
| TPM | transcripts per million |
| TPP | thiamine pyrophosphate |
| tRNA | transfer RNA |
| tRNA-His | tRNA linked to histidine |
| tRNA-Ile | tRNA linked to isoleucine |
| tRNA-LeuCUN | tRNA linked to leucine, with codon CUU, CUC, CUA, CUG (CUN) |
| tRNA-LeuUUR | tRNA linked to leucine, with codon UUA, UUG (UUR) |
| tRNA-SerAGY | tRNA linked to serine, with codon AGU, AGC (AGY) |
| TRUB2 | pseudouridine (Psi) synthase homolog 2, for COXI mt-mRNA |
| TSFM | Ts translation elongation factor, mitochondrial |
| TUFM | Tu translation elongation factor, mitochondrial |
| TWNK | Twinkle mtDNA helicase and primase = PEO1 |
| TYMP | thymidine phosphorylase protein |
| UDP | nucleotide, uridine diphosphate |
| UQCRC1 | ubiquinol-cytochrome C reductase core protein I |
| VWA8 | von Willebrand factor A domain-containing protein 8 |
| WARS2 | tryptophan tRNA ligase 2, mitochondrial |
| YMR31 | yeast mitochondrial ribosomal protein, =alpha-ketoglutarate dehydrogenase subunit |
| ZFE | Zentrale Forschungs-Einrichtung (Central research facility) |
References
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Bezawork-Geleta, A.; Brodie, E.J.; Dougan, D.A.; Truscott, K.N. LON is the master protease that protects against protein aggregation in human mitochondria through direct degradation of misfolded proteins. Sci. Rep. 2015, 5, 17397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rendon, Z.O.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell Biol. 2018, 38, e00412-17. [Google Scholar]
- Strack, P.R.; Brodie, E.J.; Zhan, H.; Schuenemann, V.J.; Valente, L.J.; Saiyed, T.; Lowth, B.R.; Angley, L.M.; Perugini, M.A.; Zeth, K.; et al. Polymerase dselta-interacting protein 38 (PDIP38) modulates the stability and activity of the mitochondrial AAA+ protease CLPXP. Commun. Biol. 2020, 3, 646. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Kohli, A.; Barcena, C.; Lopez-Otin, C.; Heidler, J.; Wittig, I.; Auburger, G. Global Proteome of LonP1+/− Mouse Embryonal Fibroblasts Reveals Impact on Respiratory Chain, but No Interdependence between Eral1 and Mitoribosomes. Int. J. Mol. Sci. 2019, 20, 4523. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; De Gaetano, A.; Mandrioli, M.; Van Tongeren, E.; Bortolotti, C.A.; Cossarizza, A.; Pinti, M. The biology of Lonp1: More than a mitochondrial protease. Int. Rev. Cell Mol. Biol. 2020, 354, 1–61. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Bianchini, E.; Borella, R.; De Biasi, S.; Nasi, M.; Boraldi, F.; Cossarizza, A.; Pinti, M. Impaired Mitochondrial Morphology and Functionality in Lonp1(wt/-) Mice. J. Clin. Med. 2020, 9, 1783. [Google Scholar] [CrossRef]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013, 4, 1397. [Google Scholar] [CrossRef] [Green Version]
- Frees, D.; Chastanet, A.; Qazi, S.; Sorensen, K.; Hill, P.; Msadek, T.; Ingmer, H. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol. Microbiol. 2004, 54, 1445–1462. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, E.C.; Glover, J.R.; Singer, M.A.; Lindquist, S. HSP100/Clp proteins: A common mechanism explains diverse functions. Trends Biochem. Sci. 1996, 21, 289–296. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife 2020, 9, e52774. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Key, J.; Torres-Odio, S.; Bach, N.C.; Gispert, S.; Koepf, G.; Reichlmeir, M.; West, A.P.; Prokisch, H.; Freisinger, P.; Newman, W.G.; et al. Inactivity of Peptidase ClpP Causes Primary Accumulation of Mitochondrial Disaggregase ClpX with Its Interacting Nucleoid Proteins, and of mtDNA. Cells 2021, 10, 3354. [Google Scholar] [CrossRef]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gersch, M.; Famulla, K.; Dahmen, M.; Gobl, C.; Malik, I.; Richter, K.; Korotkov, V.S.; Sass, P.; Rubsamen-Schaeff, H.; Madl, T.; et al. AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun. 2015, 6, 6320. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, M.; Hubner, I.; Sieber, S.A. Tailored phenyl esters inhibit ClpXP and attenuate Staphylococcus aureus alpha-hemolysin secretion. ChemBioChem 2022. [Google Scholar] [CrossRef]
- Brotz-Oesterhelt, H.; Beyer, D.; Kroll, H.P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Burton, R.E.; Burton, B.M.; Sauer, R.T.; Baker, T.A. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol. Cell 2000, 5, 639–648. [Google Scholar] [CrossRef]
