
The Clinical Chameleon of Autoinflammatory Diseases in Children



Abstract
:1. Prelude to the Concept of Autoinflammation
2. The Classical Hereditary Periodic Fevers
2.1. Familial Mediterranean Fever
2.2. Tumor Necrosis Factor Receptor-Associated Periodic Syndrome
2.3. Cryopyrin-Associated Periodic Syndrome
2.4. Mevalonate Kinase Deficiency
3. The Pyogenic Diseases with an Autoinflammatory Basis
4. An Overview of the NFKB-Related Autoinflammatory Disorders
5. Looking through Interferon-Related Autoinflammatory Disorders
6. Insights on the Polygenic and Multifactorial Autoinflammatory Disorders in Children
7. Genetic In-Depth Analyses for Autoinflammatory Disorders
8. Conclusive Remarks
- -
- Dysregulated inflammasome activity and oversecretion of interleukin-1 are the ‘incipit’ of hereditary autoinflammatory diseases, caused by mutations in genes coding for inflammasome pieces and giving rise to protean scenarios in different pediatric settings.
- -
- Familial Mediterranean fever, tumor necrosis factor receptor-associated periodic syndrome, cryopyrin-associated periodic syndrome, mevalonate kinase deficiency, but also some idiopathic pyogenic diseases, granulomatous diseases and defects of the ubiquitin-proteasome pathway are monogenic autoinflammatory diseases with a distinct molecular pathogenesis.
- -
- In each of these diseases, inflammatory episodes recur over time, often with minimal evidence of a provocative event, and patients do not display high-titer autoantibodies or antigen-specific T cells that might serve as mediators.
- -
- A further cluster of diseases of unknown etiology with either a presumed polygenic or multifactorial origin, as systemic juvenile idiopathic arthritis, Kawasaki disease and periodic fever/aphthous stomatitis/pharyngitis/cervical adenopathy syndrome, is characterized by dysregulation of innate immune responses and overexpression of inflammasome-associated genes.
- -
- Despite the recent advances in genetic testing, the diagnosis of autoinflammatory diseases is based on a thorough knowledge of the clinical phenotype, and this knowledge should assist when children present unexplained episodes of fever or inflammation.
Author Contributions
Funding
Conflicts of Interest
References
- Place, D.E.; Kanneganti, T.-D. The Innate Immune System and Cell Death in Autoinflammatory and Autoimmune Disease. Curr. Opin. Immunol. 2020, 67, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Manna, R.; Rigante, D. The Everchanging Framework of Autoinflammation. Intern. Emerg. Med. 2021, 16, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Bodar, E.J.; Drenth, J.P.H.; van der Meer, J.W.M.; Simon, A. Dysregulation of Innate Immunity: Hereditary Periodic Fever Syndromes. Br. J. Haematol. 2009, 144, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D. A Developing Portrait of Hereditary Periodic Fevers in Childhood. Expert Opin. Orphan Drugs 2018, 6, 47–55. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.K.; Fitzgerald, K.A. Mechanisms of Inflammasome Activation: Recent Advances and Novel Insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D. The Fresco of Autoinflammatory Diseases from the Pediatric Perspective. Autoimmun. Rev. 2012, 11, 348–356. [Google Scholar] [CrossRef]
- Rigante, D.; Vitale, A.; Natale, M.F.; Cantarini, L. Lights and Shadows in Autoinflammatory Syndromes from the Childhood and Adulthood Perspective. Clin. Rheumatol. 2016, 35, 565–572. [Google Scholar] [CrossRef]
- Cantarini, L.; Rigante, D.; Brizi, M.G.; Lucherini, O.M.; Sebastiani, G.D.; Vitale, A.; Gianneramo, V.; Galeazzi, M. Clinical and Biochemical Landmarks in Systemic Autoinflammatory Diseases. Ann. Med. 2012, 44, 664–673. [Google Scholar] [CrossRef]
- Caso, F.; Cantarini, L.; Lucherini, O.M.; Sfriso, P.; Fioretti, M.; Costa, L.; Vitale, A.; Atteno, M.; Galeazzi, M.; Muscari, I.; et al. Working the Endless Puzzle of Hereditary Autoinflammatory Disorders. Mod. Rheumatol. 2014, 24, 381–389. [Google Scholar] [CrossRef]
- Borges, T.; Barbosa, A.; Silva, S. Adult-Onset Systemic Autoinflammatory Disorders: A Clinical Approach. Reumatismo 2020, 71, 177–188. [Google Scholar] [CrossRef]
- Rigante, D. New Mosaic Tiles in Childhood Hereditary Autoinflammatory Disorders. Immunol. Lett. 2018, 193, 67–76. [Google Scholar] [CrossRef]
- Bernard, E.M.; Broz, P. Activation and Manipulation of Inflammasomes and Pyroptosis during Bacterial Infections. Biochem. J. 2022, 479, 867–882. [Google Scholar] [CrossRef]
- Manna, R.; Rigante, D. Familial Mediterranean Fever: Assessing the Overall Clinical Impact and Formulating Treatment Plans. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019027. [Google Scholar] [CrossRef]
- Rigante, D.; Frediani, B.; Galeazzi, M.; Cantarini, L. From the Mediterranean to the Sea of Japan: The Transcontinental Odyssey of Autoinflammatory Diseases. Biomed. Res. Int. 2013, 2013, 485103. [Google Scholar] [CrossRef] [Green Version]
- Tufan, A.; Lachmann, H.J. Familial Mediterranean Fever, from Pathogenesis to Treatment: A Contemporary Review. Turk. J. Med. Sci. 2020, 50, 1591–1610. [Google Scholar] [CrossRef]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin Inflammasome Activation and RhoA Signaling in the Autoinflammatory Diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef]
