
Epithelial–Mesenchymal Transition Suppresses AMPK and Sensitizes Cancer Cells to Pyroptosis under Energy Stress

 , , ,
, , ,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction and Real-Time RT-PCR
2.3. Western Blotting
2.4. Chromatin Immunoprecipitation (ChIP)
2.5. Bioinformatic Analysis of ChIP-Seq Datasets
2.6. Bioinformatic Analysis of RNA Expression Data
3. Results
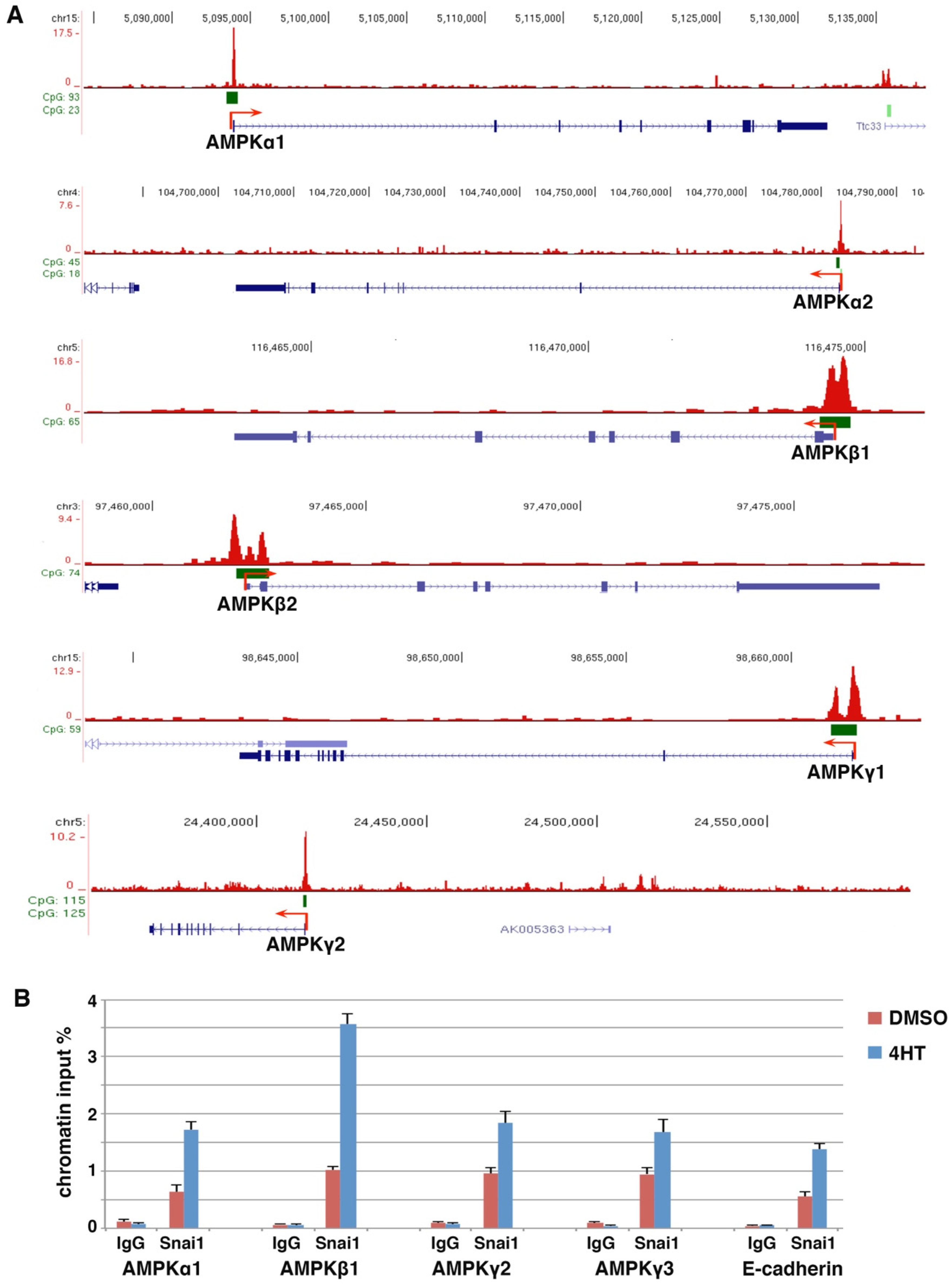
3.1. Snail Binds to the Promoters of AMPK Genes
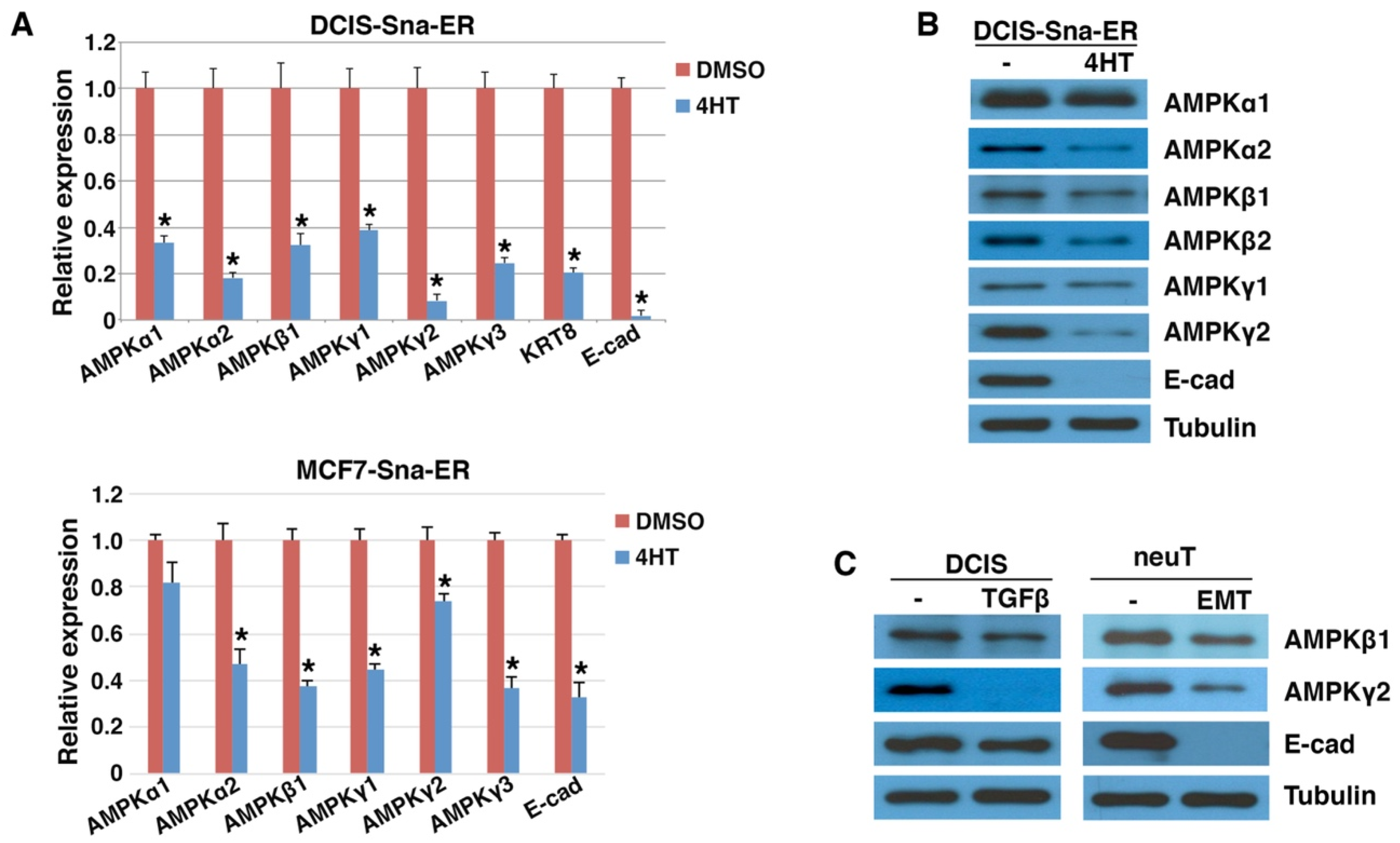
3.2. Downregulation of AMPK Genes by EMT
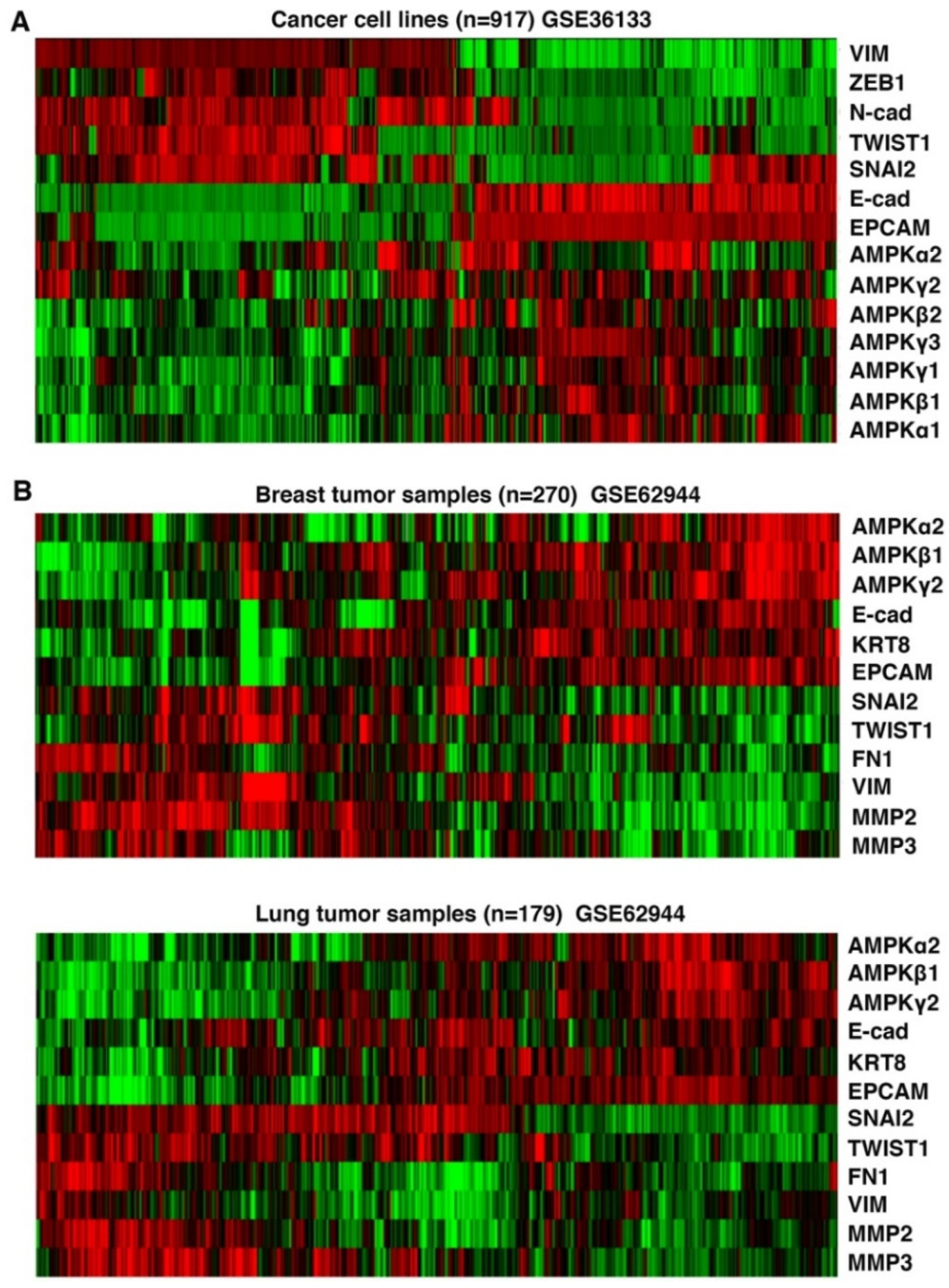
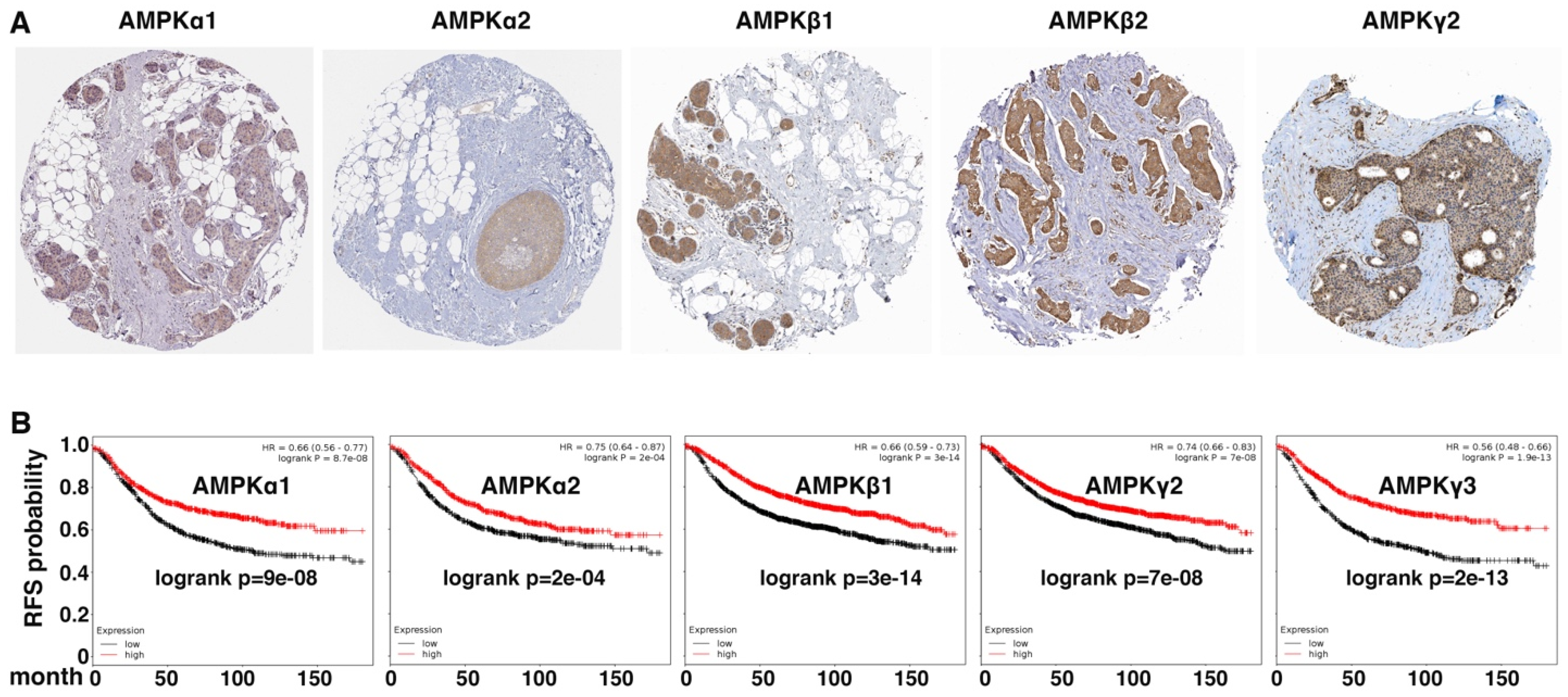
3.3. Expression of AMPK Genes in Human Cancer Cells Correlates with Epithelial Cell Markers
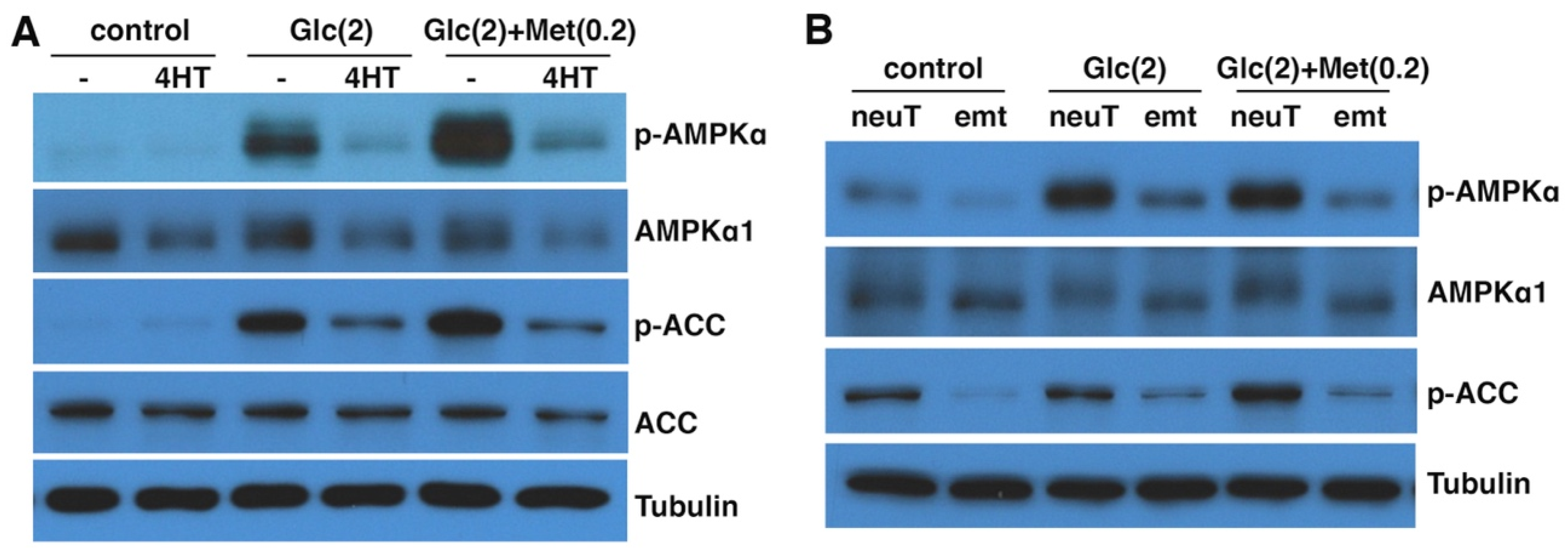
3.4. EMT Impairs Energy-Stress-Induced AMPK Activation
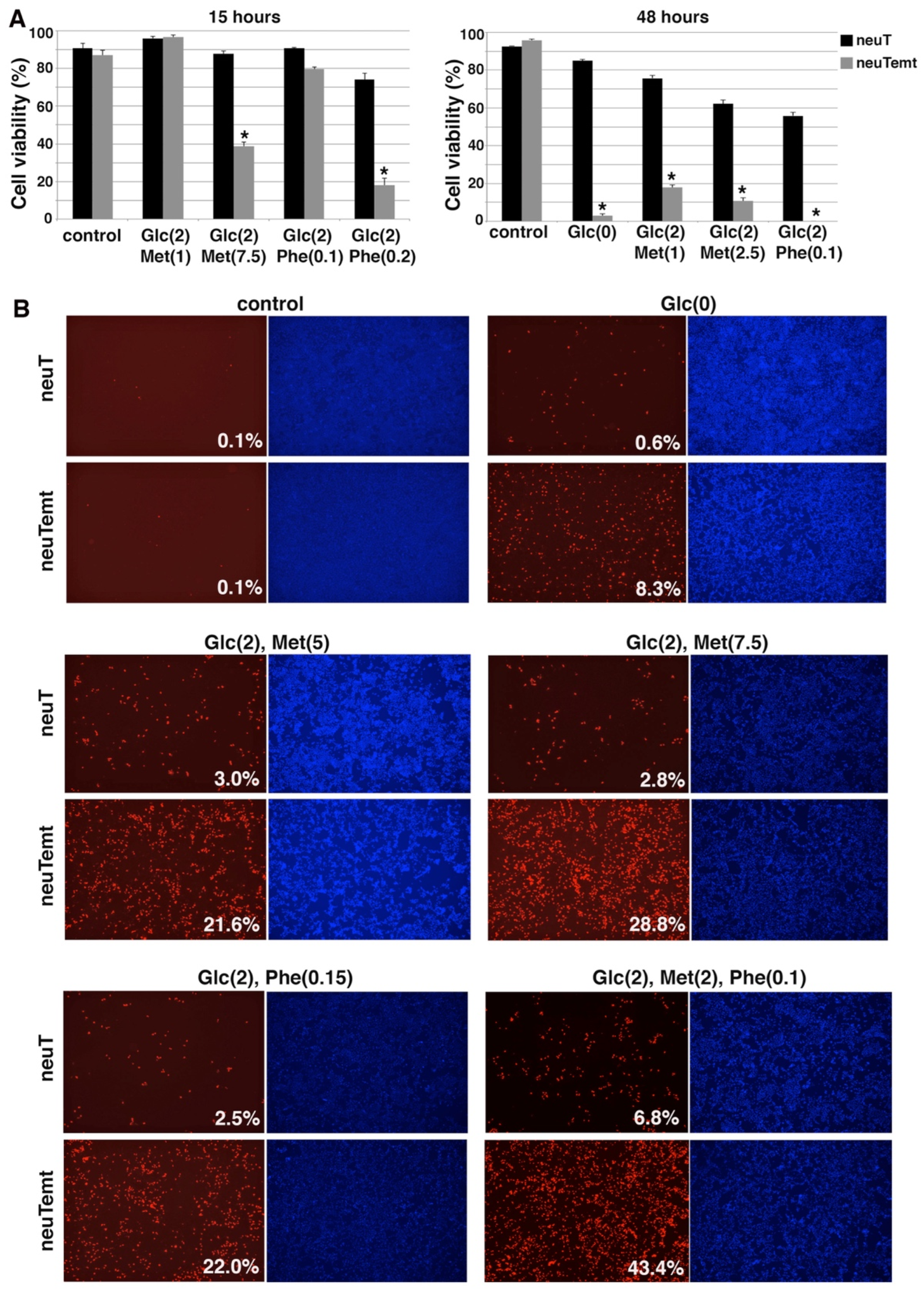
3.5. EMT Sensitizes Cells to Energy Stress
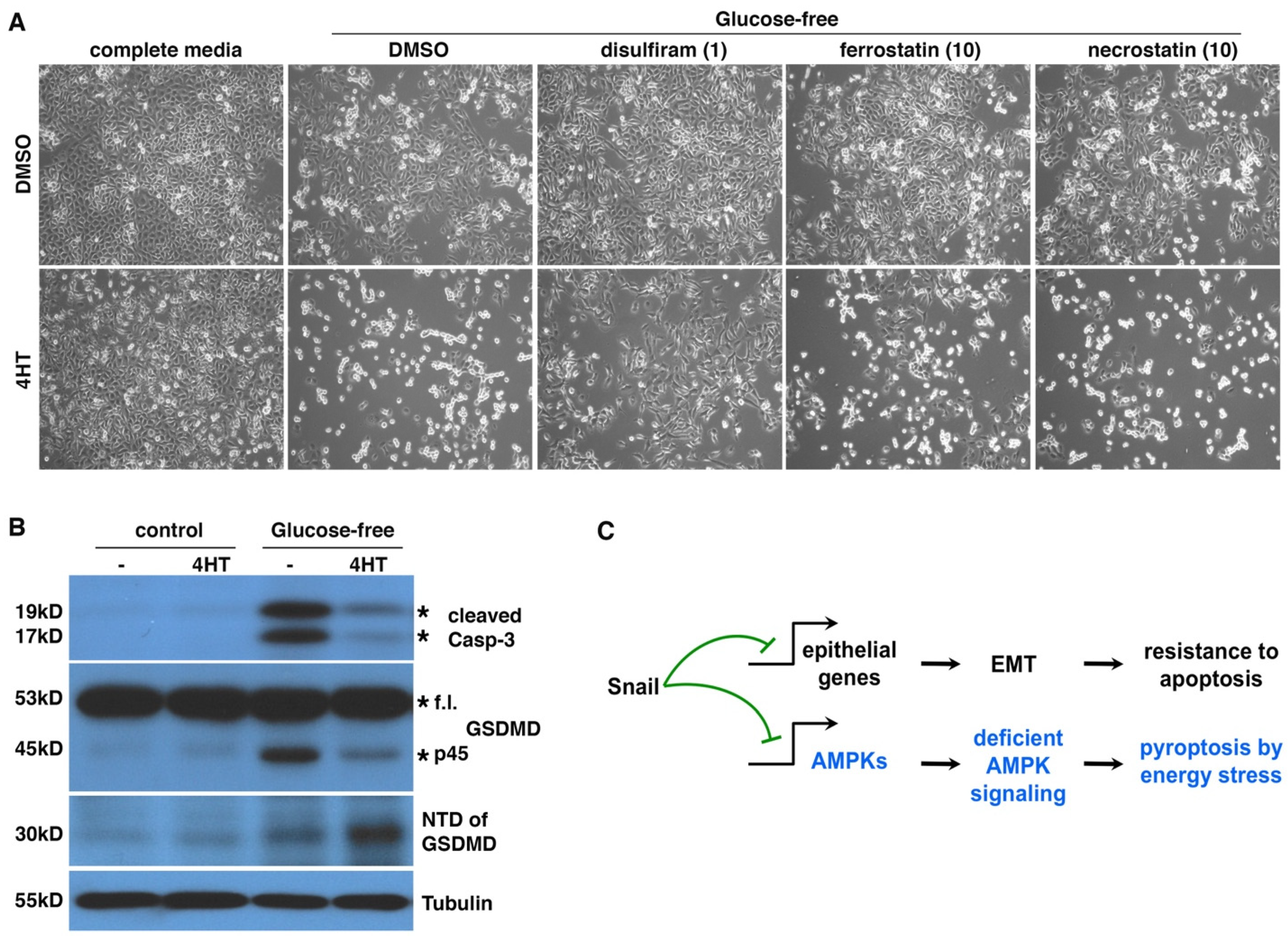
3.6. EMT Induces Pyroptotic Cell Death under Energy Stress
4. Discussion
4.1. Identifying and Targeting EMT-Induced Therapeutic Vulnerabilities
4.2. EMT Switches Energy-Stress-Induced Cell Death Forms
