
A Drug Repurposing Screen Identifies Fludarabine Phosphate as a Potential Therapeutic Agent for N-MYC Overexpressing Neuroendocrine Prostate Cancers

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines Used in This Study
2.2. Drug Screen Using Targetmol Drug Library
2.3. High-Throughput Microscope Imaging
2.4. Computational Scoring of Drug Library Screen
2.5. IncuCyte Live Cell Analysis
2.6. SDS-PAGE and Western Blot Analysis
2.7. qRT-PCR
2.8. RNASeq Analysis
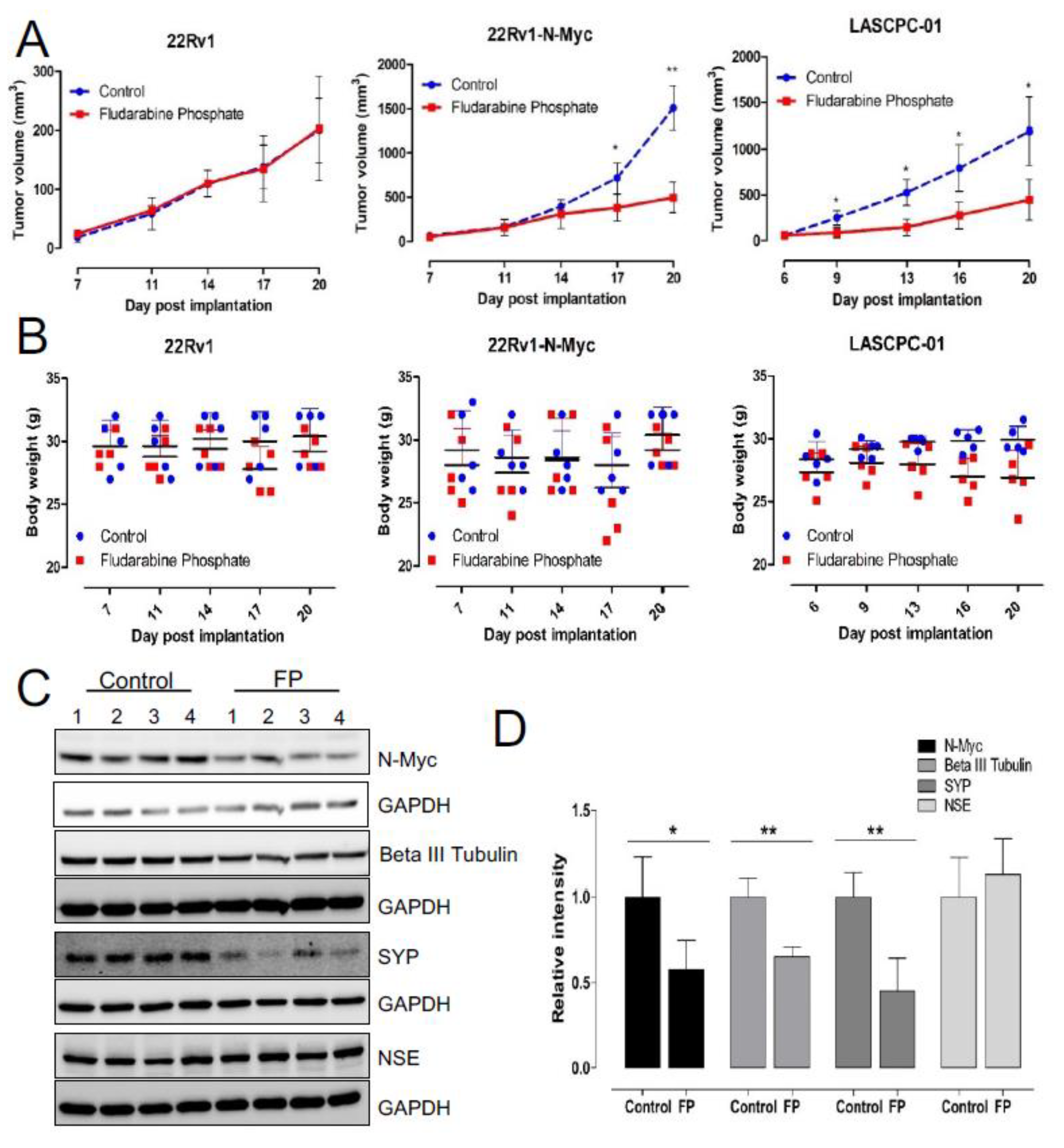
2.9. Xenograft Studies
2.10. Immunohistochemistry
2.11. Statistical Analysis
3. Results
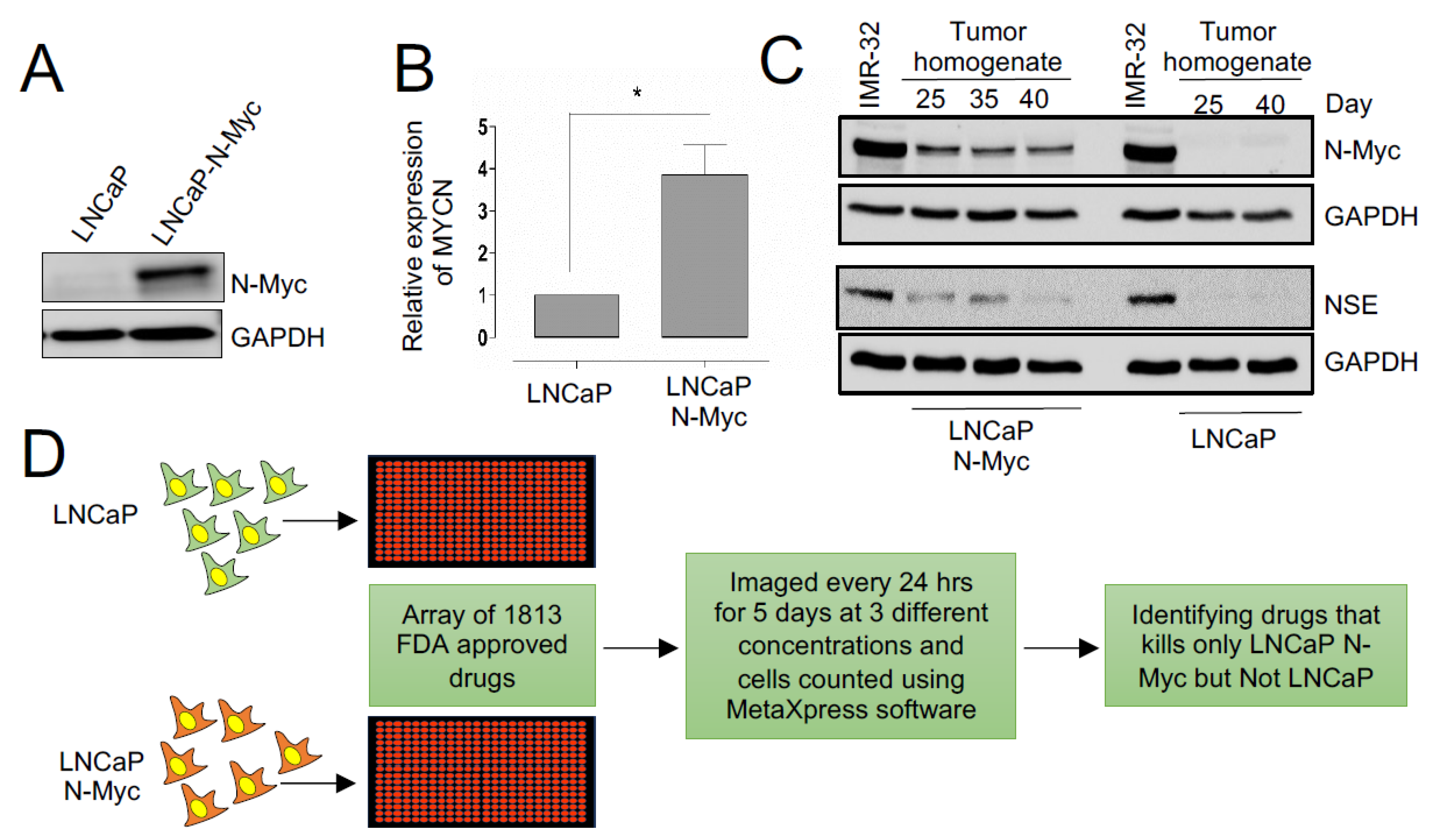
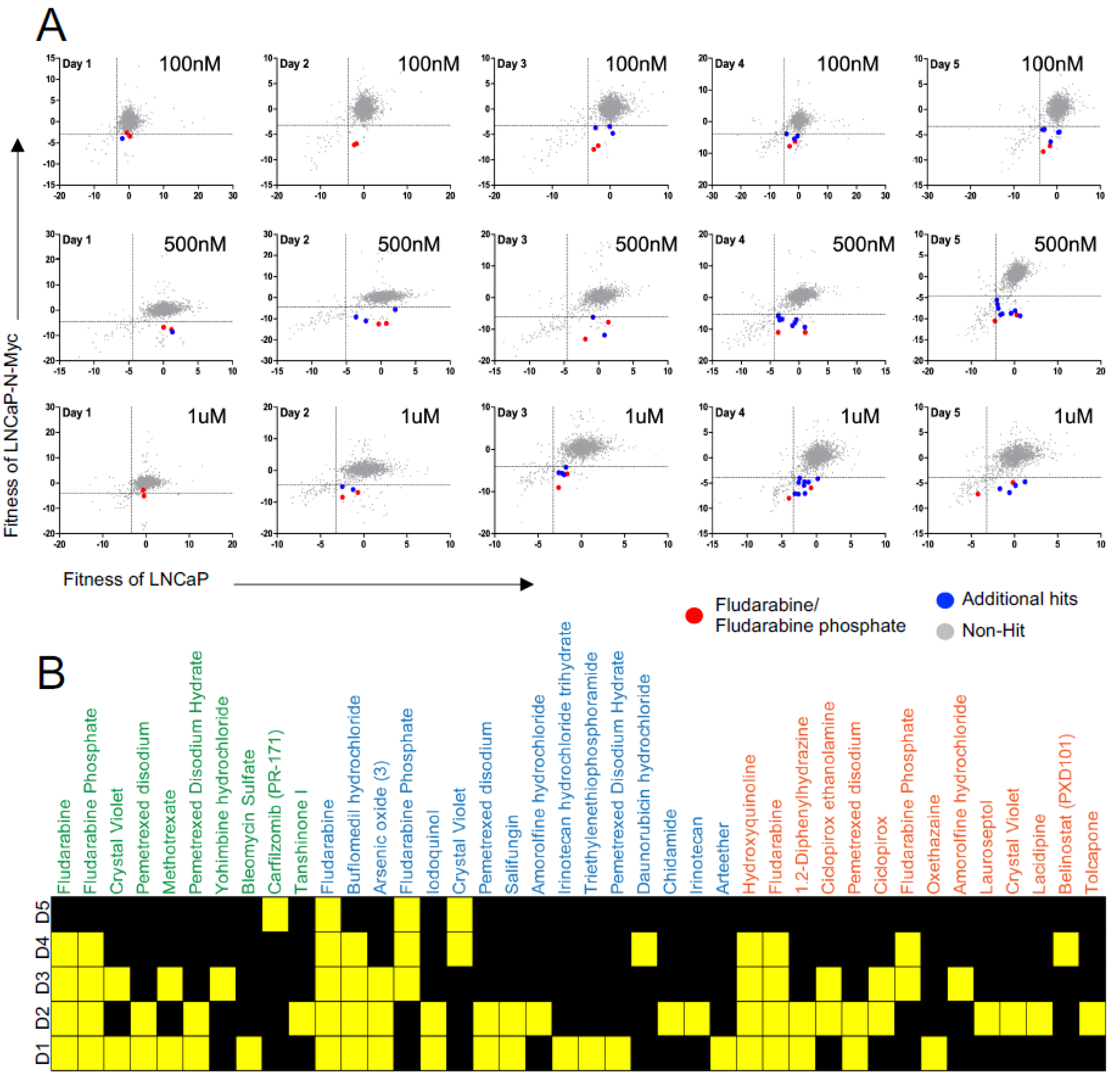
3.1. A Repurposing Drug Library Screen Identifies Compounds That Selectively Inhibit the Growth of N-MYC Overexpressing Prostate Cancer Cells
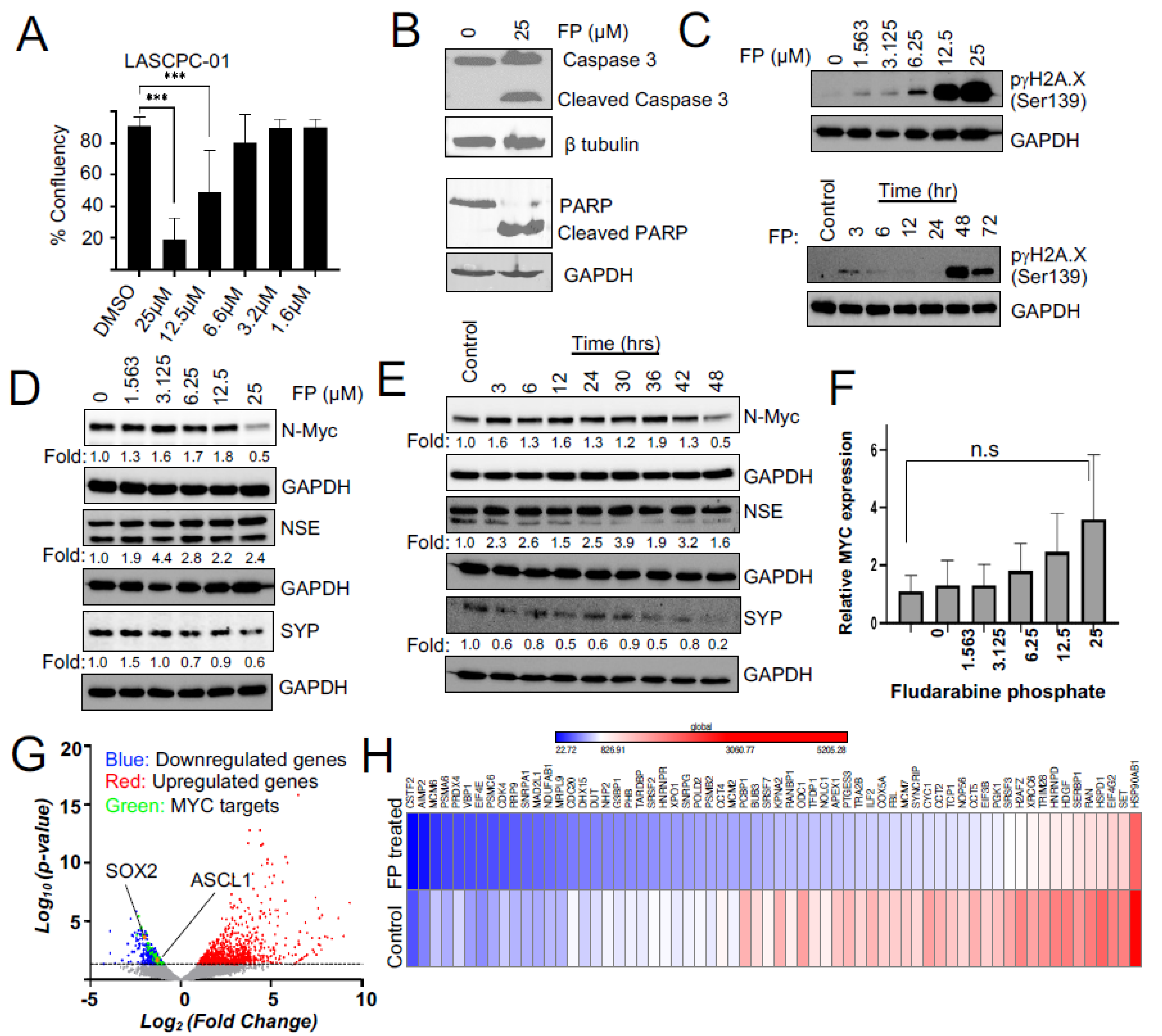
3.2. Fludarabine Phosphate Causes Apoptotic Cell Death in N-MYC Overexpressing Cells by Inducing ROS
3.3. Fludarabine Phosphate Affects N-MYC Protein Levels and N-MYC Transcriptional Targets in NEPC Cells
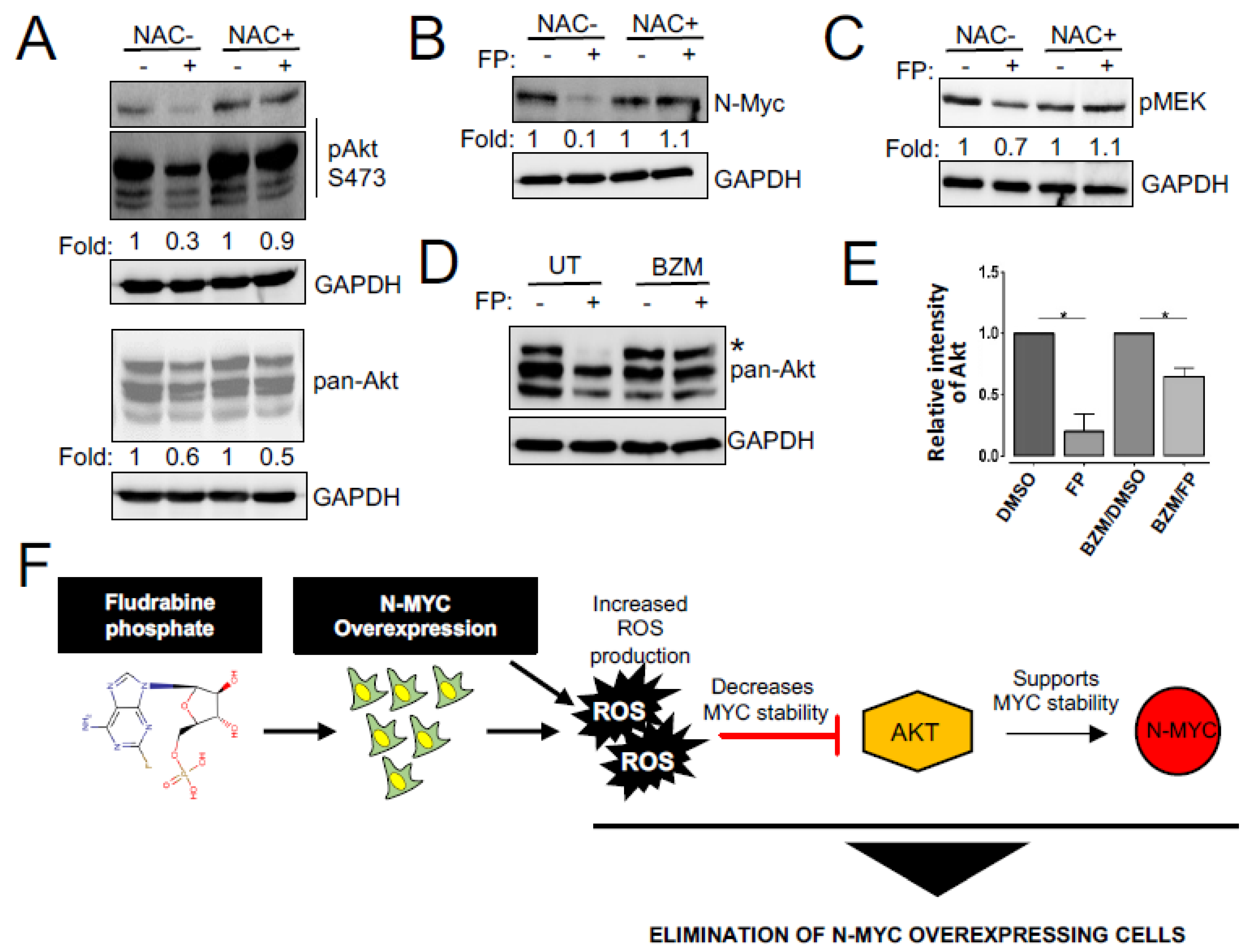
3.4. Enhanced ROS Production Destabilizes N-MYC Protein by Inhibiting AKT Signaling and Is Responsible for the Reduced Survival of NEPC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, S.L.; Bruchovsky, N.; Rennie, P.S.; Coppin, C.M. The combination of cyproterone acetate and low dose diethylstilbestrol in the treatment of advanced prostatic carcinoma. J. Urol. 1988, 140, 1460–1465. [Google Scholar] [CrossRef]
- Agus, D.B.; Cordon-Cardo, C.; Fox, W.; Drobnjak, M.; Koff, A.; Golde, D.W.; Scher, H.I. Prostate cancer cell cycle regulators: Response to androgen withdrawal and development of androgen independence. J. Natl. Cancer Inst. 1999, 91, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.; et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef]