- Magnotti, F.; Lefeuvre, L.; Benezech, S.; Malsot, T.; Waeckel, L.; Martin, A.; Kerever, S.; Chirita, D.; Desjonqueres, M.; Duquesne, A.; et al. Pyrin Dephosphorylation Is Sufficient to Trigger Inflammasome Activation in Familial Mediterranean Fever Patients. EMBO Mol. Med. 2019, 11, e10547. [Google Scholar] [CrossRef]
- Siligato, R.; Gembillo, G.; Calabrese, V.; Conti, G.; Santoro, D. Amyloidosis and Glomerular Diseases in Familial Mediterranean Fever. Medicina 2021, 57, 1049. [Google Scholar] [CrossRef]
- Rigante, D.; Lopalco, G.; Tarantino, G.; Compagnone, A.; Fastiggi, M.; Cantarini, L. Non-Canonical Manifestations of Familial Mediterranean Fever: A Changing Paradigm. Clin. Rheumatol. 2015, 34, 1503–1511. [Google Scholar] [CrossRef]
- Rigante, D.; La Torraca, I.; Avallone, L.; Pugliese, A.L.; Gaspari, S.; Stabile, A. The Pharmacologic Basis of Treatment with Colchicine in Children with Familial Mediterranean Fever. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 173–178. [Google Scholar]
- Varan, Ö.; Kucuk, H.; Babaoglu, H.; Guven, S.C.; Ozturk, M.A.; Haznedaroglu, S.; Goker, B.; Tufan, A. Efficacy and Safety of Interleukin-1 Inhibitors in Familial Mediterranean Fever Patients Complicated with Amyloidosis. Mod. Rheumatol. 2019, 29, 363–366. [Google Scholar] [CrossRef]
- Sohar, E.; Gafni, J.; Pras, M.; Heller, H. Familial Mediterranean Fever. A Survey of 470 Cases and Review of the Literature. Am. J. Med. 1967, 43, 227–253. [Google Scholar] [CrossRef]
- Livneh, A.; Langevitz, P.; Zemer, D.; Zaks, N.; Kees, S.; Lidar, T.; Migdal, A.; Padeh, S.; Pras, M. Criteria for the Diagnosis of Familial Mediterranean Fever. Arthritis Rheum. 1997, 40, 1879–1885. [Google Scholar] [CrossRef]
- Hull, K.M.; Drewe, E.; Aksentijevich, I.; Singh, H.K.; Wong, K.; McDermott, E.M.; Dean, J.; Powell, R.J.; Kastner, D.L. The TNF Receptor-Associated Periodic Syndrome (TRAPS): Emerging Concepts of an Autoinflammatory Disorder. Medicine 2002, 81, 349–368. [Google Scholar] [CrossRef]
- Todd, I.; Radford, P.M.; Daffa, N.; Bainbridge, S.E.; Powell, R.J.; Tighe, P.J. Mutant Tumor Necrosis Factor Receptor Associated with Tumor Necrosis Factor Receptor-Associated Periodic Syndrome Is Altered Antigenically and Is Retained within Patients’ Leukocytes. Arthritis Rheum. 2007, 56, 2765–2773. [Google Scholar] [CrossRef]
- Lobito, A.A.; Gabriel, T.L.; Medema, J.P.; Kimberley, F.C. Disease Causing Mutations in the TNF and TNFR Superfamilies: Focus on Molecular Mechanisms Driving Disease. Trends Mol. Med. 2011, 17, 494–505. [Google Scholar] [CrossRef]
- Rigante, D. The Broad-Ranging Panorama of Systemic Autoinflammatory Disorders with Specific Focus on Acute Painful Symptoms and Hematologic Manifestations in Children. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018067. [Google Scholar] [CrossRef]
- Rigante, D.; Lopalco, G.; Vitale, A.; Lucherini, O.M.; De Clemente, C.; Caso, F.; Emmi, G.; Costa, L.; Silvestri, E.; Andreozzi, L.; et al. Key Facts and Hot Spots on Tumor Necrosis Factor Receptor-Associated Periodic Syndrome. Clin. Rheumatol. 2014, 33, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Amatruda, M.; Carucci, N.S.; Fede, C.; Conti, G. Subclinical TRAPS Treated with Canakinumab. Reumatismo 2021, 73, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D.; Cantarini, L.; Imazio, M.; Lucherini, O.M.; Sacco, E.; Galeazzi, M.; Brizi, M.G.; Brucato, A. Autoinflammatory Diseases and Cardiovascular Manifestations. Ann. Med. 2011, 43, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Rigante, D.; Lucherini, O.M.; Cimaz, R.; Laghi Pasini, F.; Baldari, C.T.; Benucci, M.; Simonini, G.; Di Sabatino, V.; Brizi, M.G.; et al. Role of Etanercept in the Treatment of Tumor Necrosis Factor Receptor-Associated Periodic Syndrome: Personal Experience and Review of the Literature. Int. J. Immunopathol. Pharmacol. 2010, 23, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Vitale, A.; Obici, L.; Cattalini, M.; Lopalco, G.; Merlini, G.; Ricco, N.; Soriano, A.; La Torre, F.; Verrecchia, E.; Insalaco, A.; et al. Biotechnological Agents for Patients With Tumor Necrosis Factor Receptor Associated Periodic Syndrome-Therapeutic Outcome and Predictors of Response: Real-Life Data From the AIDA Network. Front. Med. 2021, 8, 668173. [Google Scholar] [CrossRef]
- Cantarini, L.; Lucherini, O.M.; Frediani, B.; Brizi, M.G.; Bartolomei, B.; Cimaz, R.; Galeazzi, M.; Rigante, D. Bridging the Gap between the Clinician and the Patient with Cryopyrin-Associated Periodic Syndromes. Int. J. Immunopathol. Pharm. 2011, 24, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef]
- Gaggiano, C.; Rigante, D.; Vitale, A.; Lucherini, O.M.; Fabbiani, A.; Capozio, G.; Marzo, C.; Gelardi, V.; Grosso, S.; Frediani, B.; et al. Hints for Genetic and Clinical Differentiation of Adult-Onset Monogenic Autoinflammatory Diseases. Mediat. Inflamm. 2019, 2019, 3293145. [Google Scholar] [CrossRef]
