
Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis


Abstract
:1. Introduction
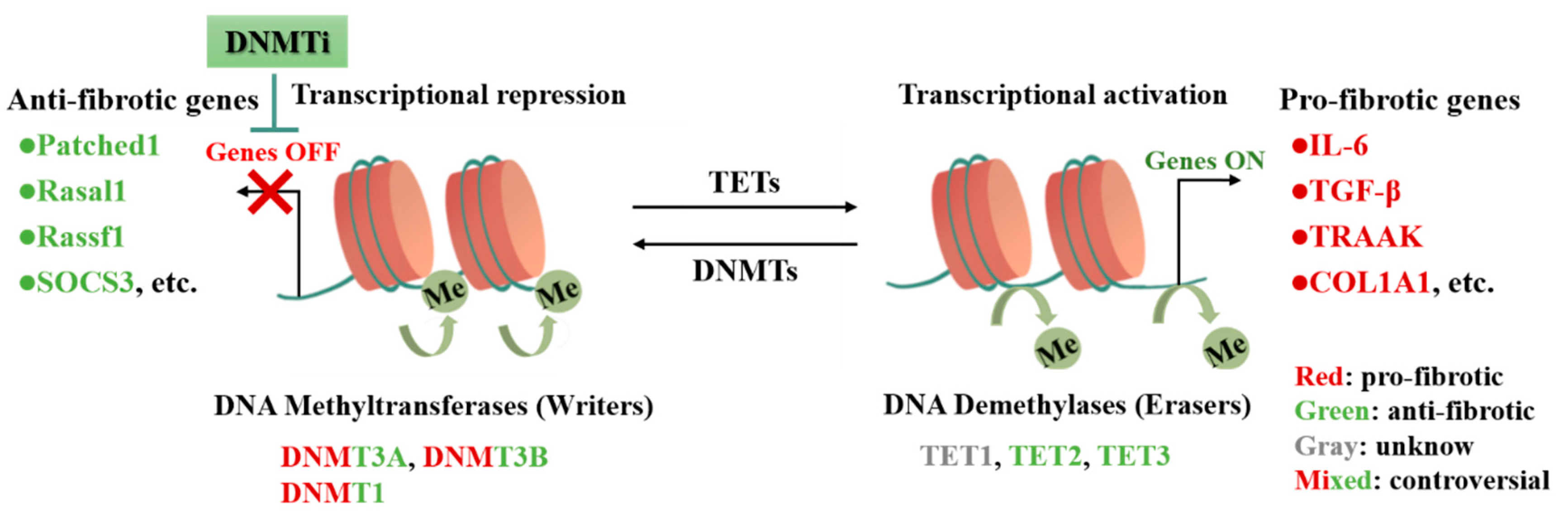
2. DNA Methylation in CFs Activation and Cardiac Fibrosis
2.1. DNA Methylation
2.2. DNMTs in CFs Activation and Cardiac Fibrosis
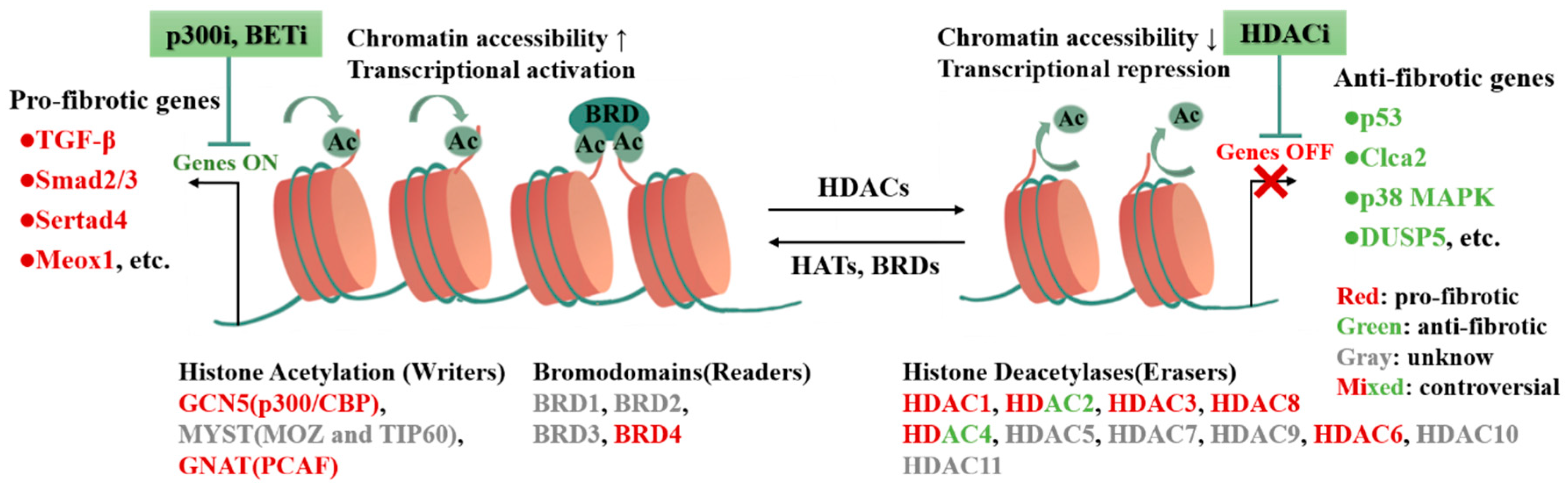
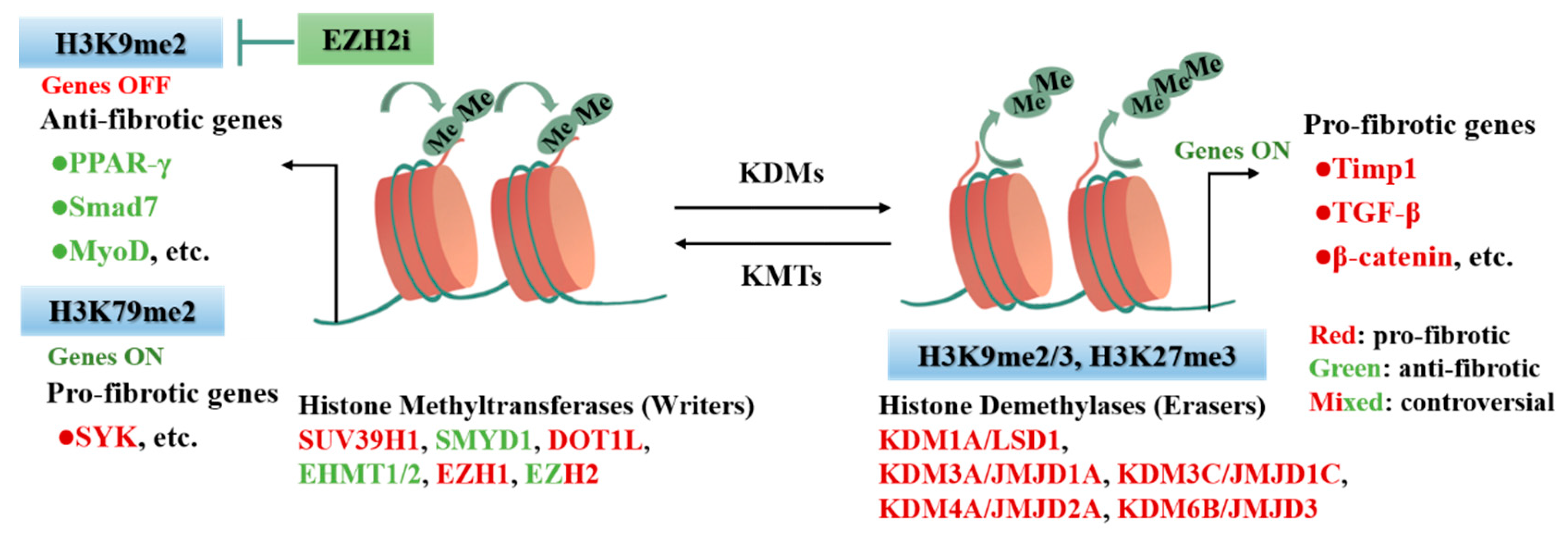
3. Histone Modification in CFs Activation and Cardiac Fibrosis
3.1. Histone Modification
3.2. Histone Acetylation in CFs Activation and Cardiac Fibrosis
3.3. Histone Methylation in CFs Activation and Cardiac Fibrosis
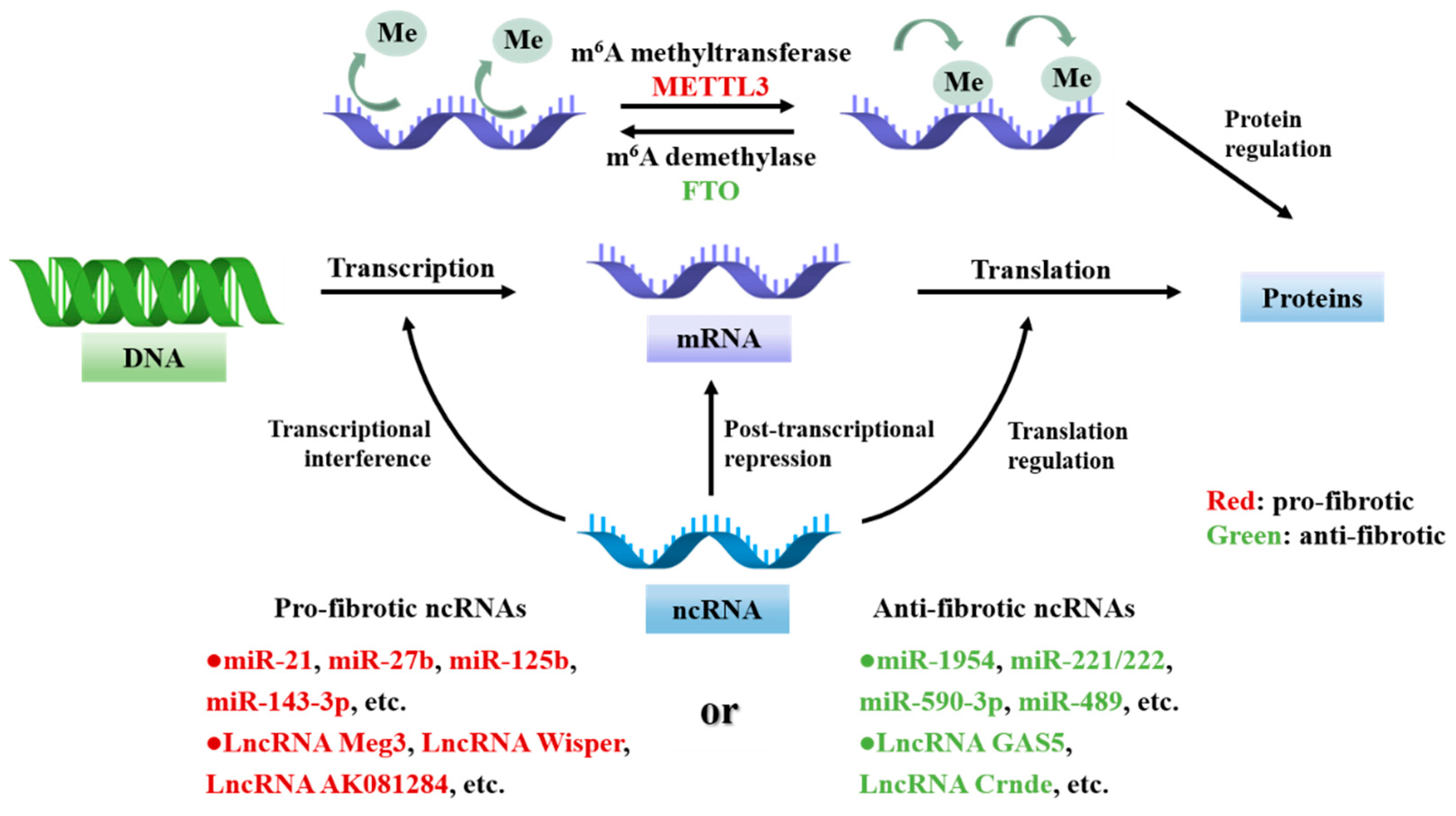
4. RNA in CFs Activation and Cardiac Fibrosis
4.1. Non-Coding RNAs in CFs Activation and Cardiac Fibrosis
4.2. RNA Modifications in CFs Activation and Cardiac Fibrosis
5. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heimlich, J.B.; Bick, A.G. Somatic Mutations in Cardiovascular Disease. Circ. Res. 2022, 130, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; López de Juan Abad, B.; Cheng, K. Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv. Drug Deliv. Rev. 2021, 173, 504–519. [Google Scholar] [CrossRef]
- Burke, R.M.; Dirkx, R.A., Jr.; Quijada, P.; Lighthouse, J.K.; Mohan, A.; O’Brien, M.; Wojciechowski, W.; Woeller, C.F.; Phipps, R.P.; Alexis, J.D.; et al. Prevention of Fibrosis and Pathological Cardiac Remodeling by Salinomycin. Circ. Res. 2021, 128, 1663–1678. [Google Scholar] [CrossRef]
- Tarbit, E.; Singh, I.; Peart, J.N.; Rose’Meyer, R.B. Biomarkers for the identification of cardiac fibroblast and myofibroblast cells. Heart Fail. Rev. 2019, 24, 1–15. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- Alexanian, M.; Przytycki, P.F.; Micheletti, R.; Padmanabhan, A.; Ye, L.; Travers, J.G.; Gonzalez-Teran, B.; Silva, A.C.; Duan, Q.; Ranade, S.S.; et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 2021, 595, 438–443. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Papait, R.; Serio, S.; Condorelli, G. Role of the Epigenome in Heart Failure. Physiol. Rev. 2020, 100, 1753–1777. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Qiu, X.; Liu, H.; Gan, C.; Tan, Z.; Xie, Y.; Wang, Y.; Ye, T. Epigenetic regulation in fibrosis progress. Pharmacol. Res. 2021, 173, 105910. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Tharp, C.A.; Rubino, M.; McKinsey, T.A. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132, e148554. [Google Scholar] [CrossRef]
- van der Harst, P.; de Windt, L.J.; Chambers, J.C. Translational Perspective on Epigenetics in Cardiovascular Disease. J. Am. Coll. Cardiol. 2017, 70, 590–606. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Nguyen, N.B.; Pezhouman, A.; Ardehali, R. Cardiac fibrosis: Potential therapeutic targets. Transl. Res. J. Lab. Clin. Med. 2019, 209, 121–137. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Parry, A.; Rulands, S.; Reik, W. Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 2021, 22, 59–66. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Song, Z.Y.; Ding, X.S.; Yang, J.J.; Shi, K.H.; Li, J. Epigenetic signatures in cardiac fibrosis, special emphasis on DNA methylation and histone modification. Heart Fail. Rev. 2018, 23, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Shi, P.; Zhao, X.D.; Xuan, H.Y.; Gong, W.H.; Ding, X.S. DNMT1 deregulation of SOCS3 axis drives cardiac fibroblast activation in diabetic cardiac fibrosis. J. Cell. Physiol. 2021, 236, 3481–3494. [Google Scholar] [CrossRef]
- Xu, S.S.; Ding, J.F.; Shi, P.; Shi, K.H.; Tao, H. DNMT1-Induced miR-152-3p Suppression Facilitates Cardiac Fibroblast Activation in Cardiac Fibrosis. Cardiovasc. Toxicol. 2021, 21, 984–999. [Google Scholar] [CrossRef]
- Zhao, K.; Weng, L.; Xu, T.; Yang, C.; Zhang, J.; Ni, G.; Guo, X.; Tu, J.; Zhang, D.; Sun, W.; et al. Low-intensity pulsed ultrasound prevents prolonged hypoxia-induced cardiac fibrosis through HIF-1alpha/DNMT3a pathway via a TRAAK-dependent manner. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1500–1514. [Google Scholar] [CrossRef]
- Tao, H.; Yang, J.J.; Chen, Z.W.; Xu, S.S.; Zhou, X.; Zhan, H.Y.; Shi, K.H. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology 2014, 323, 42–50. [Google Scholar] [CrossRef]
- Qin, R.H.; Tao, H.; Ni, S.H.; Shi, P.; Dai, C.; Shi, K.H. microRNA-29a inhibits cardiac fibrosis in Sprague-Dawley rats by downregulating the expression of DNMT3A. Anatol. J. Cardiol. 2018, 20, 198–205. [Google Scholar] [CrossRef]