- Antonicka, H.; Lin, Z.Y.; Janer, A.; Aaltonen, M.J.; Weraarpachai, W.; Gingras, A.C.; Shoubridge, E.A. A High-Density Human Mitochondrial Proximity Interaction Network. Cell Metab. 2020, 32, 479–497.e9. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Wittig, I.; Malacarne, P.F. Complexome Profiling: Assembly and Remodeling of Protein Complexes. Int. J. Mol. Sci. 2021, 22, 7809. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neher, S.B.; Villen, J.; Oakes, E.C.; Bakalarski, C.E.; Sauer, R.T.; Gygi, S.P.; Baker, T.A. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol. Cell 2006, 22, 193–204. [Google Scholar] [CrossRef]
- Courcelle, J.; Khodursky, A.; Peter, B.; Brown, P.O.; Hanawalt, P.C. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 2001, 158, 41–64. [Google Scholar] [CrossRef]
- Flynn, J.M.; Neher, S.B.; Kim, Y.I.; Sauer, R.T.; Baker, T.A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 2003, 11, 671–683. [Google Scholar] [CrossRef]
- LaBreck, C.J.; May, S.; Viola, M.G.; Conti, J.; Camberg, J.L. The Protein Chaperone ClpX Targets Native and Non-native Aggregated Substrates for Remodeling, Disassembly, and Degradation with ClpP. Front. Mol. Biosci. 2017, 4, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, N.H.; Vass, R.H.; Stoddard, P.R.; Shin, D.K.; Chien, P. Identification of ClpP substrates in Caulobacter crescentus reveals a role for regulated proteolysis in bacterial development. Mol. Microbiol. 2013, 88, 1083–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Michalik, S.; Varming, A.N.; Andersen, J.H.; Albrecht, D.; Jelsbak, L.; Krieger, S.; Ohlsen, K.; Hecker, M.; Gerth, U.; et al. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J. Proteome Res. 2013, 12, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Frees, D.; Qazi, S.N.; Hill, P.J.; Ingmer, H. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol. Microbiol. 2003, 48, 1565–1578. [Google Scholar] [CrossRef]
- Fischer, F.; Langer, J.D.; Osiewacz, H.D. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci. Rep. 2015, 5, 18375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kock, H.; Gerth, U.; Hecker, M. The ClpP peptidase is the major determinant of bulk protein turnover in Bacillus subtilis. J. Bacteriol. 2004, 186, 5856–5864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkovic, A.; Ibba, M. Elongation Factor P and the Control of Translation Elongation. Annu Rev. Microbiol. 2017, 71, 117–131. [Google Scholar] [CrossRef]
- Gerth, U.; Kock, H.; Kusters, I.; Michalik, S.; Switzer, R.L.; Hecker, M. Clp-dependent proteolysis down-regulates central metabolic pathways in glucose-starved Bacillus subtilis. J. Bacteriol. 2008, 190, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrand, A.J.; Friedman, D.B.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Proteomic analyses of iron-responsive, Clp-dependent changes in Staphylococcus aureus. Pathog. Dis. 2015, 73, ftv004. [Google Scholar] [CrossRef] [Green Version]
- Stanne, T.M.; Sjogren, L.L.; Koussevitzky, S.; Clarke, A.K. Identification of new protein substrates for the chloroplast ATP-dependent Clp protease supports its constitutive role in Arabidopsis. Biochem. J. 2009, 417, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.T.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzispyrou, I.A.; Alders, M.; Guerrero-Castillo, S.; Zapata Perez, R.; Haagmans, M.A.; Mouchiroud, L.; Koster, J.; Ofman, R.; Baas, F.; Waterham, H.R.; et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum. Mol. Genet. 2017, 26, 2541–2550. [Google Scholar] [CrossRef] [Green Version]
- Hofsetz, E.; Demir, F.; Szczepanowska, K.; Kukat, A.; Kizhakkedathu, J.N.; Trifunovic, A.; Huesgen, P.F. The Mouse Heart Mitochondria N Terminome Provides Insights into ClpXP-Mediated Proteolysis. Mol. Cell Proteom. 2020, 19, 1330–1345. [Google Scholar] [CrossRef]
- Lee, Y.G.; Kim, H.W.; Nam, Y.; Shin, K.J.; Lee, Y.J.; Park, D.H.; Rhee, H.W.; Seo, J.K.; Chae, Y.C. LONP1 and ClpP cooperatively regulate mitochondrial proteostasis for cancer cell survival. Oncogenesis 2021, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol. Cell 2016, 63, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasserre, J.P.; Beyne, E.; Pyndiah, S.; Lapaillerie, D.; Claverol, S.; Bonneu, M. A complexomic study of Escherichia coli using two-dimensional blue native/SDS polyacrylamide gel electrophoresis. Electrophoresis 2006, 27, 3306–3321. [Google Scholar] [CrossRef] [PubMed]