- Christensen, M.; Wallis, M.; Jessup, P.; Lemelle, I.; Jones, D.L. Cryopyrin-Associated Periodic Syndrome: A Treatable Genetic Inflammatory Condition. Pract. Neurol. 2021, 21, 424–426. [Google Scholar] [CrossRef]
- De Luca, E.; Guerriero, C.; Capozio, G.; Peris, K.; Rigante, D. Cold-Induced Urticaria in Children. Skinmed 2021, 19, 339–348. [Google Scholar]
- Rigante, D.; Cipolla, C.; Rossodivita, A. Recombinant Human Growth Hormone in Neonatal-Onset Multisystem Inflammatory Disease. Clin. Pediatr. Endocrinol. 2018, 27, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Federico, G.; Rigante, D.; Pugliese, A.L.; Ranno, O.; Catania, S.; Stabile, A. Etanercept Induces Improvement of Arthropathy in Chronic Infantile Neurological Cutaneous Articular (CINCA) Syndrome. Scand. J. Rheumatol. 2003, 32, 312–314. [Google Scholar] [CrossRef]
- Jesus, A.A.; Goldbach-Mansky, R. IL-1 Blockade in Autoinflammatory Syndromes. Annu. Rev. Med. 2014, 65, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D. A Systematic Approach to Autoinflammatory Syndromes: A Spelling Booklet for the Beginner. Expert Rev. Clin. Immunol. 2017, 13, 571–597. [Google Scholar] [CrossRef]
- Landmann, E.C.; Walker, U.A. Pharmacological Treatment Options for Cryopyrin-Associated Periodic Syndromes. Expert Rev. Clin. Pharmacol. 2017, 10, 855–864. [Google Scholar] [CrossRef]
- Rigante, D.; Ansuini, V.; Caldarelli, M.; Bertoni, B.; La Torraca, I.; Stabile, A. Hydrocephalus in CINCA Syndrome Treated with Anakinra. Childs Nerv. Syst. 2006, 22, 334–337. [Google Scholar] [CrossRef]
- Rigante, D.; Leone, A.; Marrocco, R.; Laino, M.E.; Stabile, A. Long-Term Response after 6-Year Treatment with Anakinra and Onset of Focal Bone Erosion in Neonatal-Onset Multisystem Inflammatory Disease (NOMID/CINCA). Rheumatol. Int. 2011, 31, 1661–1664. [Google Scholar] [CrossRef]
- Rigante, D. The Protean Visage of Systemic Autoinflammatory Syndromes: A Challenge for Inter-Professional Collaboration. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 1–18. [Google Scholar]
- Frenkel, J.; Houten, S.M.; Waterham, H.R.; Wanders, R.J.; Rijkers, G.T.; Duran, M.; Kuijpers, T.W.; van Luijk, W.; Poll-The, B.T.; Kuis, W. Clinical and Molecular Variability in Childhood Periodic Fever with Hyperimmunoglobulinaemia D. Rheumatology 2001, 40, 579–584. [Google Scholar] [CrossRef] [Green Version]
- van der Hilst, J.C.H.; Bodar, E.J.; Barron, K.S.; Frenkel, J.; Drenth, J.P.H.; van der Meer, J.W.M.; Simon, A.; International HIDS Study Group. Long-Term Follow-up, Clinical Features, and Quality of Life in a Series of 103 Patients with Hyperimmunoglobulinemia D Syndrome. Medicine 2008, 87, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D. Autoinflammatory Syndromes behind the Scenes of Recurrent Fevers in Children. Med. Sci. Monit. 2009, 15, RA179–RA187. [Google Scholar]
- Esposito, S.; Ascolese, B.; Senatore, L.; Bosis, S.; Verrecchia, E.; Cantarini, L.; Rigante, D. Current Advances in the Understanding and Treatment of Mevalonate Kinase Deficiency. Int. J. Immunopathol. Pharmacol. 2014, 27, 491–498. [Google Scholar] [CrossRef]
- Gattorno, M.; Hofer, M.; Federici, S.; Vanoni, F.; Bovis, F.; Aksentijevich, I.; Anton, J.; Arostegui, J.I.; Barron, K.; Ben-Cherit, E.; et al. Classification Criteria for Autoinflammatory Recurrent Fevers. Ann. Rheum. Dis. 2019, 78, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Tallon, B.; Corkill, M. Peculiarities of PAPA Syndrome. Rheumatology 2006, 45, 1140–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, P.J.; Chen, S.; Tayeh, M.K.; Ochoa, L.; Leal, S.M.; Pelet, A.; Munnich, A.; Lyonnet, S.; Majeed, H.A.; El-Shanti, H. Homozygous Mutations in LPIN2 Are Responsible for the Syndrome of Chronic Recurrent Multifocal Osteomyelitis and Congenital Dyserythropoietic Anaemia (Majeed Syndrome). J. Med. Genet. 2005, 42, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesus, A.A.; Osman, M.; Silva, C.A.; Kim, P.W.; Pham, T.-H.; Gadina, M.; Yang, B.; Bertola, D.R.; Carneiro-Sampaio, M.; Ferguson, P.J.; et al. A Novel Mutation of IL1RN in the Deficiency of Interleukin-1 Receptor Antagonist Syndrome: Description of Two Unrelated Cases from Brazil. Arthritis Rheum. 2011, 63, 4007–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinkel, C.; Thomsen, S.F. Autoinflammatory Syndromes Associated with Hidradenitis Suppurativa and/or Acne. Int. J. Dermatol. 2017, 56, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Wollina, U. Emerging Treatments for Pyoderma Gangrenosum. Expert Opin. Orphan Drugs 2017, 5, 827–832. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Takada, Y.; Shishodia, S.; Gutierrez, A.M.; Oommen, O.V.; Ichikawa, H.; Baba, Y.; Kumar, A. Nuclear Transcription Factor NF-Kappa B: Role in Biology and Medicine. Indian J. Exp. Biol. 2004, 42, 341–353. [Google Scholar] [PubMed]
- Sfriso, P.; Caso, F.; Tognon, S.; Galozzi, P.; Gava, A.; Punzi, L. Blau Syndrome, Clinical and Genetic Aspects. Autoimmun. Rev. 2012, 12, 44–51. [Google Scholar] [CrossRef]