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lee, D.-K.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 Expression Is Necessary for Metastatic Competence in Breast Cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; Shendure, J.; McKenna, A.; Lengner, C.J. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 2021, 39, 1150–1162.e9. [Google Scholar] [CrossRef]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221.e11. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, S.E.; Perez, D.; Pan, T.-C.; Sarkisian, C.J.; Portocarrero, C.P.; Sterner, C.J.; Notorfrancesco, K.L.; Cardiff, R.D.; Chodosh, L.A. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005, 8, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar]
- Chung, J.-H.; Rho, J.K.; Xu, X.; Lee, J.S.; Yoon, H.I.; Lee, C.T.; Choi, Y.J.; Kim, H.-R.; Kim, C.H.; Lee, J.C. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011, 73, 176–182. [Google Scholar] [CrossRef]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Katayama, R.; Nanjo, S.; Tanimoto, A.; Nishiyama, A.; Nakagawa, T.; Taniguchi, H.; Suzuki, T.; et al. Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer Res. 2019, 79, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial–mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Thornton, C.; Woods, A.; Sanders, M.J. AMP-activated protein kinase: New regulation, new roles? Biochem. J. 2012, 445, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Bedoui, S.; Herold, M.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.C.; Law, M.E.; Corsino, P.E.; Parker, N.N.; Pham, K.; Davis, B.J.; Lu, J.; Law, B.K. An in vivo model of epithelial to mesenchymal transition reveals a mitogenic switch. Cancer Lett. 2012, 326, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, A.K.; Jin, Y.; Luo, H.; Tang, M.; Pampo, C.; Shao, R.; Siemann, D.W.; Wu, L.; Heldermon, C.; Law, B.K.; et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J. Clin. Investig. 2016, 126, 4174–4186. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Yu, Q.; Liu, Y.; Tang, M.; Liang, M.; Zhang, D.; Xiao, T.S.; Wu, L.; Ruan, Y.; Bungert, J.; et al. LATS kinase–mediated CTCF phosphorylation and selective loss of genomic binding. Sci. Adv. 2020, 6, eaaw4651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, D.S.; Chen, Y.; Wang, Y.; Zhang, K.; Sen, G.L. SNAI2 Controls the Undifferentiated State of Human Epidermal Progenitor Cells. Stem Cells 2014, 32, 3209–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsankov, A.M.; Gu, H.; Akopian, V.; Ziller, M.J.; Donaghey, J.; Amit, I.; Gnirke, A.; Meissner, A. Transcription factor