- Tao, H.; Dai, C.; Ding, J.F.; Yang, J.J.; Ding, X.S.; Xu, S.S.; Shi, K.H. Epigenetic aberrations of miR-369-5p and DNMT3A control Patched1 signal pathway in cardiac fibrosis. Toxicology 2018, 410, 182–192. [Google Scholar] [CrossRef]
- Zhao, X.D.; Qin, R.H.; Yang, J.J.; Xu, S.S.; Tao, H.; Ding, X.S.; Shi, K.H. DNMT3A controls miR-200b in cardiac fibroblast autophagy and cardiac fibrosis. Inflamm. Res. 2018, 67, 681–690. [Google Scholar] [CrossRef]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, P.; Liu, W.; Shang, J.; Qiu, S.; Li, X.; Liu, W.; Shi, H.; Zhou, M.; Liu, H. Danhong Injection Alleviates Cardiac Fibrosis via Preventing the Hypermethylation of Rasal1 and Rassf1 in TAC Mice. Oxidative Med. Cell. Longev. 2020, 2020, 3158108. [Google Scholar] [CrossRef] [PubMed]
- Rajgarhia, A.; Ayasolla, K.R.; Zaghloul, N.; Lopez Da Re, J.M.; Miller, E.J.; Ahmed, M. Extracellular Superoxide Dismutase (EC-SOD) Regulates Gene Methylation and Cardiac Fibrosis During Chronic Hypoxic Stress. Front. Cardiovasc. Med. 2021, 8, 669975. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Xu, W.; Qu, W.; Gao, H.; Zhang, J.; Cheng, X.; Liu, N.; Chen, J.; Xu, G.L.; Li, X.; et al. Loss of ten-eleven translocation 2 induces cardiac hypertrophy and fibrosis through modulating ERK signaling pathway. Hum. Mol. Genet. 2021, 30, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y. Role of Mammalian DNA Methyltransferases in Development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Kusano, K.F.; Pola, R.; Murayama, T.; Curry, C.; Kawamoto, A.; Iwakura, A.; Shintani, S.; Ii, M.; Asai, J.; Tkebuchava, T.; et al. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005, 11, 1197–1204. [Google Scholar] [CrossRef]
- Wu, R.N.; Yu, T.Y.; Zhou, J.C.; Li, M.; Gao, H.K.; Zhao, C.; Dong, R.Q.; Peng, D.; Hu, Z.W.; Zhang, X.W.; et al. Targeting HMGB1 ameliorates cardiac fibrosis through restoring TLR2-mediated autophagy suppression in myocardial fibroblasts. Int. J. Cardiol. 2018, 267, 156–162. [Google Scholar] [CrossRef]
- Li, Z.M.; Xu, S.W.; Liu, P.Q. Salvia miltiorrhizaBurge (Danshen): A golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol. Sin. 2018, 39, 802–824. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.S.; Zheng, B.; Qin, Y.; Zhou, J.; Yang, Z.; Zhang, X.H.; Zhao, H.Y.; Yang, H.J.; Wen, J.K. Salvia miltiorrhiza-derived miRNAs suppress vascular remodeling through regulating OTUD7B/KLF4/NMHC IIA axis. Theranostics 2020, 10, 7787–7811. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Varela, M.; Paish, H.; Alvarez, L.; Uriarte, I.; Latasa, M.U.; Santamaria, E.; Recalde, M.; Garate, M.; Claveria, A.; Colyn, L.; et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 2021, 70, 388–400. [Google Scholar] [CrossRef] [PubMed]
- She, Q.; Shi, P.; Xu, S.S.; Xuan, H.Y.; Tao, H.; Shi, K.H.; Yang, Y. DNMT1 Methylation of LncRNA GAS5 Leads to Cardiac Fibroblast Pyroptosis via Affecting NLRP3 Axis. Inflammation 2020, 43, 1065–1076. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Chen, S.; Zhou, J.; Li, J.; Cheng, Y. Epigenetics-based therapeutics for myocardial fibrosis. Life Sci. 2021, 271, 119186. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.Y.; Hindmarch, C.C.T.; et al. Epigenetic Metabolic Reprogramming of Right Ventricular Fibroblasts in Pulmonary Arterial Hypertension: A Pyruvate Dehydrogenase Kinase-Dependent Shift in Mitochondrial Metabolism Promotes Right Ventricular Fibrosis. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Mersfelder, E.L.; Parthun, M.R. The tale beyond the tail: Histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 2006, 34, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- Spyropoulou, A.; Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G. Deregulated chromatin remodeling in the pathobiology of brain tumors. Neuromolecular Med. 2013, 15, 1–24. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nature Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’: Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef]
- Wiesel-Motiuk, N.; Assaraf, Y.G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2020, 53, 100729. [Google Scholar] [CrossRef]