- Wouters, C.H.; Maes, A.; Foley, K.P.; Bertin, J.; Rose, C.D. Blau Syndrome, the Prototypic Auto-Inflammatory Granulomatous Disease. Pediatr. Rheumatol. 2014, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D. Phenotype Variability of Autoinflammatory Disorders in the Pediatric Patient: A Pictorial Overview. J. Evid. Based Med. 2020, 13, 227–245. [Google Scholar] [CrossRef]
- Caso, F.; Costa, L.; Rigante, D.; Vitale, A.; Cimaz, R.; Lucherini, O.M.; Sfriso, P.; Verrecchia, E.; Tognon, S.; Bascherini, V.; et al. Caveats and Truths in Genetic, Clinical, Autoimmune and Autoinflammatory Issues in Blau Syndrome and Early Onset Sarcoidosis. Autoimmun. Rev. 2014, 13, 1220–1229. [Google Scholar] [CrossRef]
- Poline, J.; Fogel, O.; Pajot, C.; Miceli-Richard, C.; Rybojad, M.; Galeotti, C.; Grouteau, E.; Hachulla, E.; Brissaud, P.; Cantagrel, A.; et al. Early-Onset Granulomatous Arthritis, Uveitis and Skin Rash: Characterization of Skin Involvement in Blau Syndrome. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 340–348. [Google Scholar] [CrossRef]
- Hospach, T.; Glowatzki, F.; Blankenburg, F.; Conzelmann, D.; Stirnkorb, C.; Müllerschön, C.S.; von den Driesch, P.; Köhler, L.M.; Rohlfs, M.; Klein, C.; et al. Scoping Review of Biological Treatment of Deficiency of Interleukin-36 Receptor Antagonist (DITRA) in Children and Adolescents. Pediatr. Rheumatol. Online J. 2019, 17, 37. [Google Scholar] [CrossRef] [Green Version]
- Frare, C.P.; Blumstein, A.J.; Paller, A.S.; Pieretti, L.; Choate, K.A.; Bowcock, A.M.; Larralde, M. CARD14-Associated Papulosquamous Eruption (CAPE) in Pediatric Patients: Three Additional Cases and Review of the Literature. Pediatr. Dermatol. 2021, 38, 1237–1242. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Zhou, Q. NF-ΚB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 399. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic Hypomorphic Mutations in a Linear Deubiquitinase Define Otulipenia, an Early-Onset Autoinflammatory Disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Deng, M.; Gan, C.; Wang, L.; Mao, H.; Li, Q. A Novel Missense Mutation in TNFAIP3 Causes Haploinsufficiency of A20. Cell Immunol. 2022, 371, 104453. [Google Scholar] [CrossRef]
- Poulter, J.A.; Collins, J.C.; Cargo, C.; De Tute, R.M.; Evans, P.; Ospina Cardona, D.; Bowen, D.T.; Cunnington, J.R.; Baguley, E.; Quinn, M.; et al. Novel Somatic Mutations in UBA1 as a Cause of VEXAS Syndrome. Blood 2021, 137, 3676–3681. [Google Scholar] [CrossRef]
- Vitale, A.; Rigante, D.; Maggio, M.C.; Emmi, G.; Romano, M.; Silvestri, E.; Lucherini, O.M.; Emmi, L.; Gerloni, V.; Cantarini, L. Rare NLRP12 Variants Associated with the NLRP12-Autoinflammatory Disorder Phenotype: An Italian Case Series. Clin. Exp. Rheumatol. 2013, 31, 155–156. [Google Scholar]
- McDermott, A.; Jacks, J.; Kessler, M.; Emanuel, P.D.; Gao, L. Proteasome-Associated Autoinflammatory Syndromes: Advances in Pathogeneses, Clinical Presentations, Diagnosis, and Management. Int. J. Dermatol. 2015, 54, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Bienias, M.; Brück, N.; Griep, C.; Wolf, C.; Kretschmer, S.; Kind, B.; Tüngler, V.; Berner, R.; Lee-Kirsch, M.A. Therapeutic Approaches to Type I Interferonopathies. Curr. Rheumatol. Rep. 2018, 20, 32. [Google Scholar] [CrossRef]
- Torrelo, A. CANDLE Syndrome As a Paradigm of Proteasome-Related Autoinflammation. Front. Immunol. 2017, 8, 927. [Google Scholar] [CrossRef] [Green Version]
- Araújo-Vilar, D.; Santini, F. Diagnosis and Treatment of Lipodystrophy: A Step-by-Step Approach. J. Endocrinol. Invest. 2019, 42, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, F.; Zhang, X. STING-Associated Vasculopathy with Onset in Infancy: A Familial Case Series Report and Literature Review. Ann. Transl. Med. 2021, 9, 176. [Google Scholar] [CrossRef]
- Crow, Y.J.; Vanderver, A.; Orcesi, S.; Kuijpers, T.W.; Rice, G.I. Therapies in Aicardi-Goutières Syndrome. Clin. Exp. Immunol. 2014, 175, 1–8. [Google Scholar] [CrossRef]
- Cetin Gedik, K.; Lamot, L.; Romano, M.; Demirkaya, E.; Piskin, D.; Torreggiani, S.; Adang, L.A.; Armangue, T.; Barchus, K.; Cordova, D.R.; et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology Points to Consider for Diagnosis and Management of Autoinflammatory Type I Interferonopathies: CANDLE/PRAAS, SAVI and AGS. Ann. Rheum. Dis. 2022, 81, 601–613. [Google Scholar] [CrossRef]
- Wekell, P.; Karlsson, A.; Berg, S.; Fasth, A. Review of Autoinflammatory Diseases, with a Special Focus on Periodic Fever, Aphthous Stomatitis, Pharyngitis and Cervical Adenitis Syndrome. Acta Paediatr. 2016, 105, 1140–1151. [Google Scholar] [CrossRef]
- Malcova, H.; Milota, T.; Strizova, Z.; Cebecauerova, D.; Striz, I.; Sediva, A.; Horvath, R. Interleukin-1 Blockade in Polygenic Autoinflammatory Disorders: Where Are We Now? Front. Pharmacol. 2020, 11, 619273. [Google Scholar] [CrossRef] [PubMed]
- Stabile, A.; Bertoni, B.; Ansuini, V.; La Torraca, I.; Sallì, A.; Rigante, D. The Clinical Spectrum and Treatment Options of Macrophage Activation Syndrome in the Pediatric Age. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 53–59. [Google Scholar]