- Butland, G.; Peregrin-Alvarez, J.M.; Li, J.; Yang, W.; Yang, X.; Canadien, V.; Starostine, A.; Richards, D.; Beattie, B.; Krogan, N.; et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 2005, 433, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Arifuzzaman, M.; Maeda, M.; Itoh, A.; Nishikata, K.; Takita, C.; Saito, R.; Ara, T.; Nakahigashi, K.; Huang, H.C.; Hirai, A.; et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome. Res. 2006, 16, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Janga, S.C.; Babu, M.; Diaz-Mejia, J.J.; Butland, G.; Yang, W.; Pogoutse, O.; Guo, X.; Phanse, S.; Wong, P.; et al. Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 2009, 7, e96. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.C.; Hornig-Do, H.T.; Bruni, F.; Chang, J.H.; Jourdain, A.A.; Martinou, J.C.; Falkenberg, M.; Spahr, H.; Larsson, N.G.; Lewis, R.J.; et al. A human mitochondrial poly(A) polymerase mutation reveals the complexities of post-transcriptional mitochondrial gene expression. Hum. Mol. Genet. 2014, 23, 6345–6355. [Google Scholar] [CrossRef] [Green Version]
- Kasashima, K.; Sumitani, M.; Endo, H. Maintenance of mitochondrial genome distribution by mitochondrial AAA+ protein ClpX. Exp. Cell Res. 2012, 318, 2335–2343. [Google Scholar] [CrossRef]
- Capo-Chichi, J.M.; Boissel, S.; Brustein, E.; Pickles, S.; Fallet-Bianco, C.; Nassif, C.; Patry, L.; Dobrzeniecka, S.; Liao, M.; Labuda, D.; et al. Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J. Med. Genet. 2015, 52, 303–311. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Zietkiewicz, S.; Kousi, M.; Szklarczyk, R.; Haack, T.B.; Gersting, S.W.; Muntau, A.C.; Rakovic, A.; Renkema, G.H.; Rodenburg, R.J.; et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am. J. Hum. Genet. 2015, 96, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Kanabus, M.; Shahni, R.; Saldanha, J.W.; Murphy, E.; Plagnol, V.; Hoff, W.V.; Heales, S.; Rahman, S. Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation. J. Inherit. Metab Dis. 2015, 38, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Senft, K.; Heidler, J.; Herholz, M.; Kukat, A.; Hohne, M.N.; Hofsetz, E.; Becker, C.; Kaspar, S.; Giese, H.; et al. A salvage pathway maintains highly functional respiratory complex I. Nat. Commun. 2020, 11, 1643. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Wevers, R.A. CLPB Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Pronicka, E.; Ropacka-Lesiak, M.; Trubicka, J.; Pajdowska, M.; Linke, M.; Ostergaard, E.; Saunders, C.; Horsch, S.; van Karnebeek, C.; Yaplito-Lee, J.; et al. A scoring system predicting the clinical course of CLPB defect based on the foetal and neonatal presentation of 31 patients. J. Inherit. Metab. Dis. 2017, 40, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, S.B.; Zietkiewicz, S.; Guerrero-Castillo, S.; Feichtinger, R.G.; Wagner, M.; Russell, J.; Ellaway, C.; Mroz, D.; Wyszkowski, H.; Weis, D.; et al. Neutropenia and intellectual disability are hallmarks of biallelic and de novo CLPB deficiency. Genet. Med. 2021, 23, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Huang, Y.; Lyu, Y.; Gao, M.; Ma, J.; Gai, Z.; Liu, Y. Clinical and genetic analysis of an infant with 3-methylglutaconic aciduria type VII. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2020, 37, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, A.; Garncarz, W.; Karakoc-Aydiner, E.; Ozen, A.; Kiykim, E.; Yesil, G.; Boztug, K.; Baris, S. Novel CLPB mutation in a patient with 3-methylglutaconic aciduria causing severe neurological involvement and congenital neutropenia. Clin. Immunol. 2016, 165, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.T.; Cupo, R.R.; Wattanasirakul, P.; Spencer, D.H.; Locke, A.E.; Makaryan, V.; Bolyard, A.A.; Kelley, M.L.; Kingston, N.L.; Shorter, J.; et al. Heterozygous variants of CLPB are a cause of severe congenital neutropenia. Blood 2022, 139, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Debray, F.G.; Morin, C.; Janvier, A.; Villeneuve, J.; Maranda, B.; Laframboise, R.; Lacroix, J.; Decarie, J.C.; Robitaille, Y.; Lambert, M.; et al. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J. Med. Genet. 2011, 48, 183–189. [Google Scholar] [CrossRef]