- Pezaro, C.; Omlin, A.; Lorente, D.; Rodrigues, D.N.; Ferraldeschi, R.; Bianchini, D.; Mukherji, D.; Riisnaes, R.; Altavilla, A.; Crespo, M.; et al. Visceral disease in castration-resistant prostate cancer. Eur. Urol. 2014, 65, 270–273. [Google Scholar] [CrossRef]
- Beltran, H.; Tagawa, S.T.; Park, K.; MacDonald, T.; Milowsky, M.I.; Mosquera, J.M.; Rubin, M.A.; Nanus, D.M. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J. Clin. Oncol. 2012, 30, e386–e389. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Wang, H.T.; Yao, Y.H.; Li, B.G.; Tang, Y.; Chang, J.W.; Zhang, J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: Factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J. Clin. Oncol. 2014, 32, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. MCR 2014, 12, 815–822. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. G34, another connection between MYCN and a pediatric tumor. Cancer Discov. 2013, 3, 484–486. [Google Scholar] [CrossRef][Green Version]
- Wong, H.L.; Yang, K.C.; Shen, Y.; Zhao, E.Y.; Loree, J.M.; Kennecke, H.F.; Kalloger, S.E.; Karasinska, J.M.; Lim, H.J.; Mungall, A.J.; et al. Molecular characterization of metastatic pancreatic neuroendocrine tumors (PNETs) using whole-genome and transcriptome sequencing. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002329. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef]
- Kim, J.H.; Dhanasekaran, S.M.; Mehra, R.; Tomlins, S.A.; Gu, W.; Yu, J.; Kumar-Sinha, C.; Cao, X.; Dash, A.; Wang, L.; et al. Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res. 2007, 67, 8229–8239. [Google Scholar] [CrossRef]