- Rigante, D.; Capoluongo, E.; Bertoni, B.; Ansuini, V.; Chiaretti, A.; Piastra, M.; Pulitanò, S.; Genovese, O.; Compagnone, A.; Stabile, A. First Report of Macrophage Activation Syndrome in Hyperimmunoglobulinemia D with Periodic Fever Syndrome. Arthritis Rheum. 2007, 56, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D.; Emmi, G.; Fastiggi, M.; Silvestri, E.; Cantarini, L. Macrophage Activation Syndrome in the Course of Monogenic Autoinflammatory Disorders. Clin. Rheumatol. 2015, 34, 1333–1339. [Google Scholar] [CrossRef]
- Grevich, S.; Shenoi, S. Update on the Management of Systemic Juvenile Idiopathic Arthritis and Role of IL-1 and IL-6 Inhibition. Adolesc. Health Med. Ther. 2017, 8, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Martini, A.; Ravelli, A.; Avcin, T.; Beresford, M.W.; Burgos-Vargas, R.; Cuttica, R.; Ilowite, N.T.; Khubchandani, R.; Laxer, R.M.; Lovell, D.J.; et al. Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J. Rheumatol. 2019, 46, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcini, F.; Capannini, S.; Rigante, D. Kawasaki Syndrome: An Intriguing Disease with Numerous Unsolved Dilemmas. Pediatr. Rheumatol. Online J. 2011, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, A.; Rigante, D.; Cimaz, R.; Ravelli, A.; Tarissi de Jacobis, I.; Rimini, A.; Cardinale, F.; Cattalini, M.; De Zorzi, A.; Dellepiane, R.M.; et al. Revised Recommendations of the Italian Society of Pediatrics about the General Management of Kawasaki Disease. Ital. J. Pediatr. 2021, 47, 16. [Google Scholar] [CrossRef]
- Rigante, D. Kawasaki Disease as the Immune-Mediated Echo of a Viral Infection. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020039. [Google Scholar] [CrossRef]
- Rigante, D.; Valentini, P.; Rizzo, D.; Leo, A.; De Rosa, G.; Onesimo, R.; De Nisco, A.; Angelone, D.F.; Compagnone, A.; Delogu, A.B. Responsiveness to Intravenous Immunoglobulins and Occurrence of Coronary Artery Abnormalities in a Single-Center Cohort of Italian Patients with Kawasaki Syndrome. Rheumatol. Int. 2010, 30, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D.; Andreozzi, L.; Fastiggi, M.; Bracci, B.; Natale, M.F.; Esposito, S. Critical Overview of the Risk Scoring Systems to Predict Non-Responsiveness to Intravenous Immunoglobulin in Kawasaki Syndrome. Int. J. Mol. Sci. 2016, 17, 278. [Google Scholar] [CrossRef] [Green Version]
- Bordea, M.A.; Costache, C.; Grama, A.; Florian, A.I.; Lupan, I.; Samasca, G.; Deleanu, D.; Makovicky, P.; Makovicky, P.; Rimarova, K. Cytokine Cascade in Kawasaki Disease versus Kawasaki-like Syndrome. Physiol. Res. 2022, 71, 17–27. [Google Scholar] [CrossRef]
- Koné-Paut, I.; Tellier, S.; Belot, A.; Brochard, K.; Guitton, C.; Marie, I.; Meinzer, U.; Cherqaoui, B.; Galeotti, C.; Boukhedouni, N.; et al. Phase II Open Label Study of Anakinra in Intravenous Immunoglobulin-Resistant Kawasaki Disease. Arthritis Rheumatol. 2021, 73, 151–161. [Google Scholar] [CrossRef]
- Gentileschi, S.; Vitale, A.; Frediani, B.; Galeazzi, M.; Rigante, D.; Cantarini, L. Challenges and New Horizons in the Periodic Fever, Aphthous Stomatitis, Pharyngitis and Adenitis (PFAPA) Syndrome. Expert Opin. Orphan Drugs 2017, 5, 165–171. [Google Scholar] [CrossRef]
- Esposito, S.; Bianchini, S.; Fattizzo, M.; Baggi, E.; Marchisio, P.; Rigante, D. The Enigma of Periodic Fever, Aphthous Stomatitis, Pharyngitis and Adenitis Syndrome. Pediatr. Infect. Dis. J. 2014, 33, 650–652. [Google Scholar] [CrossRef]
- Lazzareschi, I.; Rossi, E.; Curatola, A.; Capozio, G.; Benacquista, L.; Iezzi, L.; Rigante, D. Assessment of Congenital Neutropenia in Children: Common Clinical Sceneries and Clues for Management. Mediterr. J. Hematol. Infect. Dis. 2022, 14, e2022008. [Google Scholar] [CrossRef]
- Rigante, D.; Vitale, A.; Natale, M.F.; Lopalco, G.; Andreozzi, L.; Frediani, B.; D’Errico, F.; Iannone, F.; Cantarini, L. A Comprehensive Comparison between Pediatric and Adult Patients with Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Cervical Adenopathy (PFAPA) Syndrome. Clin. Rheumatol. 2017, 36, 463–468. [Google Scholar] [CrossRef]
- Sicignano, L.L.; Rigante, D.; Moccaldi, B.; Massaro, M.G.; Delli Noci, S.; Patisso, I.; Capozio, G.; Verrecchia, E.; Manna, R. Children and Adults with PFAPA Syndrome: Similarities and Divergences in a Real-Life Clinical Setting. Adv. Ther. 2021, 38, 1078–1093. [Google Scholar] [CrossRef]
- Rigante, D.; Corina, L. Periodic Fever, Aphthous Stomatitis, Pharyngitis and Cervical Adenitis (PFAPA) Syndrome: A Debate about Diagnosis and Treatment in Children Continues. Int. J. Pediatr. Otorhinolaryngol. 2020, 130, 109830. [Google Scholar] [CrossRef]
- Gaggiano, C.; Rigante, D.; Sota, J.; Grosso, S.; Cantarini, L. Treatment Options for Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Cervical Adenitis (PFAPA) Syndrome in Children and Adults: A Narrative Review. Clin. Rheumatol. 2019, 38, 11–17. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Azzarà, A.; Molinario, C.; Pietrobono, R.; Rigante, D.; Verrecchia, E.; Sicignano, L.L.; Genuardi, M.; Gurrieri, F.; Manna, R. Rare Missense Variants in the ALPK1 Gene May Predispose to Periodic Fever, Aphthous Stomatitis, Pharyngitis and Adenitis (PFAPA) Syndrome. Eur. J. Hum. Genet. 2019, 27, 1361–1368. [Google Scholar] [CrossRef]