- Pang, M.; Zhuang, S. Histone deacetylase: A potential therapeutic target for fibrotic disorders. J. Pharmacol. Exp. Ther. 2010, 335, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugyei-Twum, A.; Advani, A.; Advani, S.L.; Zhang, Y.; Thai, K.; Kelly, D.J.; Connelly, K.A. High glucose induces Smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc. Diabetol. 2014, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Rai, R.; Sun, T.; Ramirez, V.; Lux, E.; Eren, M.; Vaughan, D.E.; Ghosh, A.K. Acetyltransferase p300 inhibitor reverses hypertension-induced cardiac fibrosis. J. Cell. Mol. Med. 2019, 23, 3026–3031. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Sunagawa, Y.; Funamoto, M.; Wakabayashi, H.; Genpei, M.; Miyazaki, Y.; Katanasaka, Y.; Sari, N.; Shimizu, S.; Katayama, A.; et al. The Synthetic Curcumin Analogue GO-Y030 Effectively Suppresses the Development of Pressure Overload-induced Heart Failure in Mice. Sci. Rep. 2020, 10, 7172. [Google Scholar] [CrossRef]
- Lim, Y.; Jeong, A.; Kwon, D.H.; Lee, Y.U.; Kim, Y.K.; Ahn, Y.; Kook, T.; Park, W.J.; Kook, H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int. J. Mol. Sci. 2021, 22, 9944. [Google Scholar] [CrossRef]
- Shao, T.; Xue, Y.; Fang, M. Epigenetic Repression of Chloride Channel Accessory 2 Transcription in Cardiac Fibroblast: Implication in Cardiac Fibrosis. Front. Cell Dev. Biol. 2021, 9, 771466. [Google Scholar] [CrossRef]
- Deng, M.; Yang, S.; Ji, Y.; Lu, Y.; Qiu, M.; Sheng, Y.; Sun, W.; Kong, X. Overexpression of peptidase inhibitor 16 attenuates angiotensin II-induced cardiac fibrosis via regulating HDAC1 of cardiac fibroblasts. J. Cell. Mol. Med. 2020, 24, 5249–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.V.; Rethi, L.; Chung, C.C.; Yeh, Y.H.; Kao, Y.H.; Chen, Y.J. Class I HDAC modulates angiotensin II-induced fibroblast migration and mitochondrial overactivity. Eur. J. Clin. Investig. 2022, 52, e13712. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kook, T.; Min, H.K.; Kwon, D.H.; Cho, Y.K.; Kim, M.; Shin, S.; Joung, H.; Jeong, S.H.; Lee, S.; et al. PP2A negatively regulates the hypertrophic response by dephosphorylating HDAC2 S394 in the heart. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Yang, X.; Yu, T.; Zhang, S. MicroRNA-489 suppresses isoproterenol-induced cardiac fibrosis by downregulating histone deacetylase 2. Exp. Ther. Med. 2020, 19, 2229–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, L.; Pin, L.; Zhu, S.; Zhong, X.; Zhang, Y.; Shun, M.; Liu, Y.; Hou, M. Plantamajoside attenuates isoproterenol-induced cardiac hypertrophy associated with the HDAC2 and AKT/GSK-3β signaling pathway. Chem. -Biol. Interact. 2019, 307, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.B.; Chen, S.Y.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Kee, H.J.; Bai, L.; Kim, M.K.; Kee, S.J.; Jeong, M.H. Selective HDAC8 Inhibition Attenuates Isoproterenol-Induced Cardiac Hypertrophy and Fibrosis via p38 MAPK Pathway. Front. Pharmacol. 2021, 12, 677757. [Google Scholar] [CrossRef]
- Zhang, L.X.; Du, J.; Zhao, Y.T.; Wang, J.; Zhang, S.; Dubielecka, P.M.; Wei, L.; Zhuang, S.; Qin, G.; Chin, Y.E.; et al. Transgenic overexpression of active HDAC4 in the heart attenuates cardiac function and exacerbates remodeling in infarcted myocardium. J. Appl. Physiol. 2018, 125, 1968–1978. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Deng, M.; Lu, A.; Chen, Y.; Chen, Y.; Wu, C.; Tan, Z.; Boini, K.M.; Yang, T.; Zhu, Q.; et al. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J. Cell. Mol. Med. 2019, 23, 8139–8150. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.S.; Bagchi, R.A.; Felisbino, M.B.; Hirsch, R.A.; Smith, H.E.; Riching, A.S.; Enyart, B.Y.; Koch, K.A.; Cavasin, M.A.; Alexanian, M.; et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ. Res. 2019, 125, 662–677. [Google Scholar] [CrossRef]
- He, Z.; Jiao, H.; An, Q.; Zhang, X.; Zengyangzong, D.; Xu, J.; Liu, H.; Ma, L.; Zhao, W. Discovery of novel 4-phenylquinazoline-based BRD4 inhibitors for cardiac fibrosis. Acta Pharm. Sin. B 2022, 12, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, Y.; Ren, J. Acetylation in cardiovascular diseases: Molecular mechanisms and clinical implications. Biochim. Biophy. Acta—Mol. Basis Dis. 2020, 1866, 165836. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K. FAT-free p300 is good for scar-free tissue repair. J. Cell. Biochem. 2014, 115, 1486–1489. [Google Scholar] [CrossRef]