- Lessel, D.; Schob, C.; Kury, S.; Reijnders, M.R.F.; Harel, T.; Eldomery, M.K.; Coban-Akdemir, Z.; Denecke, J.; Edvardson, S.; Colin, E.; et al. De Novo Missense Mutations in DHX30 Impair Global Translation and Cause a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 101, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.O.; Shaybekyan, H.; Zhao, Y.; Kang, X.; Fishbein, G.A.; Khanlou, N.; Alejos, J.C.; Halnon, N.; Satou, G.; Biniwale, R.; et al. Case Report: Whole Exome Sequencing Identifies Compound Heterozygous Variants in TSFM Gene Causing Juvenile Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 798985. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.; Drabkin, M.; Wormser, O.; Yogev, Y.; Dolgin, V.; Shorer, Z.; Gradstein, L.; Shelef, I.; Flusser, H.; Birk, O.S. Phenotypic variability and mutation hotspot in COX15-related Leigh syndrome. Am. J. Med. Genet. A 2020, 182, 1506–1512. [Google Scholar] [CrossRef]
- Ducamp, S.; Luscieti, S.; Ferrer-Cortes, X.; Nicolas, G.; Manceau, H.; Peoc’h, K.; Yien, Y.Y.; Kannengiesser, C.; Gouya, L.; Puy, H.; et al. A mutation in the iron-responsive element of ALAS2 is a modifier of disease severity in a patient suffering from CLPX associated erythropoietic protoporphyria. Haematologica 2021, 106, 2030–2033. [Google Scholar] [CrossRef]
- Kardon, J.R.; Yien, Y.Y.; Huston, N.C.; Branco, D.S.; Hildick-Smith, G.J.; Rhee, K.Y.; Paw, B.H.; Baker, T.A. Mitochondrial ClpX Activates a Key Enzyme for Heme Biosynthesis and Erythropoiesis. Cell 2015, 161, 858–867. [Google Scholar] [CrossRef] [Green Version]
- Kullar, P.J.; Gomez-Duran, A.; Gammage, P.A.; Garone, C.; Minczuk, M.; Golder, Z.; Wilson, J.; Montoya, J.; Hakli, S.; Karppa, M.; et al. Heterozygous SSBP1 start loss mutation co-segregates with hearing loss and the m.1555A>G mtDNA variant in a large multigenerational family. Brain 2018, 141, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, S.; Thompson, K.; Campistol, J.; Colomer, J.; Hardy, S.A.; He, L.; Fernandez-Marmiesse, A.; Palacios, L.; Jou, C.; Jimenez-Mallebrera, C.; et al. Long-term survival in a child with severe encephalopathy, multiple respiratory chain deficiency and GFM1 mutations. Front. Genet. 2015, 6, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasgow, R.I.C.; Thompson, K.; Barbosa, I.A.; He, L.; Alston, C.L.; Deshpande, C.; Simpson, M.A.; Morris, A.A.M.; Neu, A.; Lobel, U.; et al. Novel GFM2 variants associated with early-onset neurological presentations of mitochondrial disease and impaired expression of OXPHOS subunits. Neurogenetics 2017, 18, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; AlSayed, M.; Alhassnan, Z.; Alotaibi, M.; Kayyali, H.; Al-Owain, M.; Shah, A.; Rahbeeni, Z.; Al-Muhaizea, M.A.; et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 2017, 136, 921–939. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wei, X.; Sha, Y.; Liu, W.; Gao, H.; Lin, J.; Li, Y.; Tang, Y.; Wang, Y.; Wang, Y.; et al. Whole-exome sequencing in patients with premature ovarian insufficiency: Early detection and early intervention. J. Ovarian Res. 2020, 13, 114. [Google Scholar] [CrossRef]
- Ahmed, S.; Jelani, M.; Alrayes, N.; Mohamoud, H.S.; Almramhi, M.M.; Anshasi, W.; Ahmed, N.A.; Wang, J.; Nasir, J.; Al-Aama, J.Y. Exome analysis identified a novel missense mutation in the CLPP gene in a consanguineous Saudi family expanding the clinical spectrum of Perrault Syndrome type-3. J. Neurol. Sci. 2015, 353, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.; Urquhart, J.E.; O’Sullivan, J.; Williams, S.G.; Bhaskar, S.S.; Jenkinson, E.M.; Lourenco, C.M.; Heiberg, A.; Pearce, S.H.; Shalev, S.A.; et al. Expanding the genotypic spectrum of Perrault syndrome. Clin. Genet. 2017, 91, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Dursun, F.; Mohamoud, H.S.; Karim, N.; Naeem, M.; Jelani, M.; Kirmizibekmez, H. A Novel Missense Mutation in the CLPP Gene Causing Perrault Syndrome Type 3 in a Turkish Family. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Lerat, J.; Jonard, L.; Loundon, N.; Christin-Maitre, S.; Lacombe, D.; Goizet, C.; Rouzier, C.; Van Maldergem, L.; Gherbi, S.; Garabedian, E.N.; et al. An Application of NGS for Molecular Investigations in Perrault Syndrome: Study of 14 Families and Review of the Literature. Hum. Mutat. 2016, 37, 1354–1362. [Google Scholar] [CrossRef]
- Theunissen, T.E.; Szklarczyk, R.; Gerards, M.; Hellebrekers, D.M.; Mulder-Den Hartog, E.N.; Vanoevelen, J.; Kamps, R.; de Koning, B.; Rutledge, S.L.; Schmitt-Mechelke, T.; et al. Specific MRI Abnormalities Reveal Severe Perrault Syndrome due to CLPP Defects. Front. Neurol. 2016, 7, 203. [Google Scholar] [CrossRef]