binding dynamics during human ES cell differentiation. Nature 2015, 518, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.A.; Aakre, M.E.; Gorska, A.E.; Price, J.O.; Eltom, S.E.; Pietenpol, J.A.; Moses, H.L. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004, 6, R215–R231. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [Green Version]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 3 Pt 348, 607–614. [Google Scholar] [CrossRef]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3beta-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e5. [Google Scholar] [CrossRef]
- Wang, J.; Ye, C.; Chen, C.; Xiong, H.; Xie, B.; Zhou, J.; Chen, Y.; Zheng, S.; Wang, L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 16875–16886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. ChemMedChem 2016, 11, 2261–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.-Y.; Temkin, S.M.; Fang, X. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.-H.; Park, E.S.; Liang, J.; Dennison, J.B.; Tsavachidou, D.; Nguyen-Charles, C.; Cheng, K.W.; Hall, H.; Zhang, D.; Lu, Y.; et al. Dual Inhibition of Tumor Energy Pathway by 2-Deoxyglucose and Metformin Is Effective against a Broad Spectrum of Preclinical Cancer Models. Mol. Cancer Ther. 2011, 10, 2350–2362. [Google Scholar] [CrossRef] [Green Version]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting Cancer Cell Metabolism: The Combination of Metformin and 2-Deoxyglucose Induces p53-Dependent Apoptosis in Prostate Cancer Cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, V.; Raimondo, L.; Formisano, L.; Giuliano, M.; De Placido, S.; Rosa, R.; Bianco, R. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat. Rev. 2015, 41, 877–883. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-S.; Heinrichs, S.; Xu, D.; Garrison, S.P.; Zambetti, G.P.; Adams, J.M.; Look, A.T. Slug Antagonizes p53-Mediated Apoptosis of Hematopoietic Progenitors by Repressing puma. Cell 2005, 123, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.; Chen, D.; Ao, J.; Zhang, W.; Yi, J.; Ren, X.; Fei, J.; Li, F.; Niu, M.; Chen, H.; et al. Transcriptional suppression of AMPKα1 promotes breast cancer metastasis upon oncogene activation. Proc. Natl. Acad. Sci. USA 2020, 117, 8013–8021. [Google Scholar] [CrossRef] [Green Version]
- Russell, F.M.; Hardie, D.G. AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer? Int. J. Mol. Sci. 2020, 22, 186. [Google Scholar] [CrossRef]