- Ghosh, A.K. Acetyltransferase p300 Is a Putative Epidrug Target for Amelioration of Cellular Aging-Related Cardiovascular Disease. Cells 2021, 10, 2839. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Varga, J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell. Physiol. 2007, 213, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Sui, X.; Wang, J.; Zhang, J.; Pei, Y.; Guo, L.; Liang, Z. Secretory products from epicardial adipose tissue induce adverse myocardial remodeling after myocardial infarction by promoting reactive oxygen species accumulation. Cell Death Dis. 2021, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Schuetze, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J. Mol. Cell. Cardiol. 2014, 67, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Murphy, S.B.; Kishore, R.; Vaughan, D.E. Global gene expression profiling in PAI-1 knockout murine heart and kidney: Molecular basis of cardiac-selective fibrosis. PLoS ONE 2013, 8, e63825. [Google Scholar] [CrossRef] [Green Version]
- Eom, G.H.; Cho, Y.K.; Ko, J.H.; Shin, S.; Choe, N.; Kim, Y.; Joung, H.; Kim, H.S.; Nam, K.I.; Kee, H.J.; et al. Casein kinase-2α1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation 2011, 123, 2392–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kee, H.J.; Ryu, Y.; Seok, Y.M.; Choi, S.Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin. Hypertens. 2019, 25, 13. [Google Scholar] [CrossRef]
- Barbosa, D.M.; Fahlbusch, P.; Herzfeld de Wiza, D.; Jacob, S.; Kettel, U.; Al-Hasani, H.; Krüger, M.; Ouwens, D.M.; Hartwig, S.; Lehr, S.; et al. Rhein, a novel Histone Deacetylase (HDAC) inhibitor with antifibrotic potency in human myocardial fibrosis. Sci. Rep. 2020, 10, 4888. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Hou, X.; Li, J.; Li, T.; Qiu, A.; Liu, N.; Zhuang, S. Histone deacetylase 6 inhibition mitigates renal fibrosis by suppressing TGF-β and EGFR signaling pathways in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2020, 319, F1003–F1014. [Google Scholar] [CrossRef]
- Yang, Y.; Bae, M.; Park, Y.K.; Lee, Y.; Pham, T.X.; Rudraiah, S.; Manautou, J.; Koo, S.I.; Lee, J.Y. Histone deacetylase 9 plays a role in the antifibrogenic effect of astaxanthin in hepatic stellate cells. J. Nutr. Biochem. 2017, 40, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.L.; Haak, A.J.; Caporarello, N.; Choi, K.M.; Ye, Z.; Yan, H.; Varelas, X.; Ordog, T.; Ligresti, G.; Tschumperlin, D.J. TGFβ-induced fibroblast activation requires persistent and targeted HDAC-mediated gene repression. J. Cell Sci. 2019, 132, jcs233486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.S.; Wen, H.C.; Weng, C.M.; Lee, H.S.; Chen, B.C.; Lin, C.H. Histone deacetylase 7 mediates endothelin-1-induced connective tissue growth factor expression in human lung fibroblasts through p300 and activator protein-1 activation. J. Biomed. Sci. 2021, 28, 38. [Google Scholar] [CrossRef] [PubMed]
- Schuetze, K.B.; McKinsey, T.A.; Long, C.S. Targeting cardiac fibroblasts to treat fibrosis of the heart: Focus on HDACs. J. Mol. Cell. Cardiol. 2014, 70, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, D.H.; Eom, G.H.; Ko, J.H.; Shin, S.; Joung, H.; Choe, N.; Nam, Y.S.; Min, H.K.; Kook, T.; Yoon, S.; et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 2016, 7, 10492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Spiltoir, J.I.; Stratton, M.S.; Cavasin, M.A.; Demos-Davies, K.; Reid, B.G.; Qi, J.; Bradner, J.E.; McKinsey, T.A. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013, 63, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 13. [Google Scholar] [CrossRef]
- Schumacher, D.; Peisker, F.; Kramann, R. MEOX1: A novel druggable target that orchestrates the activation of fibroblasts in cardiac fibrosis. Signal Transduct. Target. Ther. 2021, 6, 440. [Google Scholar] [CrossRef]
- Kim, S.Y.; Zhang, X.; Schiattarella, G.G.; Altamirano, F.; Ramos, T.A.R.; French, K.M.; Jiang, N.; Szweda, P.A.; Evers, B.M.; May, H.I.; et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation 2020, 142, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 32. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Blin, G.; Liand, M.; Mauduit, C.; Chehade, H.; Benahmed, M.; Simeoni, U.; Siddeek, B. Maternal Exposure to High-Fat Diet Induces Long-Term Derepressive Chromatin Marks in the Heart. Nutrients 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Zhang, R.; Mo, B.; Chen, L.; Liu, L.; Yu, Y.; Cao, W.; Fang, G.; Wan, Y.; Gu, Y.; et al. EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J. Mol. Cell. Cardiol. 2019, 135, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Feng, B.; Chakrabarti, S. ANRIL regulates production of extracellular matrix proteins and vasoactive factors in diabetic complications. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E191–E200. [Google Scholar] [CrossRef]