- Tucker, E.J.; Rius, R.; Jaillard, S.; Bell, K.; Lamont, P.J.; Travessa, A.; Dupont, J.; Sampaio, L.; Dulon, J.; Vuillaumier-Barrot, S.; et al. Genomic sequencing highlights the diverse molecular causes of Perrault syndrome: A peroxisomal disorder (PEX6), metabolic disorders (CLPP, GGPS1), and mtDNA maintenance/translation disorders (LARS2, TFAM). Hum. Genet. 2020, 139, 1325–1343. [Google Scholar] [CrossRef] [PubMed]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New insights into Perrault syndrome, a clinically and genetically heterogeneous disorder. Hum. Genet. 2022, 141, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Forli, F.; Bruschini, L.; Franciosi, B.; Battini, R.; Marinella, G.; Berrettini, S.; Lazzerini, F. A Rare Case of Perrault Syndrome with Auditory Neuropathy Spectrum Disorder: Cochlear Implantation Treatment and Literature Review. Audiol. Res. 2021, 11, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Pallister, P.D.; Opitz, J.M. The Perrault syndrome: Autosomal recessive ovarian dysgenesis with facultative, non-sex-limited sensorineural deafness. Am. J. Med. Genet. 1979, 4, 239–246. [Google Scholar] [CrossRef]
- Gottschalk, M.E.; Coker, S.B.; Fox, L.A. Neurologic anomalies of Perrault syndrome. Am. J. Med. Genet. 1996, 65, 274–276. [Google Scholar] [CrossRef]
- Amor, D.J.; Delatycki, M.B.; Gardner, R.J.; Storey, E. New variant of familial cerebellar ataxia with hypergonadotropic hypogonadism and sensorineural deafness. Am. J. Med. Genet. 2001, 99, 29–33. [Google Scholar] [CrossRef]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef] [PubMed]
- Morino, H.; Pierce, S.B.; Matsuda, Y.; Walsh, T.; Ohsawa, R.; Newby, M.; Hiraki-Kamon, K.; Kuramochi, M.; Lee, M.K.; Klevit, R.E.; et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014, 83, 2054–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldak, M.; Ozieblo, D.; Pollak, A.; Stepniak, I.; Lazniewski, M.; Lechowicz, U.; Kochanek, K.; Furmanek, M.; Tacikowska, G.; Plewczynski, D.; et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J. Transl. Med. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Ruiz, M.; Garcia-Martinez, A.; Corral-Juan, M.; Perez-Alvarez, A.I.; Plasencia, A.M.; Villamar, M.; Moreno-Pelayo, M.A.; Matilla-Duenas, A.; Menendez-Gonzalez, M.; Del Castillo, I. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: Overlap of TWNK-related recessive disorders. J. Transl. Med. 2019, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Fekete, B.; Pentelenyi, K.; Rudas, G.; Gal, A.; Grosz, Z.; Illes, A.; Idris, J.; Csukly, G.; Domonkos, A.; Molnar, M.J. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med. Genet. 2019, 20, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, K.; Morino, H.; Miyamoto, R.; Matsuda, Y.; Ohsawa, R.; Kanaya, Y.; Tada, Y.; Kurashige, T.; Kawakami, H. Middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant: A case report. BMC Med. Genet. 2020, 21, 68. [Google Scholar] [CrossRef]
- Gotta, F.; Lamp, M.; Geroldi, A.; Trevisan, L.; Origone, P.; Fugazza, G.; Fabbri, S.; Nesti, C.; Rubegni, A.; Morani, F.; et al. A novel mutation of Twinkle in Perrault syndrome: A not rare diagnosis? Ann. Hum. Genet. 2020, 84, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tang, S.; Li, H.; Xu, X.; Lyu, J. Analysis of TWNK variant in a family affected with Perrault syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2020, 37, 739–742. [Google Scholar] [CrossRef]
- Wei, L.; Hou, L.; Ying, Y.Q.; Luo, X.P. A Novel Missense Mutation in TWNK Gene Causing Perrault Syndrome Type 5 in a Chinese Family and Review of the Literature. Pharmgenomics Pers. Med. 2022, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef] [Green Version]
- Karstensen, H.G.; Rendtorff, N.D.; Hindbaek, L.S.; Colombo, R.; Stein, A.; Birkebaek, N.H.; Hartmann-Petersen, R.; Lindorff-Larsen, K.; Hojland, A.T.; Petersen, M.B.; et al. Novel HARS2 missense variants identified in individuals with sensorineural hearing impairment and Perrault syndrome. Eur. J. Med. Genet. 2020, 63, 103733. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.M.; Gerkes, E.H.; Smith, R.J.H.; Molina-Ramirez, L.P.; O’Keefe, R.T.; Newman, W.G. A recurrent missense variant in HARS2 results in variable sensorineural hearing loss in three unrelated families. J. Hum. Genet. 2020, 65, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Jiang, W.; Cao, L.; Na, X.; Yang, J. Two novel likely pathogenic variants of HARS2 identified in a Chinese family with sensorineural hearing loss. Hereditas 2020, 157, 47. [Google Scholar] [CrossRef] [PubMed]