- Boutros, P.C.; Fraser, M.; Harding, N.J.; de Borja, R.; Trudel, D.; Lalonde, E.; Meng, A.; Hennings-Yeomans, P.H.; McPherson, A.; Sabelnykova, V.Y.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Gleave, M.E. Targeting the adaptive molecular landscape of castration-resistant prostate cancer. EMBO Mol. Med. 2015, 7, 878–894. [Google Scholar] [CrossRef]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Pourtier-Manzanedo, A.; Gil, J.; Beach, D.H. Myc confers androgen-independent prostate cancer cell growth. J. Clin. Investig. 2003, 112, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Kallioniemi, A.H.; Syvanen, A.C.; Hyytinen, E.R.; Karhu, R.; Tammela, T.; Isola, J.J.; Kallioniemi, O.P. Genetic changes in primary and recurrent prostate cancer by comparative genomic hybridization. Cancer Res. 1995, 55, 342–347. [Google Scholar] [PubMed]
- Mosquera, J.M.; Beltran, H.; Park, K.; MacDonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef]
- Tuthill, M.C.; Wada, R.K.; Arimoto, J.M.; Sugino, C.N.; Kanemaru, K.K.; Takeuchi, K.K.; Sidell, N. N-myc oncogene expression in neuroblastoma is driven by Sp1 and Sp3. Mol. Genet. Metab. 2003, 80, 272–280. [Google Scholar] [CrossRef]
- Strieder, V.; Lutz, W. E2F proteins regulate MYCN expression in neuroblastomas. J. Biol. Chem. 2003, 278, 2983–2989. [Google Scholar] [CrossRef]
- Zhao, Z.; Shelton, S.D.; Oviedo, A.; Baker, A.L.; Bryant, C.P.; Omidvarnia, S.; Du, L. The PLAGL2/MYCN/miR-506-3p interplay regulates neuroblastoma cell fate and associates with neuroblastoma progression. J. Exp. Clin. Cancer Res. 2020, 39, 41. [Google Scholar] [CrossRef]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef]
- Kenney, A.M.; Widlund, H.R.; Rowitch, D.H. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 2004, 131, 217–228. [Google Scholar] [CrossRef]
- Chesler, L.; Schlieve, C.; Goldenberg, D.D.; Kenney, A.; Kim, G.; McMillan, A.; Matthay, K.K.; Rowitch, D.; Weiss, W.A. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006, 66, 8139–8146. [Google Scholar] [CrossRef]