- Ge, Z.; Yin, C.; Li, Y.; Tian, D.; Xiang, Y.; Li, Q.; Tang, Y.; Zhang, Y. Long noncoding RNA NEAT1 promotes cardiac fibrosis in heart failure through increased recruitment of EZH2 to the Smad7 promoter region. J. Transl. Med. 2022, 20, 7. [Google Scholar] [CrossRef]
- Zhu, W.S.; Tang, C.M.; Xiao, Z.; Zhu, J.N.; Lin, Q.X.; Fu, Y.H.; Hu, Z.Q.; Zhang, Z.; Yang, M.; Zheng, X.L.; et al. Targeting EZH1 and EZH2 contributes to the suppression of fibrosis-associated genes by miR-214-3p in cardiac myofibroblasts. Oncotarget 2016, 7, 78331–78342. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhang, C.; Li, J.; Che, J.; Yang, X.; Xian, Y.; Li, X.; Cao, C. Long non-coding RNA MALAT1 promotes cardiac remodeling in hypertensive rats by inhibiting the transcription of MyoD. Aging 2019, 11, 8792–8809. [Google Scholar] [CrossRef]
- Li, F.; Li, L.; Zhang, J.; Yang, X.; Liu, Y. Histone methyltransferase DOT1L mediates the TGF-β1/Smad3 signaling pathway through epigenetic modification of SYK in myocardial infarction. Hum. Cell 2022, 35, 98–110. [Google Scholar] [CrossRef]
- Huo, J.L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.J.; Tran, T.A.T.; Wang, M.; Ranek, M.J.; Kokkonen-Simon, K.M.; Gao, J.; Luo, X.; Tan, W.; Kyrychenko, V.; Liao, L.; et al. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat. Commun. 2018, 9, 5230. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lu, Y.; Jiang, C. Inhibition of histone demethylase JMJD1C attenuates cardiac hypertrophy and fibrosis induced by angiotensin II. J. Recept. Signal Transduct. Res. 2020, 40, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Wang, Q.; Yang, D.; Zhu, M.; Wang, J.; Zhu, Y.; Liu, X. Targeting JMJD3 histone demethylase mediates cardiac fibrosis and cardiac function following myocardial infarction. Biochem. Biophys. Res. Commun. 2020, 528, 671–677. [Google Scholar] [CrossRef]
- Luo, M. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev. 2018, 118, 6656–6705. [Google Scholar] [CrossRef]
- Yuan, J.L.; Yin, C.Y.; Li, Y.Z.; Song, S.; Fang, G.J.; Wang, Q.S. EZH2 as an Epigenetic Regulator of Cardiovascular Development and Diseases. J. Cardiovasc. Pharmacol. 2021, 78, 192–201. [Google Scholar] [CrossRef]
- Jiang, Y.; Xiang, C.; Zhong, F.; Zhang, Y.; Wang, L.; Zhao, Y.; Wang, J.; Ding, C.; Jin, L.; He, F.; et al. Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics 2021, 11, 361–378. [Google Scholar] [CrossRef]
- Delgado-Olguín, P.; Huang, Y.; Li, X.; Christodoulou, D.; Seidman, C.E.; Seidman, J.G.; Tarakhovsky, A.; Bruneau, B.G. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 2012, 44, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhu, N.; Xu, J. miR-101a-3p overexpression prevents acetylcholine-CaCl(2)-induced atrial fibrillation in rats via reduction of atrial tissue fibrosis, involving inhibition of EZH2. Mol. Med. Rep. 2021, 24, 740. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Liu, Z.P. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics 2015, 7, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Guénantin, A.C.; Jebeniani, I.; Leschik, J.; Watrin, E.; Bonne, G.; Vignier, N.; Pucéat, M. Targeting the histone demethylase LSD1 prevents cardiomyopathy in a mouse model of laminopathy. J. Clin. Investig. 2021, 131, e136488. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Chen, H.Z.; Wang, L.; Liu, D.P.; Hill, J.A.; Liu, Z.P. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J. Clin. Investig. 2011, 121, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Investig. 2017, 127, 335–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, B.; González, A.; Díez, J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation 2010, 121, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Szulik, M.W.; Davis, K.; Bakhtina, A.; Azarcon, P.; Bia, R.; Horiuchi, E.; Franklin, S. Transcriptional regulation by methyltransferases and their role in the heart: Highlighting novel emerging functionality. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H847–H865. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, S.; Shang, J.; Jiang, Y.; Dai, Y.; Xu, B.; Yu, Y.; Liang, Z.; Yang, Y. LncRNA: Shedding light on mechanisms and opportunities in fibrosis and aging. Ageing Res. Rev. 2019, 52, 17–31. [Google Scholar] [CrossRef]