- Souissi, A.; Ben Said, M.; Frikha, F.; Elloumi, I.; Masmoudi, S.; Megarbane, A. Expanding the Clinical and Molecular Spectrum of HARS2-Perrault Syndrome: Identification of a Novel Homozygous Missense Variant in the HARS2 gene. Genet. Test. Mol. Biomark. 2021, 25, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Wang, L.; Peng, H.; Liu, H.; Liu, H.; Yuan, Q.; Lin, Y.; Xu, J.; Pang, X.; Wu, H.; et al. Disruption of Hars2 in Cochlear Hair Cells Causes Progressive Mitochondrial Dysfunction and Hearing Loss in Mice. Front. Cell Neurosci. 2021, 15, 804345. [Google Scholar] [CrossRef]
- Pierce, S.B.; Gersak, K.; Michaelson-Cohen, R.; Walsh, T.; Lee, M.K.; Malach, D.; Klevit, R.E.; King, M.C.; Levy-Lahad, E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013, 92, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Solda, G.; Caccia, S.; Robusto, M.; Chiereghin, C.; Castorina, P.; Ambrosetti, U.; Duga, S.; Asselta, R. First independent replication of the involvement of LARS2 in Perrault syndrome by whole-exome sequencing of an Italian family. J. Hum. Genet. 2016, 61, 295–300. [Google Scholar] [CrossRef]
- Zerkaoui, M.; Demain, L.A.M.; Cherkaoui Jaouad, I.; Ratbi, I.; Amjoud, K.; Urquhart, J.E.; O’Sullivan, J.; Newman, W.G.; Sefiani, A. Marfanoid habitus is a nonspecific feature of Perrault syndrome. Clin. Dysmorphol. 2017, 26, 200–204. [Google Scholar] [CrossRef]
- Kosaki, R.; Horikawa, R.; Fujii, E.; Kosaki, K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am. J. Med. Genet. A 2018, 176, 404–408. [Google Scholar] [CrossRef]
- Al-Jaroudi, D.; Enabi, S.; AlThagafi, M.S. Perrault syndrome with amenorrhea, infertility, Tarlov cyst, and degenerative disc. Gynecol. Endocrinol. 2019, 35, 1037–1039. [Google Scholar] [CrossRef]
- Carminho-Rodrigues, M.T.; Klee, P.; Laurent, S.; Guipponi, M.; Abramowicz, M.; Cao-van, H.; Guinand, N.; Paoloni-Giacobino, A. LARS2-Perrault syndrome: A new case report and literature review. BMC Med. Genet. 2020, 21, 109. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Rudinger-Thirion, J.; Frugier, M.; Wilson, M.; Luig, M.; Alahakoon, T.I.; Nixon, C.Y.; Kirk, E.P.; Roscioli, T.; Lunke, S.; et al. The expanding LARS2 phenotypic spectrum: HLASA, Perrault syndrome with leukodystrophy, and mitochondrial myopathy. Hum. Mutat. 2020, 41, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Xu, H.; Tian, Y.; Liu, D.; Liu, H.; Li, R.; Dou, Q.; Zuo, B.; Zhai, R.; Tang, W.; et al. Perrault syndrome: Clinical report and retrospective analysis. Mol. Genet. Genom. Med. 2020, 8, e1445. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.M.; Antunes, D.; O’Sullivan, J.; Bhaskhar, S.S.; O’Keefe, R.T.; Newman, W.G. A known pathogenic variant in the essential mitochondrial translation gene RMND1 causes a Perrault-like syndrome with renal defects. Clin. Genet. 2018, 94, 276–277. [Google Scholar] [CrossRef]
- Ozieblo, D.; Pazik, J.; Stepniak, I.; Skarzynski, H.; Oldak, M. Two Novel Pathogenic Variants Confirm RMND1 Causative Role in Perrault Syndrome with Renal Involvement. Genes 2020, 11, 1060. [Google Scholar] [CrossRef]
- Hochberg, I.; Demain, L.A.M.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodriguez-Palmero, A.; Schluter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 2021, 108, 2195–2204. [Google Scholar] [CrossRef]
- Usami, S.; Nishio, S. Nonsyndromic Hearing Loss and Deafness, Mitochondrial. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhen, X.; Wu, B.; Wang, J.; Lu, C.; Gao, H.; Qiao, J. Increased Incidence of Mitochondrial Cytochrome C Oxidase 1 Gene Mutations in Patients with Primary Ovarian Insufficiency. PLoS ONE 2015, 10, e0132610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demain, L.A.; Conway, G.S.; Newman, W.G. Genetics of mitochondrial dysfunction and infertility. Clin. Genet. 2017, 91, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Tiosano, D.; Guran, T.; Baris, H.N.; Bayram, Y.; Mory, A.; Shapiro-Kulnane, L.; Hodges, C.A.; Akdemir, Z.C.; Turan, S.; et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum. Mol. Genet. 2018, 27, 1913–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Xia, B.H.; Zhuo, G.C.; Zhang, C.J.; Leng, J.H. Premature ovarian insufficiency may be associated with the mutations in mitochondrial tRNA genes. Endocr. J. 2019, 66, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Del’Olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Kauppila, T.E.S.; Motori, E.; Li, X.; Atanassov, I.; Folz-Donahue, K.; Bonekamp, N.A.; Albarran-Gutierrez, S.; Stewart, J.B.; Larsson, N.G. Increased Total mtDNA Copy Number Cures Male Infertility Despite Unaltered mtDNA Mutation Load. Cell Metab 2017, 26, 429–436 e424. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Sato, A.; Yoshida, K.; Morita, T.; Tanaka, H.; Inoue, S.; Yonekawa, H.; Hayashi, J. Mitochondria-related male infertility. Proc. Natl. Acad. Sci. USA 2006, 103, 15148–15153. [Google Scholar] [CrossRef] [Green Version]