- Liang, J.; Mills, G.B. AMPK: A Contextual Oncogene or Tumor Suppressor? Cancer Res. 2013, 73, 2929–2935. [Google Scholar] [CrossRef] [Green Version]
- Morales, D.R.; Morris, A.D. Metformin in Cancer Treatment and Prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022, 327, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, C.; Wang, Z.; Zhang, J.; Wang, L.; Zhang, S.; Cao, J.; Tao, Z.; Li, T.; Wang, B.; et al. A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer. Oncotarget 2017, 8, 84224–84236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Chen, X.; Wang, L.; Yang, B.; Cai, S. Metformin Adjunct With Antineoplastic Agents for the Treatment of Lung Cancer: A Meta-Analysis of Randomized Controlled Trials and Observational Cohort Studies. Front. Pharmacol. 2021, 12, 639016. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.N.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.-Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.-J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Eguchi, Y.; Srinivasan, A.; Tomaselli, K.J.; Shimizu, S.; Tsujimoto, Y. ATP-dependent steps in apoptotic signal trans-duction. Cancer Res. 1999, 59, 2174–2181. [Google Scholar]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Zamaraeva, M.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells die with increased cytosolic ATP during apoptosis: A bioluminescence study with intracellular luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [Green Version]
- Eharijith, A.; Ebenezer, D.L.; Enatarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Wang, L.; Negro, R.; Wu, H. TRPM2, linking oxidative stress and Ca2+ permeation to NLRP3 inflammasome activation. Curr. Opin. Immunol. 2020, 62, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.-F.; Yu, T.; Chu, X.-M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Dominic, A.; Le, N.-T.; Takahashi, M. Loop Between NLRP3 Inflammasome and Reactive Oxygen Species. Antioxid. Redox Signal. 2022, 36, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Hafner-Bratkovič, I.; Devant, P.; D’andrea, J.M.; Ngwa, E.M.; Boršić, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 2021, 184, 4495–4511.e19. [Google Scholar] [CrossRef]
- Ren, Y.; Shen, H.-M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019, 25, 101154. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.J.; Konstantinou, M.; Goode, E.F.; Meier, P. The Diversification of Cell Death and Immunity: Memento Mori. Mol. Cell 2019, 76, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial–Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, M.; Li, J.W.; Luo, H.; Lulu, S.; Calbay, O.; Shenoy, A.; Tan, M.; Law, B.K.; Huang, S.; Xiao, T.S.; et al. Epithelial–Mesenchymal Transition Suppresses AMPK and Sensitizes Cancer Cells to Pyroptosis under Energy Stress. Cells 2022, 11, 2208. https://doi.org/10.3390/cells11142208
Liang M, Li JW, Luo H, Lulu S, Calbay O, Shenoy A, Tan M, Law BK, Huang S, Xiao TS, et al. Epithelial–Mesenchymal Transition Suppresses AMPK and Sensitizes Cancer Cells to Pyroptosis under Energy Stress. Cells. 2022; 11(14):2208. https://doi.org/10.3390/cells11142208
Chicago/Turabian StyleLiang, Mingwei, Jennifer W. Li, Huacheng Luo, Sarah Lulu, Ozlem Calbay, Anitha Shenoy, Ming Tan, Brian K. Law, Shuang Huang, Tsan Sam Xiao, and et al. 2022. "Epithelial–Mesenchymal Transition Suppresses AMPK and Sensitizes Cancer Cells to Pyroptosis under Energy Stress" Cells 11, no. 14: 2208. https://doi.org/10.3390/cells11142208
APA StyleLiang, M., Li, J. W., Luo, H., Lulu, S., Calbay, O., Shenoy, A., Tan, M., Law, B. K., Huang, S., Xiao, T. S., Chen, H., Wu, L., Chang, J., & Lu, J. (2022). Epithelial–Mesenchymal Transition Suppresses AMPK and Sensitizes Cancer Cells to Pyroptosis under Energy Stress. Cells, 11(14), 2208. https://doi.org/10.3390/cells11142208