- Fu, Q.; Lu, Z.; Fu, X.; Ma, S.; Lu, X. MicroRNA 27b promotes cardiac fibrosis by targeting the FBW7/Snail pathway. Aging 2019, 11, 11865–11879. [Google Scholar] [CrossRef]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.H.; Zhang, Y.H.; Ding, Y.Q.; Bi, X.Y.; Yuan, J.; Zhou, H.; Wang, P.X.; Zhang, L.L.; Ye, J.T. MicroRNA-99b-3p promotes angiotensin II-induced cardiac fibrosis in mice by targeting GSK-3β. Acta Pharmacol. Sin. 2021, 42, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; Xue, K.; Zhang, J.; Wang, C.; Zhang, Q.; Chen, X.; Gao, C.; Yu, X.; Sun, L. MicroRNA-143-3p promotes human cardiac fibrosis via targeting sprouty3 after myocardial infarction. J. Mol. Cell. Cardiol. 2019, 129, 281–292. [Google Scholar] [CrossRef]
- Ramanujam, D.; Schön, A.P.; Beck, C.; Vaccarello, P.; Felician, G.; Dueck, A.; Esfandyari, D.; Meister, G.; Meitinger, T.; Schulz, C.; et al. MicroRNA-21-Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload. Circulation 2021, 143, 1513–1525. [Google Scholar] [CrossRef]
- Li, G.; Shao, Y.; Guo, H.C.; Zhi, Y.; Qiao, B.; Ma, K.; Lai, Y.Q.; Du, J.; Li, Y. MicroRNA-27b-3p downregulates FGF1 and aggravates pathological cardiac remodelling. Cardiovasc. Res. 2021, 118, cvab248. [Google Scholar] [CrossRef]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J. Cell. Mol. Med. 2020, 24, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Verjans, R.; Peters, T.; Beaumont, F.J.; van Leeuwen, R.; van Herwaarden, T.; Verhesen, W.; Munts, C.; Bijnen, M.; Henkens, M.; Diez, J.; et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload-Induced Heart Failure. Hypertension 2018, 71, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, V.; Takano, A.P.C.; Guleria, R.S.; Gupta, S. Deficiency of MicroRNA miR-1954 Promotes Cardiac Remodeling and Fibrosis. J. Am. Heart Assoc. 2019, 8, e012880. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.Y.; Li, T.T.; Wang, J.; Jiang, Y.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Yang, Y.; Zhang, X.F.; et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J. Mol. Cell. Cardiol. 2018, 115, 64–72. [Google Scholar] [CrossRef]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Micheletti, R.; Plaisance, I.; Abraham, B.J.; Sarre, A.; Ting, C.C.; Alexanian, M.; Maric, D.; Maison, D.; Nemir, M.; Young, R.A.; et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 2017, 9, eaai9118. [Google Scholar] [CrossRef] [Green Version]
- Hao, K.; Lei, W.; Wu, H.; Wu, J.; Yang, Z.; Yan, S.; Lu, X.A.; Li, J.; Xia, X.; Han, X.; et al. LncRNA-Safe contributes to cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial infarction. Theranostics 2019, 9, 7282–7297. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhuang, Y.; Zhu, H.; Wu, H.; Li, D.; Zhan, L.; Yang, W.; Yuan, Y.; Xie, Y.; Yang, S.; et al. LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J. Mol. Cell. Cardiol. 2019, 133, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fu, X.; Kataoka, M.; Liu, N.; Wang, Y.; Gao, F.; Liang, T.; Dong, X.; Pei, J.; Hu, X.; et al. Long noncoding RNA Cfast regulates cardiac fibrosis. Mol. Therapy. Nucleic Acids 2021, 23, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Zhang, J.G.; Qin, R.H.; Dai, C.; Shi, P.; Yang, J.J.; Deng, Z.Y.; Shi, K.H. LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology 2017, 386, 11–18. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, Y.; Hu, Y.; Guan, J.; Xu, L.; Xiao, W.; Zhong, Q.; Ren, C.; Lu, J.; Liang, J.; et al. Long noncoding RNA Crnde attenuates cardiac fibrosis via Smad3-Crnde negative feedback in diabetic cardiomyopathy. FEBS J. 2019, 286, 1645–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, C.; Lyu, D.; He, C.; Li, R.; Lu, Q. Dioscin elevates lncRNA MANTIS in therapeutic angiogenesis for heart diseases. Aging Cell 2021, 20, e13392. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhuang, Y.; Yang, W.; Xie, Y.; Shang, W.; Su, S.; Dong, X.; Wu, J.; Jiang, W.; Zhou, Y.; et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21162. [Google Scholar] [CrossRef]