- Knoepfler, P.S.; Kenney, A.M. Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle 2006, 5, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Yue, M.; Su, H.; Ren, P.; Jiang, J.; Li, F.; Hu, Y.; Du, H.; Liu, H.; Qing, G. Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival. Mol. Cell 2016, 64, 493–506. [Google Scholar] [CrossRef]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.; Prochownik, E.V.; Metallo, S.J. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef]
- Larsson, L.; Hook, P.; Pircher, P. Regulation of human muscle contraction at the cellular and molecular levels. Ital. J. Neurol. Sci. 1999, 20, 413–422. [Google Scholar] [CrossRef]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed]
- Timme, N.; Han, Y.; Liu, S.; Yosief, H.O.; Garcia, H.D.; Bei, Y.; Klironomos, F.; MacArthur, I.C.; Szymansky, A.; von Stebut, J.; et al. Small-Molecule Dual PLK1 and BRD4 Inhibitors are Active Against Preclinical Models of Pediatric Solid Tumors. Transl. Oncol. 2020, 13, 221–232. [Google Scholar] [CrossRef]
- Ton, A.T.; Singh, K.; Morin, H.; Ban, F.; Leblanc, E.; Lee, J.; Lallous, N.; Cherkasov, A. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 8277. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Brideau, C.; Gunter, B.; Pikounis, B.; Liaw, A. Improved statistical methods for hit selection in high-throughput screening. J. Biomol. Screen. 2003, 8, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Malo, N.; Hanley, J.A.; Cerquozzi, S.; Pelletier, J.; Nadon, R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006, 24, 167–175. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Gewirtz, D.A.; Randolph, J.K.; Chawla, J.; Orr, M.S.; Fornari, F.A. Induction of DNA damage, inhibition of DNA synthesis and suppression of c-myc expression by the anthracycline analog, idarubicin (4-demethoxy-daunorubicin) in the MCF-7 breast tumor cell line. Cancer Chemother. Pharmacol. 1998, 41, 361–369. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, J.; Zhao, Y.; Fei, C.; Zheng, Q.; Li, X.; Chang, C. Chidamide, a novel histone deacetylase inhibitor, inhibits the viability of MDS and AML cells by suppressing JAK2/STAT3 signaling. Am. J. Transl. Res. 2016, 8, 3169–3178. [Google Scholar]
- Yang, J.; Milasta, S.; Hu, D.; AlTahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting Histone Demethylases in MYC-Driven Neuroblastomas with Ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Di Nisio, V.; Pietrantoni, I.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Ciccarelli, C.; Gravina, G.L.; Festuccia, C.; Del Fattore, A.; et al. Pro-differentiating and radiosensitizing effects of inhibiting HDACs by PXD-101 (Belinostat) in in vitro and in vivo models of human rhabdomyosarcoma cell lines. Cancer Lett. 2019, 461, 90–101. [Google Scholar] [CrossRef]
- Keating, M.J.; McLaughlin, P.; Plunkett, W.; Robertson, L.E.; O’Brien, S.; Gandhi, V.; Gregoire, V.; Yang, L.; Cabanillas, F. Fludarabine--present status and future developments in chronic lymphocytic leukemia and lymphoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 1994, 5 (Suppl. S2), 79–83. [Google Scholar] [CrossRef]
- Brockman, R.W.; Cheng, Y.C.; Schabel, F.M., Jr.; Montgomery, J.A. Metabolism and chemotherapeutic activity of 9-beta-D-arabinofuranosyl-2-fluoroadenine against murine leukemia L1210 and evidence for its phosphorylation by deoxycytidine kinase. Cancer Res. 1980, 40, 3610–3615. [Google Scholar] [PubMed]
- Huang, P.; Chubb, S.; Plunkett, W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 1990, 265, 16617–16625. [Google Scholar] [CrossRef]