- Williams, L.B.; Javed, A.; Sabri, A.; Morgan, D.J.; Huff, C.D.; Grigg, J.R.; Heng, X.T.; Khng, A.J.; Hollink, I.H.I.M.; Morrison, M.A.; et al. ALPK1 Missense Pathogenic Variant in Five Families Leads to ROSAH Syndrome, an Ocular Multisystem Autosomal Dominant Disorder. Genet. Med. 2019, 21, 2103–2115. [Google Scholar] [CrossRef] [Green Version]
- Marshall, G.S.; Edwards, K.M.; Butler, J.; Lawton, A.R. Syndrome of Periodic Fever, Pharyngitis, and Aphthous Stomatitis. J. Pediatr. 1987, 110, 43–46. [Google Scholar] [CrossRef]
- Cantarini, L.; Vitale, A.; Sicignano, L.L.; Emmi, G.; Verrecchia, E.; Patisso, I.; Cerrito, L.; Fabiani, C.; Cevenini, G.; Frediani, B.; et al. Diagnostic Criteria for Adult-Onset Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Cervical Adenitis (PFAPA) Syndrome. Front. Immunol. 2017, 8, 1018. [Google Scholar] [CrossRef] [Green Version]
- Rama, M.; Mura, T.; Koné-Paut, I.; Boursier, G.; Aouinti, S.; Touitou, I.; Sarrabay, G. Is Gene Panel Sequencing More Efficient than Clinical-Based Gene Sequencing to Diagnose Autoinflammatory Diseases? A Randomized Study. Clin. Exp. Immunol. 2021, 203, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D. When, How, and Why Do Fevers Hold Children Hostage? J. Evid. Based Med. 2020, 13, 85–88. [Google Scholar] [CrossRef] [PubMed]
- De Rose, D.U.; Coppola, M.; Gallini, F.; Maggio, L.; Vento, G.; Rigante, D. Overview of the Rarest Causes of Fever in Newborns: Handy Hints for the Neonatologist. J. Perinatol. 2021, 41, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Lopalco, G.; Selmi, C.; Napodano, S.; De Rosa, G.; Caso, F.; Costa, L.; Iannone, F.; Rigante, D. Autoimmunity and Autoinflammation as the Yin and Yang of Idiopathic Recurrent Acute Pericarditis. Autoimmun. Rev. 2015, 14, 90–97. [Google Scholar] [CrossRef]
- Cantarini, L.; Iacoponi, F.; Lucherini, O.M.; Obici, L.; Brizi, M.G.; Cimaz, R.; Rigante, D.; Benucci, M.; Sebastiani, G.D.; Brucato, A.; et al. Validation of a Diagnostic Score for the Diagnosis of Autoinflammatory Diseases in Adults. Int. J. Immunopathol. Pharmacol. 2011, 24, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Piram, M.; Koné-Paut, I.; Lachmann, H.J.; Frenkel, J.; Ozen, S.; Kuemmerle-Deschner, J.; Stojanov, S.; Simon, A.; Finetti, M.; Sormani, M.P.; et al. Validation of the Auto-Inflammatory Diseases Activity Index (AIDAI) for Hereditary Recurrent Fever Syndromes. Ann. Rheum. Dis. 2014, 73, 2168–2173. [Google Scholar] [CrossRef]
- Ter Haar, N.M.; Annink, K.V.; Al-Mayouf, S.M.; Amaryan, G.; Anton, J.; Barron, K.S.; Benseler, S.M.; Brogan, P.A.; Cantarini, L.; Cattalini, M.; et al. Development of the Autoinflammatory Disease Damage Index (ADDI). Ann. Rheum. Dis. 2017, 76, 821–830. [Google Scholar] [CrossRef]
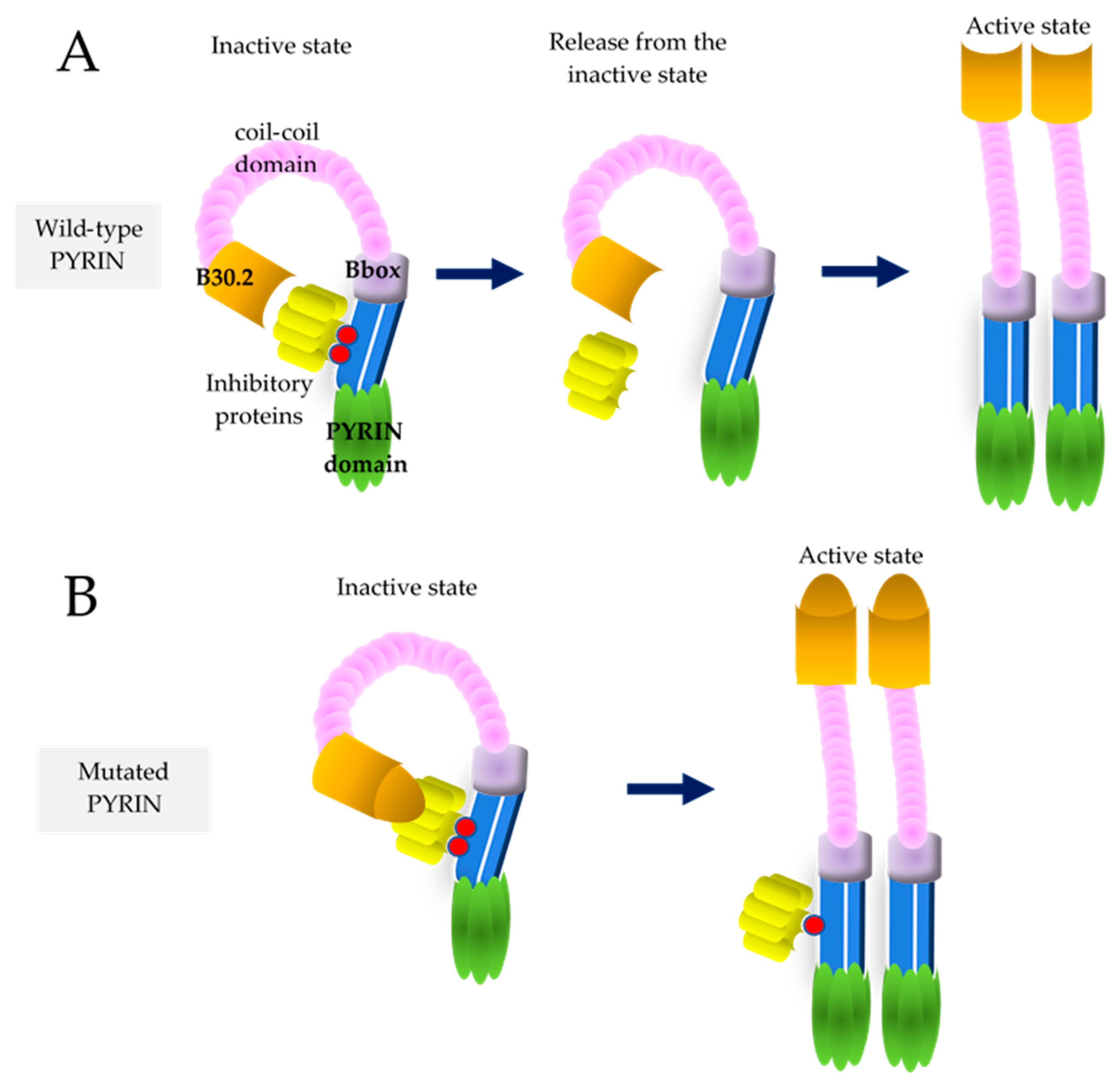

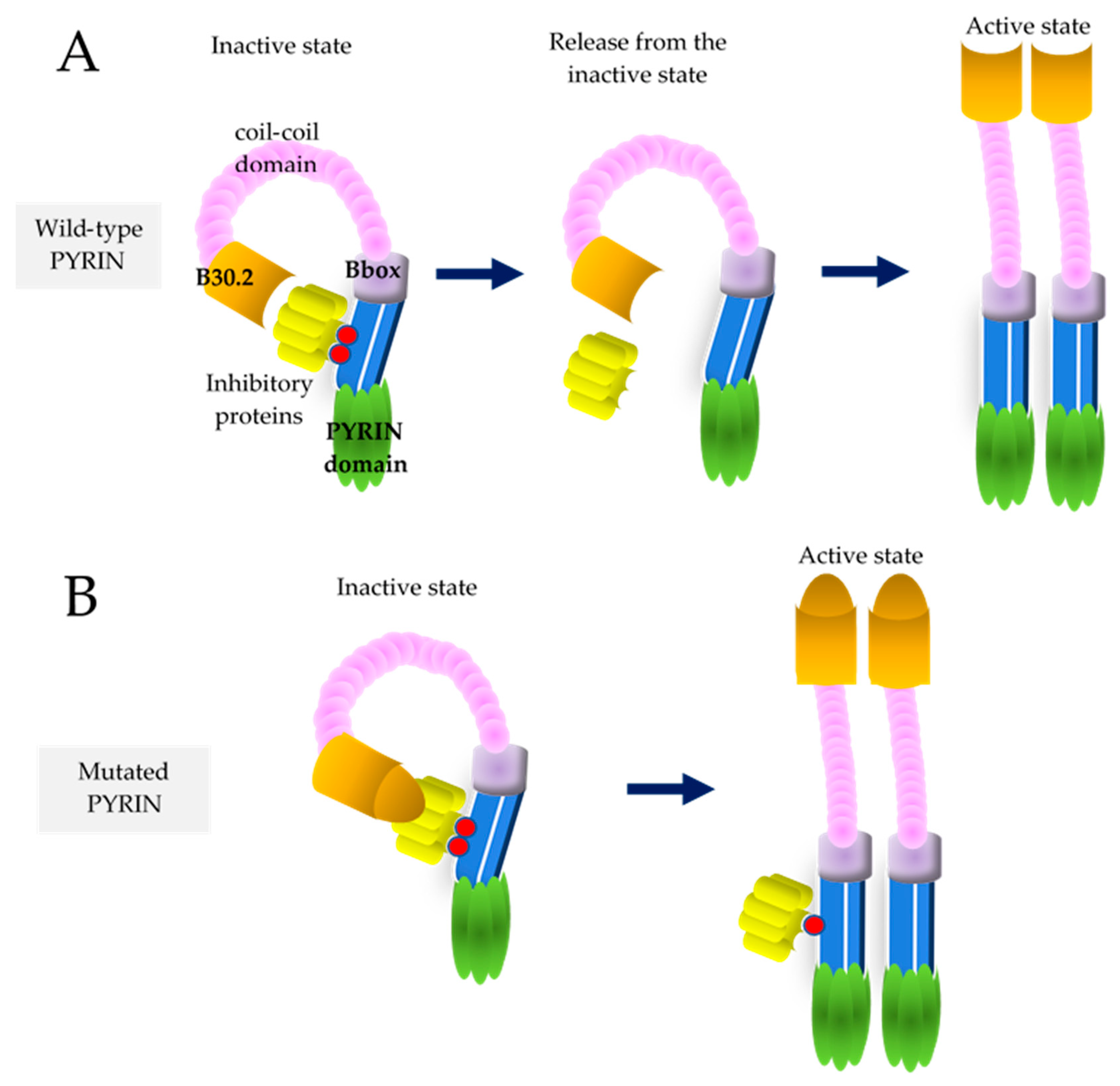{kind=link}
| Gene Locus | Protein | Inheritance | Main Manifestations and Complications | Available Treatments | |
|---|---|---|---|---|---|
| FMF | MEFV 16p13.3 | PYRIN or marenostrin | AR | serositis, limb pain or transient arthritis, erysipelas-like eruption on the legs, nonspecific skin manifestations (like urticaria, angioedema, erythema nodosum, vasculitis), risk of amyloidosis | Colchicine, canakinumab, anakinra |
| TRAPS | TNFRSF1A 12p13 | TNFRSF1A, TNF receptor | AD | severe migrating muscle pain, arthralgia or arthritis, serositis, painful orbital edema, painful conjunctivitis, risk of amyloidosis | Canakinumab, anakinra, corticosteroids |
| FCAS | NLRP3 1q44 | CRYOPYRIN | AD | cold-induced urticaria-like rashes, conjunctivitis, arthralgia | Anakinra, rilonacept, canakinumab |
| MWS | cold-induced urticaria-like rash, conjunctivitis, episcleritis, arthralgia, neurosensorial deafness, risk of amyloidosis | ||||
| CINCA | migrating non-itchy urticaria-like rash, uveitis, papilledema, deforming arthritis involving large joints, aseptic chronic meningopathy, retinal dystrophy, neurosensorial deafness, risk of amyloidosis | ||||
| MKD | MVK 12q24 | MEVALONATE KINASE | AR | fatigue, painful generalized lymph node enlargement, vomiting, diarrhea, abdominal pain, arthralgia, skin rashes of varying severity, oral and/or genital aphthosis, splenomegaly during flares | Anti-inflammatory drugs, corticosteroids, anakinra ‘on demand’, canakinumab |
| PAPA | PSTPIP1 15q24–25 | PSTPIP1 (proline-serine- threonine phosphatase interacting protein 1) | AD | sterile pyogenic arthritis, pyoderma gangrenosum, severe acne, skin abscesses, recurrent non-healing sterile ulcers | Corticosteroids, infliximab, anakinra, immunosuppressive agents |
| MS | LPIN2 18p11.31 | LIPIN2 (phosphatidate phosphatase) | AR | recurrent multifocal osteomyelitis, neutrophilic dermatosis, dyserythropoietic anemia | Corticosteroids, bisphosphonates, TNF-α inhibitors, IL-1 antagonists (anakinra) |
| DIRA | IL1RN 2q14.1 | IL1RN (interleukin-1 receptor antagonist) | AR | sterile multifocal osteomyelitis starting in the neonatal period, skin pustulosis, osteitis | Anakinra |