- Ju, W.; Liu, K.; Ouyang, S.; Liu, Z.; He, F.; Wu, J. Changes in N6-Methyladenosine Modification Modulate Diabetic Cardiomyopathy by Reducing Myocardial Fibrosis and Myocyte Hypertrophy. Front. Cell Dev. Biol. 2021, 9, 702579. [Google Scholar] [CrossRef]
- Mathiyalagan, P.; Adamiak, M.; Mayourian, J.; Sassi, Y.; Liang, Y.; Agarwal, N.; Jha, D.; Zhang, S.; Kohlbrenner, E.; Chepurko, E.; et al. FTO-Dependent N(6)-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation 2019, 139, 518–532. [Google Scholar] [CrossRef]
- Landry, N.M.; Rattan, S.G.; Filomeno, K.L.; Meier, T.W.; Meier, S.C.; Foran, S.J.; Meier, C.F.; Koleini, N.; Fandrich, R.R.; Kardami, E.; et al. SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res. Cardiol. 2021, 116, 25. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Y.; Zhu, H.; Hu, J.; Xie, Z. MiR-34a/miR-93 target c-Ski to modulate the proliferaton of rat cardiac fibroblasts and extracellular matrix deposition in vivo and in vitro. Cell. Signal. 2018, 46, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, J.; Niu, G.; Weng, J.; Zhang, Q.; Xie, M.; Li, C.; Sun, K. Apigenin inhibits isoproterenol-induced myocardial fibrosis and Smad pathway in mice by regulating oxidative stress and miR-122-5p/155-5p expressions. Drug Dev. Res. 2022, 83, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Leisegang, M.S.; Fork, C.; Josipovic, I.; Richter, F.M.; Preussner, J.; Hu, J.; Miller, M.J.; Epah, J.; Hofmann, P.; Günther, S.; et al. Long Noncoding RNA MANTIS Facilitates Endothelial Angiogenic Function. Circulation 2017, 136, 65–79. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.X.; Mu, B.; Li, X.; Bie, Z.D. circCELF1 Inhibits Myocardial Fibrosis by Regulating the Expression of DKK2 Through FTO/m(6)A and miR-636. J. Cardiovasc. Transl. Res. 2022. [CrossRef]
- de Oliveira Camargo, R.; Abual’anaz, B.; Rattan, S.G.; Filomeno, K.L.; Dixon, I.M.C. Novel factors that activate and deactivate cardiac fibroblasts: A new perspective for treatment of cardiac fibrosis. Wound Repair Regen. Off. Publ. Wound Health Soc. Eur. Tissue Repair Soc. 2021, 29, 667–677. [Google Scholar] [CrossRef]
- Hill, J.A. When the CAR Targets Scar. N. Engl. J. Med. 2019, 381, 2475–2476. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Méndez Fernández, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Paoletti, C.; Chiono, V. Bioengineering Methods in MicroRNA-Mediated Direct Reprogramming of Fibroblasts Into Cardiomyocytes. Front. Cardiovasc. Med. 2021, 8, 750438. [Google Scholar] [CrossRef]
- Pascale, E.; Caiazza, C.; Paladino, M.; Parisi, S.; Passaro, F.; Caiazzo, M. MicroRNA Roles in Cell Reprogramming Mechanisms. Cells 2022, 11, 940. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclass | Modifier | Fibrosis Model | Target | Output | Refs. |
|---|---|---|---|---|---|
| DNA methylation | |||||
| DNMTs | DNMT1 | DCM | SOCS3 | Pro-fibrotic, CFs activation | [24] |
| ISO | microRNA- 152-3p | Pro-fibrotic, CFs activation and proliferation | [25] | ||
| DNMT3a | TAC | TRAAK | Pro-fibrotic, CFs activation | [26] | |
| ISO | RASSF1A, ERK1/2 | Pro-fibrotic, CFs activation | [27] | ||
| Ras/ERK1/2 | Pro-fibrotic, CFs activation and proliferation | [28] | |||
| AAC | Patched1 | Pro-fibrotic, CFs proliferation | [29] | ||
| miR-200b | Pro-fibrotic, CFs autophagy | [30] | |||
| DNMT3b | Hypoxia | HIF-1α | Pro-fibrotic, CFs activation | [31] | |
| TAC | Rasal1, Rassf1 | Pro-fibrotic. CFs activation | [32] | ||
| DNMT1, DNMT3b | Hypoxia | RASSF1A, ERK1/2 | Pro-fibrotic, CFs proliferation | [33] | |
| DNA demethylation | |||||
| TETs | TET2 | Ang-II | IL-6 | Anti-fibrotic, suppression of inflammatory response | [34] |
| TET2 KO | Hspa1b | Anti-fibrotic, protection of cardiomyocytes | [35] | ||
| TET3 | TAC | Rasal1 | Anti-fibrotic, EndMT | [36] | |
| Subclass | Modifier | Fibrosis Model | Target | Output | Refs. |
|---|---|---|---|---|---|
| Histone acetylation | |||||
| HATs | p300 | high glucose | Smad2 | Pro-fibrotic, collagen production | [56] |
| Ang-II | H3K9 | Pro-fibrotic, CFs activation and type I collagen synthesis | [57] | ||
| TAC | GATA4 | Pro-fibrotic, collagen production | [58] | ||
| PCAF | ISO | Smad2 | Pro-fibrotic, CFs activation | [59] | |
| Histone deacetylation | |||||
| HDACs Class I | HDAC1 | LAD, TAC | Clca2 | Pro-fibrotic, CFs activation | [60] |
| Ang-II | p53 | Pro-fibrotic, CFs activation and proliferation | [61] | ||
| mitochondria | Pro-fibrotic, CFs migration | [62] | |||