- Crimi, M.; Galbiati, S.; Perini, M.P.; Bordoni, A.; Malferrari, G.; Sciacco, M.; Biunno, I.; Strazzer, S.; Moggio, M.; Bresolin, N.; et al. A mitochondrial tRNA(His) gene mutation causing pigmentary retinopathy and neurosensorial deafness. Neurology 2003, 60, 1200–1203. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G tRNALeu (UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef] [Green Version]
- Shah, Z.H.; Toompuu, M.; Hakkinen, T.; Rovio, A.T.; van Ravenswaay, C.; De Leenheer, E.M.; Smith, R.J.; Cremers, F.P.; Cremers, C.W.; Jacobs, H.T. Novel coding-region polymorphisms in mitochondrial seryl-tRNA synthetase (SARSM) and mitoribosomal protein S12 (RPMS12) genes in DFNA4 autosomal dominant deafness families. Hum. Mutat. 2001, 17, 433–434. [Google Scholar] [CrossRef]
- Wu, S.; Hei, Z.; Zheng, L.; Zhou, J.; Liu, Z.; Wang, J.; Fang, P. Structural analyses of a human lysyl-tRNA synthetase mutant associated with autosomal recessive nonsyndromic hearing impairment. Biochem. Biophys. Res. Commun. 2021, 554, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Hobbie, S.N.; Akshay, S.; Kalapala, S.K.; Bruell, C.M.; Shcherbakov, D.; Bottger, E.C. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 20888–20893. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.H.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. A 2015, 167, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Innes, A.M.; Chudley, A.E.; Reed, M.H.; Shuckett, E.P.; Hildes-Ripstein, G.E.; Greenberg, C.R. Third case of cerebral, ocular, dental, auricular, skeletal anomalies (CODAS) syndrome, further delineating a new malformation syndrome: First report of an affected male and review of literature. Am. J. Med. Genet. 2001, 102, 44–47. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, D.; Zhang, D.; Li, P.; Gao, Y. Mitochondrial Protein Translation: Emerging Roles and Clinical Significance in Disease. Front. Cell Dev. Biol. 2021, 9, 675465. [Google Scholar] [CrossRef] [PubMed]
- Krafczyk, R.; Qi, F.; Sieber, A.; Mehler, J.; Jung, K.; Frishman, D.; Lassak, J. Proline codon pair selection determines ribosome pausing strength and translation efficiency in bacteria. Commun. Biol. 2021, 4, 589. [Google Scholar] [CrossRef] [PubMed]
- Huter, P.; Arenz, S.; Bock, L.V.; Graf, M.; Frister, J.O.; Heuer, A.; Peil, L.; Starosta, A.L.; Wohlgemuth, I.; Peske, F.; et al. Structural Basis for Polyproline-Mediated Ribosome Stalling and Rescue by the Translation Elongation Factor EF-P. Mol. Cell 2017, 68, 515–527.e6. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Bell, T.A.; Barkow, S.R.; Baker, T.A.; Sauer, R.T. Structural basis of ClpXP recognition and unfolding of ssrA-tagged substrates. Elife 2020, 9, e61496. [Google Scholar] [CrossRef] [PubMed]
- Lytvynenko, I.; Paternoga, H.; Thrun, A.; Balke, A.; Muller, T.A.; Chiang, C.H.; Nagler, K.; Tsaprailis, G.; Anders, S.; Bischofs, I.; et al. Alanine Tails Signal Proteolysis in Bacterial Ribosome-Associated Quality Control. Cell 2019, 178, 76–90 e22. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.; Cluet, D.; Ricci, E.P. Ribosome dynamics and mRNA turnover, a complex relationship under constant cellular scrutiny. Wiley Interdiscip Rev. RNA 2021, 12, e1658. [Google Scholar] [CrossRef]
- Kester, J.C.; Kandror, O.; Akopian, T.; Chase, M.R.; Zhu, J.; Rubin, E.J.; Goldberg, A.L.; Fortune, S.M. ClpX Is Essential and Activated by Single-Strand DNA Binding Protein in Mycobacteria. J. Bacteriol. 2021, 203, e00608-20. [Google Scholar] [CrossRef]
- Torres-Odio, S.; Lei, Y.; Gispert, S.; Maletzko, A.; Key, J.; Menissy, S.S.; Wittig, I.; Auburger, G.; West, A.P. Loss of Mitochondrial Protease CLPP Activates Type I IFN Responses through the Mitochondrial DNA-cGAS-STING Signaling Axis. J. Immunol. 2021, 206, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Maletzko, A.; Key, J.; Wittig, I.; Gispert, S.; Koepf, G.; Canet-Pons, J.; Torres-Odio, S.; West, A.P.; Auburger, G. Increased presence of nuclear DNAJA3 and upregulation of cytosolic STAT1 and of nucleic acid sensors trigger innate immunity in the ClpP-null mouse. Neurogenetics 2021, 22, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Rhine, K.; Vidaurre, V.; Myong, S. RNA Droplets. Annu. Rev. Biophys. 2020, 49, 247–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glab, T.K.; Boratynski, J. Potential of Casein as a Carrier for Biologically Active Agents. Top. Curr. Chem. 2017, 375, 71. [Google Scholar] [CrossRef] [Green Version]