- Li, L.; Keating, M.J.; Plunkett, W.; Yang, L.Y. Fludarabine-mediated repair inhibition of cisplatin-induced DNA lesions in human chronic myelogenous leukemia-blast crisis K562 cells: Induction of synergistic cytotoxicity independent of reversal of apoptosis resistance. Mol. Pharmacol. 1997, 52, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Moufarij, M.A.; Sampath, D.; Keating, M.J.; Plunkett, W. Fludarabine increases oxaliplatin cytotoxicity in normal and chronic lymphocytic leukemia lymphocytes by suppressing interstrand DNA crosslink removal. Blood 2006, 108, 4187–4193. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front. Biosci. A J. Virtual Libr. 1998, 3, d250–d268. [Google Scholar] [CrossRef]
- Littler, S.; Sloss, O.; Geary, B.; Pierce, A.; Whetton, A.D.; Taylor, S.S. Oncogenic MYC amplifies mitotic perturbations. Open Biol. 2019, 9, 190136. [Google Scholar] [CrossRef]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tong, G.; Zhang, Y.; Liang, S.; Tang, K.; Yang, Q. PGK1 Drives Hepatocellular Carcinoma Metastasis by Enhancing Metabolic Process. Int. J. Mol. Sci. 2017, 18, 1630. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.Y.; Ishdorj, G.; Graham, B.A.; Johnston, J.B.; Gibson, S.B. Valproic acid enhances fludarabine-induced apoptosis mediated by ROS and involving decreased AKT and ATM activation in B-cell-lymphoid neoplastic cells. Apoptosis 2014, 19, 191–200. [Google Scholar] [CrossRef]
- Pessetto, Z.Y.; Ma, Y.; Hirst, J.J.; von Mehren, M.; Weir, S.J.; Godwin, A.K. Drug repurposing identifies a synergistic combination therapy with imatinib mesylate for gastrointestinal stromal tumor. Mol. Cancer Ther. 2014, 13, 2276–2287. [Google Scholar] [CrossRef]