| HDAC2 | ISO | PPP2CA | Pro-fibrotic, α-SMA synthesis | [63] | |
| α-SMA | Pro-fibrotic, CFs activation | [64] | |||
| AKT/ GSK-3β | Pro-fibrotic, collagen production | [65] | |||
| HDAC3 | DCM | DUSP5 | Pro-fibrotic, fibrosis markers and collagen accumulation | [66] | |
| HDAC8 | ISO | p38 MAPK | Pro-fibrotic, markers of fibrosis | [67] | |
| Class II | HDAC4 | MI | N.A. | Pro-fibrotic, cardiokines reduction | [68] |
| HDAC5/ HDAC6 | Ang-II | COX2/ PGE2 | Pro-fibrotic, cardiac hypertrophy | [69] | |
| BRDs | BRD4 | TAC | Sertad4 | Pro-fibrotic, CFs activation and proliferation | [70] |
| TAC | Meox1 | Pro-fibrotic, CFs activation | [9] | ||
| c-MYC | Pro-fibrotic, CFs activation | [71] | |||
| Subclass | Modifier | Fibrosis Model | Target | Output | Refs. |
|---|---|---|---|---|---|
| Histone methylation | |||||
| KMTs | EZH2 | High-Fat | H3K27me2/3 | Anti-fibrotic, suppression of pro-fibrotic genes | [105] |
| Ang-II | ACTA2 | Pro-fibrotic, CFs activation and migration | [106] | ||
| DCM | lncRNA- ANRIL | Pro-fibrotic, increased expression of FN, Col1α4 | [107] | ||
| TAC | Smad7 | Pro-fibrotic, CFs activation | [108] | ||
| EZH1/2 | Ang-II | PPAR-γ | Pro-fibrotic, Col1a1 and Col3a1 synthesis | [109] | |
| SUV39H1 | SHRs | MyoD | Pro-fibrotic, CFs proliferation and collagen accumulation | [110] | |
| DOT1L | MI | SYK | Pro-fibrotic, CFs activation | [111] | |
| Histone demethylation | |||||
| KDMs | LSD1 | TAC | TGF-β | Pro-fibrotic, CFs activation and collagen secretion | [112] |
| KDM3A | Timp1 | Pro-fibrotic, CFs activation | [113] | ||
| KDM3C | Ang-II | Timp1 | Pro-fibrotic, CFs activation | [114] | |
| KDM6B | β-catenin | Pro-fibrotic, ECM deposition | [115] | ||
| Subclass | Modifier | Fibrosis Model | Target | Output | Refs. |
|---|---|---|---|---|---|
| Non-coding RNAs | |||||
| miRNAs | miR-27b | Ang-II | FBW7 | pro-fibrotic, CFs proliferation and collagen production | [129] |
| miR-125b | Apelin, p53 | Pro-fibrotic, CFs proliferation | [130] | ||
| miR-99b -3p | GSK-3β | Pro-fibrotic, CFs proliferation and migration | [131] | ||
| miR-143 -3p | MI | SPRY3 | Pro-fibrotic, CFs activation, proliferation, and migration | [132] | |
| miR-21 | TAC | N.A. | Pro-fibrotic, CFs activation | [133] | |
| miR-27b -3p | TAC/ Ang-II | FGF1 | Pro-fibrotic, mitochondrial oxidative phosphorylation | [134] | |
| miR-590 -3p | MI | ZEB1 | Anti-fibrotic, CFs activation, proliferation, and migration | [135] | |
| miR-221/ 222 | Ang-II | SMAD2 | Anti-fibrotic, CFs activation, and proliferation | [136] | |
| miR-1954 | THBS1 | Anti-fibrotic, attenuation inflammation | [137] | ||
| lncRNAs | lncRNA AK081284 | DCM | IL-17 | Pro-fibrotic, CFs proliferation, and collagen production | [138] |
| lncRNA Meg3 | TAC | MMP-2 | Pro-fibrotic, ECM deposition | [139] | |
| lncRNA Wisper | MI | TIA1-related protein | Pro-fibrotic, CFs proliferation | [140] | |
| lncRNA AK137033 | Sfrp2 | Pro-fibrotic, CFs activation, and proliferation | [141] | ||
| lncRNA PCFL | miR-378 | Pro-fibrotic, CFs proliferation, and collagen production | [142] | ||
| lncRNA AK048087 | MI/ Ang-II | COTL1 | Pro-fibrotic, CFs activation, and proliferation | [143] | |
| lncRNA GAS5 | ISO | miR-21 | Anti-fibrotic, CFs proliferation | [144] | |
| lncRNA Crnde | DCM | Smad3 | Anti-fibrotic, CFs activation | [145] | |
| lncRNA MANTIS | MI | Sox18, Smad6, etc. | Anti-fibrotic, vascular neogenesis | [146] | |
| RNA modifications | |||||
| m6A | METTL3 | MI | Fibrosis-related genes | Pro-fibrotic, CFs activation, and proliferation | [147] |
| FTO | DCM | CD36, Slc5a33 | Anti-fibrotic, collagen deposition suppression | [148] | |
| MI | Serca2a | Anti-fibrotic, CFs activation, proliferation, and migration | [149] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, J.; Liu, J.; Zuo, S. Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis. Cells 2022, 11, 2347. https://doi.org/10.3390/cells11152347
Shao J, Liu J, Zuo S. Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis. Cells. 2022; 11(15):2347. https://doi.org/10.3390/cells11152347
Chicago/Turabian StyleShao, Jingrong, Jiao Liu, and Shengkai Zuo. 2022. "Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis" Cells 11, no. 15: 2347. https://doi.org/10.3390/cells11152347
APA StyleShao, J., Liu, J., & Zuo, S. (2022). Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis. Cells, 11(15), 2347. https://doi.org/10.3390/cells11152347