| BS | NOD2 (CARD15) 16q12.1–13 | NOD2 (nucleotide binding oligomerization domain containing 2) | AD | non-erosive granulomatous polyarthritis (‘boggy synovitis’ with painless effusion and cyst-like swelling of joints), granulomatous panuveitis, skin granulomatous rash | Corticosteroids, TNF-α inhibitors (infliximab), IL-1 antagonists, JAK inhibitors (tofacitinib) |
| DITRA | IL36RN 2q14.1 | IL36RN (interleukin-36 receptor antagonist) | AR | severe pustular psoriasis (generalized or limited to the distal part of limbs) | TNF-α inhibitors (adalimumab), IL-12/23 antagonists, IL-17 antagonists |
| CAMPS | CARD14 17q25.3 | CARD14 (caspase recruitment domain- containing protein 14) | AD | psoriasis in a wide range of phenotypes | Methotrexate, corticosteroids, cyclosporine, phototherapy, acitretin, vitamin D analogs, TNF-α inhibitors, IL-12/23 antagonists, IL-17 antagonists |
| ORAS | OTULIN 5p15.2 | OTULIN (deubiquitinase) | AR | fever starting in the neonatal period, neutrophilic dermatosis associated with panniculitis, growth retardation | TNF-α inhibitors |
| HA20 | TNFAIP3 6q23.3 | TNFAIP3 (tumor necrosis factor alpha-induced protein 3, A20) | AD | recurrent mucosal ulcerations of the oral cavity, gastrointestinal tube and urogenital tract, skin rashes, polyarthritis, uveitis, vasculitides, recurrent fevers, association with different autoimmune disorders (systemic lupus erythematosus, psoriatic arthritis, juvenile idiopathic arthritis, autoimmune hepatitis and Hashimoto thyroiditis) | TNF-α inhibitors, colchicine |
| FCAS2 | NLRP12 19q13.42 | NLRP12 (nucleotide- binding oligomerization domain, leucine rich repeat and pyrin domain containing 12) | AD | cold-induced rashes, joint pain, abdominal pain, sensorineural deafness, headache | Anakinra, TNF-α inhibitors, IL-6 antagonists (tocilizumab) |
| PRAAS | PSMB8 | PSMB8 (proteasome 20s subunit beta 8) | AR | chronic atypical neutrophilic dermatosis, lipodystrophy, erythema nodosum-like panniculitis, abnormal growth of lips, muscular weakness and atrophy, severe joint contractures, basal ganglia calcifications, ear and nose chondritis, aseptic meningitis, conjunctivitis, hepatosplenomegaly, lymph node enlargement, arthralgia | Corticosteroids, immunosuppressive agents, anakinra, IL-6 antagonists (tocilizumab), TNF-α inhibitors, dapsone, JAK inhibitors (baricitinib) |
| SAVI | STING1 (TMEM173) 5q31.2 | STING1 (stimulator of interferon genes protein 1) | AD | vasculopathy causing severe skin lesions on face, ears, nose and digits, resulting in ulcerations, necrosis or amputations, chronic interstitial lung disease | JAK inhibitors (ruxolitinib) |
| AGS | TREX1, RNASEH2B, RNASEH2C, RNASEH2A, SAMHD1, ADAR, IFIH1 3p21.31, 13q14.3, 11q13.1, 19p13.13, 20q11.23, 1q21.3, 2q24.2 | Enzymes involved in the duplication, repair and recombination of nucleic acids | AR (AD for IFIH1) | leukoencephalopathy (mimicking transplacental infections), calcifications in cerebral and basal ganglia, dystonia, microcephaly, cognitive impairment, abnormal eye movements, glaucoma, livedo reticularis, digital chilblain lesions on hands and feet, hepatosplenomegaly, jaundice, silent positivity of autoantibodies | No cure is available, corticosteroids and intravenous immunoglobulin may control systemic and organ inflammation |
| Tel Hashomer Criteria | Livneh’s Criteria |
|---|---|
| Major | Major |
| Recurrent fevers + peritonitis/pleurisy/serositis | Typical attack of peritonitis |
| AA-amyloidosis | Typical attack of unilateral pleuritic or pericarditis |
| Favourable response to prophylaxis with colchicine | Typical attack of monoarthritis |
| Minor | Fever (rectal temperature of 38 °C or higher) alone |
| Recurrent fevers | Minor |
| Erysipelas-like erythema | Incomplete attack involving the abdomen |
| Family history of FMF in a first-degree relative | Incomplete attack involving the chest |
| Incomplete attack involving one large joint | |
| Exertional leg pain | |
| Favourable response to prophylaxis with colchicine | |
| Supportive | |
| Family history of familial Mediterranean fever | |
| Typical ethnic origin (Armenian, Turkish, Arabian, Sephardic Jew) | |
| Age less than 20 years at disease onset | |
| Severity of attacks requiring bed rest | |
| Spontaneous remission of attacks | |
| Symptom-free intervals between attacks | |
| Transient increase of inflammatory parameters during attacks | |
| Episodic proteinuria or hematuria | |
| Surgical removal of a “white” appendix | |
| Consanguinity of parents |
| Familial Mediterranean Fever (FMF) | Autosomal Dominant Familial Periodic Fever (Tumor Necrosis Factor Receptor-Associated Periodic Syndrome, TRAPS) | Cryopyrin-Associated Periodic Syndrome (CAPS) | Mevalonate Kinase Deficiency (MKD, Hyper-IgD Syndrome) |
|---|---|---|---|