- Kikuchi, E.; Menendez, S.; Ozu, C.; Ohori, M.; Cordon-Cardo, C.; Logg, C.R.; Kasahara, N.; Bochner, B.H. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 2007, 14, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Voeks, D.; Martiniello-Wilks, R.; Madden, V.; Smith, K.; Bennetts, E.; Both, G.W.; Russell, P.J. Gene therapy for prostate cancer delivered by ovine adenovirus and mediated by purine nucleoside phosphorylase and fludarabine in mouse models. Gene Ther. 2002, 9, 759–768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hong, J.S.; Waud, W.R.; Levasseur, D.N.; Townes, T.M.; Wen, H.; McPherson, S.A.; Moore, B.A.; Bebok, Z.; Allan, P.W.; Secrist, J.A., 3rd; et al. Excellent in vivo bystander activity of fludarabine phosphate against human glioma xenografts that express the escherichia coli purine nucleoside phosphorylase gene. Cancer Res. 2004, 64, 6610–6615. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, Oxidative Stress, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Felsher, D.W. Tumor dormancy and MYC inactivation: Pushing cancer to the brink of normalcy. Cancer Res. 2005, 65, 4471–4474. [Google Scholar] [CrossRef] [PubMed]
- Moussay, E.; Palissot, V.; Vallar, L.; Poirel, H.A.; Wenner, T.; El Khoury, V.; Aouali, N.; Van Moer, K.; Leners, B.; Bernardin, F.; et al. Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol. Cancer 2010, 9, 115. [Google Scholar] [CrossRef] [PubMed]
- Oscier, D.; Orchard, J.A.; Culligan, D.; Cunningham, D.; Johnson, S.; Parker, A.; Klein, M.; Gieschen, H. The bioavailability of oral fludarabine phosphate is unaffected by food. Hematol. J. 2001, 2, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Lukenbill, J.; Kalaycio, M. Fludarabine: A review of the clear benefits and potential harms. Leuk. Res. 2013, 37, 986–994. [Google Scholar] [CrossRef]