- Middendorf, D.; Bindrich, U.; Siemer, C.; Topfl, S.; Heinz, V. Affecting Casein Micelles by Pulsed Electrical Field (PEF) for Inclusion of Lipophilic Organic Compounds. Appl. Sci. 2021, 11, 4611. [Google Scholar] [CrossRef]
- Thomas, M.G.; Martinez Tosar, L.J.; Desbats, M.A.; Leishman, C.C.; Boccaccio, G.L. Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. J. Cell Sci. 2009, 122, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, J.; Hernandez-Alicea, L.; Vass, R.H.; Chien, P. A Phosphosignaling Adaptor Primes the AAA+ Protease ClpXP to Drive Cell Cycle-Regulated Proteolysis. Mol. Cell 2015, 59, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Kardon, J.R.; Moroco, J.A.; Engen, J.R.; Baker, T.A. Mitochondrial ClpX activates an essential biosynthetic enzyme through partial unfolding. Elife 2020, 9, e54387. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trap assays | Proteome profiles | ||||||||||
| E. coli | C. crescentus | S. aureus | P. anserina | H. sapiens AML | B. subtilis | S. aureus | A. thaliana | M. musculus heart | H. sapiens fibroblasts | ||
| PubMed-Ids | 12667450, 16630889 | 23647068 | 12791139 | 26679294 | 26058080 | 15317791, 17981983 | 25743475 | 18754756 | 27797820, 32467259 | 34943861 | |
| nucleotide cofactors/substrates? | |||||||||||
| ACADSB | + | + | + | FAD | |||||||
| ACO2 | + | + | [4Fe-4S] cluster | ||||||||
| AhpC/At3g11630/PRDX | + | + | + | NADH | |||||||
| ClpB | + | + | ATP | ||||||||
| ClpX | + | + | + | + | + | + | + | ATP | |||
| CodY | + | + | GTP | ||||||||
| CS | + | + | - | ||||||||
| DeaD/At5g26742 | + | + | ATP | ||||||||
| DnaJ/DNAJA3 | + | + | ATP | ||||||||
| DnaK/HSPA9 | + | + | + | + | + | ATP | |||||
| FabG/HSD17B4/8 | + | + | + | + | NAD+ | ||||||
| FtsA | + | + | ATP | ||||||||
| FtsZ | + | + | + | GTP | |||||||
| FusA/GFM1 | + | + | + | + | + | + | GTP, RNA-binding | ||||
| GatA | + | + | - | ||||||||
| GatB | + | + | - | ||||||||
| GroEL/At2g28000/HSPD1 | + | + | + | ATP | |||||||
| GyrB | + | + | + | ATP | |||||||
| InfB/MTIF2 | + | + | GTP, RNA-binding | ||||||||
| KatE | + | + | heme | ||||||||
| LipA/LIAS | + | + | [4Fe-4S] cluster, SAM | ||||||||
| McsB | + | + | ATP | ||||||||
| MecA | + | + | - | ||||||||
| MetK | + | + | SAM | ||||||||
| MurA | + | + | UDP | ||||||||
| MurC | + | + | UDP, NADP+ | ||||||||
| NrdA/E/H | + | + | + | ATP, ADP, CDP, UDP, GDP, dGTP, dTTP | |||||||
| OAT | + | + | P5P | ||||||||
| OmpA | + | + | - | ||||||||
| PdhC/DLAT/DLST | + | + | + | FAD, AcCoA, lipoate | |||||||
| PdhD/DLD | + | + | + | NAD+, FAD | |||||||
| Pnp/PNPT1 | + | + | + | ATP, (nucleoside 5′-monophosphate)n | |||||||
| PpiB/At3g62030 | + | + | - | ||||||||
| RecA/DMC1 | + | + | + | ATP, ssDNA-binding | |||||||
| RfaG/GspA | + | + | nucleotide-binding | ||||||||
| Rho | + | + | ATP, RNA-binding | ||||||||
| RplN | + | + | 16S rRNA-binding | ||||||||
| RplS | + | + | 16S rRNA-binding | ||||||||
| RplU | + | + | rRNA-binding | ||||||||
| RpoS/SigB | + | + | DNA/RNA-binding | ||||||||
| SdhB/SDHA/C | + | + | FAD, quinone, FeS-cluster | ||||||||
| SecA | + | + | + | + | ATP, RNP-complex-binding | ||||||
| SHMT2 | + | + | THF, P5P | ||||||||
| Tkt | + | + | TPP | ||||||||
| Tpx | + | + | - | ||||||||
| Tsf/TSFM | + | GTP, RNA-binding | |||||||||
| TufB/A/TUFM | + | + | + | + | GTP, RNA-binding | ||||||
| UvrA | + | + | ATP, ssDNA-binding | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auburger, G.; Key, J.; Gispert, S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells 2022, 11, 2370. https://doi.org/10.3390/cells11152370
Auburger G, Key J, Gispert S. The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells. 2022; 11(15):2370. https://doi.org/10.3390/cells11152370
Chicago/Turabian StyleAuburger, Georg, Jana Key, and Suzana Gispert. 2022. "The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes" Cells 11, no. 15: 2370. https://doi.org/10.3390/cells11152370
APA StyleAuburger, G., Key, J., & Gispert, S. (2022). The Bacterial ClpXP-ClpB Family Is Enriched with RNA-Binding Protein Complexes. Cells, 11(15), 2370. https://doi.org/10.3390/cells11152370