| Presence of confirmatory MEFV genotype and at least 1 among: - Attacks lasting 1–3 days - Arthritis - Chest pain - Abdominal pain or Presence of a non-confirmatory MEFV genotype and at least 2 among: - Attacks lasting 1–3 days - Arthritis - Chest pain - Abdominal pain | Presence of confirmatory TNFRSF1A genotype and at least 1 among: - Attacks lasting ≥7 days - Myalgia - Migratory skin rash - Positive family history or Presence of a non-confirmatory TNFRSF1A genotype and at least 2 among: - Attacks lasting ≥7 days - Myalgia - Migratory skin rash - Positive family history | Presence of confirmatory NLRP3 genotype and at least 1 among: - Urticaria-like rash - Eye inflammation - Sensorineural hearing loss or Presence of a non-confirmatory NLRP3 genotype and at least 2 among: - Urticaria-like rash - Eye inflammation - Sensorineural hearing loss | Presence of confirmatory MVK genotype and at least 1 among: - Gastrointestinal symptoms - Cervical lymphadenopathy - Aphthous stomatitis |
| Onset | Clinical Signs Observed in Children at Onset | |
|---|---|---|
| Pyogenic arthritis/pyoderma gangrenosum/acne (PAPA) syndrome | First infancy | Skin ulcerations, pyoderma gangrenosum often associated with cystic acne, sterile pyogenic oligoarthritides |
| Majeed syndrome (MS) | First 2 years of life | Diffuse neutrophilic dermatosis, chronic non-bacterial osteomyelitis, dyserythropoietic anemia |
| Deficiency of the interleukin-1 receptor antagonist (DIRA) | Neonatal period | Pustular rash, nail abnormalities, ichthyosis-like changes of the skin, multifocal osteomyelitis, multi-organ failure |
| Onset | Clinical signs observed in children at onset | |
|---|---|---|
| Blau syndrome (BS) | First infancy | Brown-coloured scaly and ichthyosis-like or lichenoid rashes, recurrent polyarthritis, granulomatous uveitis (anterior, posterior or intermediate), risk of ocular sequelae (synechiae, cataracts, band keratopathy) |
| Deficiency of the interleukin-36 receptor antagonist (DITRA) | Variable (many cases may start in the first infancy) | Generalized severe pustular psoriasis, acute generalized exanthematous pustulosis, pustulosis of palms and soles, disseminated subcorneal pustules, Hallopeau’s acrodermatitis continua, recurrent fevers |
| CARD14-mediated psoriasis (CAMPS) | Variable (many cases may start in the first infancy) | Plaque psoriasis, pityriasis rubra pilaris, pustular psoriasis, joint pain, recurrent fevers |
| OTULIN-related autoinflammatory syndrome (ORAS) | First infancy | Erythematous skin rash with nodules, joint and abdominal pain, diarrhea, lymph node enlargement, stunted growth, recurrent fevers |
| Haploinsufficiency of A20 (HA20) | First or second decade | Early-onset Behçet’s-like disease signs as aphthous stomatitis, oral and genital ulcers and/or uveitis combined with diffuse lymphadenopathy, arthritis, recurrent fevers |
| Onset | Clinical Signs Observed in Children at Onset | |
|---|---|---|
| Proteasome-associated autoinflammatory syndromes (PRAAS) | Early childhood | Recurrent fevers, nodular skin rashes and annular violaceous plaques evolving to panniculitis-induced lipodystrophy (loss of adipose tissue), large nose, lips and ears, eyelid swelling, muscle weakness and atrophy, joint contractures, disproportionately long and thick fingers, hepatosplenomegaly, basal ganglia calcifications |
| STING-associated vasculopathy with infancy onset (SAVI) | Neonatal period | Skin vasculitis with violaceous scaling or pernio-like lesions on fingers, toes or nose, usually exacerbated by cold exposure, that progress to ulcerations of extremities or to autoamputation phenomena, chronic interstitial lung disease |
| Aicardi-Goutières syndrome (AGS) | Neonatal or prenatal period | Subacute encephalopathy, cerebral and basal ganglia calcifications, spasticity, dystonia, microcephaly, eye abnormalities, livedo reticularis, digital vasculitis with chilblain lesions on hands and feet, hepatosplenomegaly, silent positivity of autoantibodies |
| Cardinal Sign | Major Criteria | Minor Criteria |
|---|---|---|
| Fever of unknown origin that is documented to be daily (until 39 °C once a day with intermittent course) for at least 3 consecutive days and reoccurring over an observation period of at least two weeks |
|
|
| PFAPA syndrome in children | Periodically recurring high fevers (with “clockwork” periodism at intervals of 4–6 weeks) + Onset before 5 years + Child’s complete wellness between attacks (with normal growth and no sequelae) | At least 1 among:
| To rule out:
|
| PFAPA syndrome in adults (at least 16-year-old) | Recurrent fevers + Increased inflammatory parameters during febrile attacks + Symptom-free intervals | At least 1 between:
| To rule out:
|
| Eurofever/PRINTO classification criteria for PFAPA syndrome | At least 7 out of the following 8 signs (either positive [from a to d] or negative [from e to h]): |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sangiorgi, E.; Rigante, D. The Clinical Chameleon of Autoinflammatory Diseases in Children. Cells 2022, 11, 2231. https://doi.org/10.3390/cells11142231
Sangiorgi E, Rigante D. The Clinical Chameleon of Autoinflammatory Diseases in Children. Cells. 2022; 11(14):2231. https://doi.org/10.3390/cells11142231
Chicago/Turabian StyleSangiorgi, Eugenio, and Donato Rigante. 2022. "The Clinical Chameleon of Autoinflammatory Diseases in Children" Cells 11, no. 14: 2231. https://doi.org/10.3390/cells11142231
APA StyleSangiorgi, E., & Rigante, D. (2022). The Clinical Chameleon of Autoinflammatory Diseases in Children. Cells, 11(14), 2231. https://doi.org/10.3390/cells11142231