- Muthalagu, N.; Monteverde, T.; Raffo-Iraolagoitia, X.; Wiesheu, R.; Whyte, D.; Hedley, A.; Laing, S.; Kruspig, B.; Upstill-Goddard, R.; Shaw, R.; et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2020, 10, 872–887. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elhasasna, H.; Khan, R.; Bhanumathy, K.K.; Vizeacoumar, F.S.; Walke, P.; Bautista, M.; Dahiya, D.K.; Maranda, V.; Patel, H.; Balagopal, A.; et al. A Drug Repurposing Screen Identifies Fludarabine Phosphate as a Potential Therapeutic Agent for N-MYC Overexpressing Neuroendocrine Prostate Cancers. Cells 2022, 11, 2246. https://doi.org/10.3390/cells11142246
Elhasasna H, Khan R, Bhanumathy KK, Vizeacoumar FS, Walke P, Bautista M, Dahiya DK, Maranda V, Patel H, Balagopal A, et al. A Drug Repurposing Screen Identifies Fludarabine Phosphate as a Potential Therapeutic Agent for N-MYC Overexpressing Neuroendocrine Prostate Cancers. Cells. 2022; 11(14):2246. https://doi.org/10.3390/cells11142246
Chicago/Turabian StyleElhasasna, Hussain, Raymond Khan, Kalpana K. Bhanumathy, Frederick S. Vizeacoumar, Prachi Walke, Maricris Bautista, Dinesh K. Dahiya, Vincent Maranda, Hardikkumar Patel, Amrutha Balagopal, and et al. 2022. "A Drug Repurposing Screen Identifies Fludarabine Phosphate as a Potential Therapeutic Agent for N-MYC Overexpressing Neuroendocrine Prostate Cancers" Cells 11, no. 14: 2246. https://doi.org/10.3390/cells11142246
APA StyleElhasasna, H., Khan, R., Bhanumathy, K. K., Vizeacoumar, F. S., Walke, P., Bautista, M., Dahiya, D. K., Maranda, V., Patel, H., Balagopal, A., Alli, N., Krishnan, A., Freywald, A., & Vizeacoumar, F. J. (2022). A Drug Repurposing Screen Identifies Fludarabine Phosphate as a Potential Therapeutic Agent for N-MYC Overexpressing Neuroendocrine Prostate Cancers. Cells, 11(14), 2246. https://doi.org/10.3390/cells11142246