
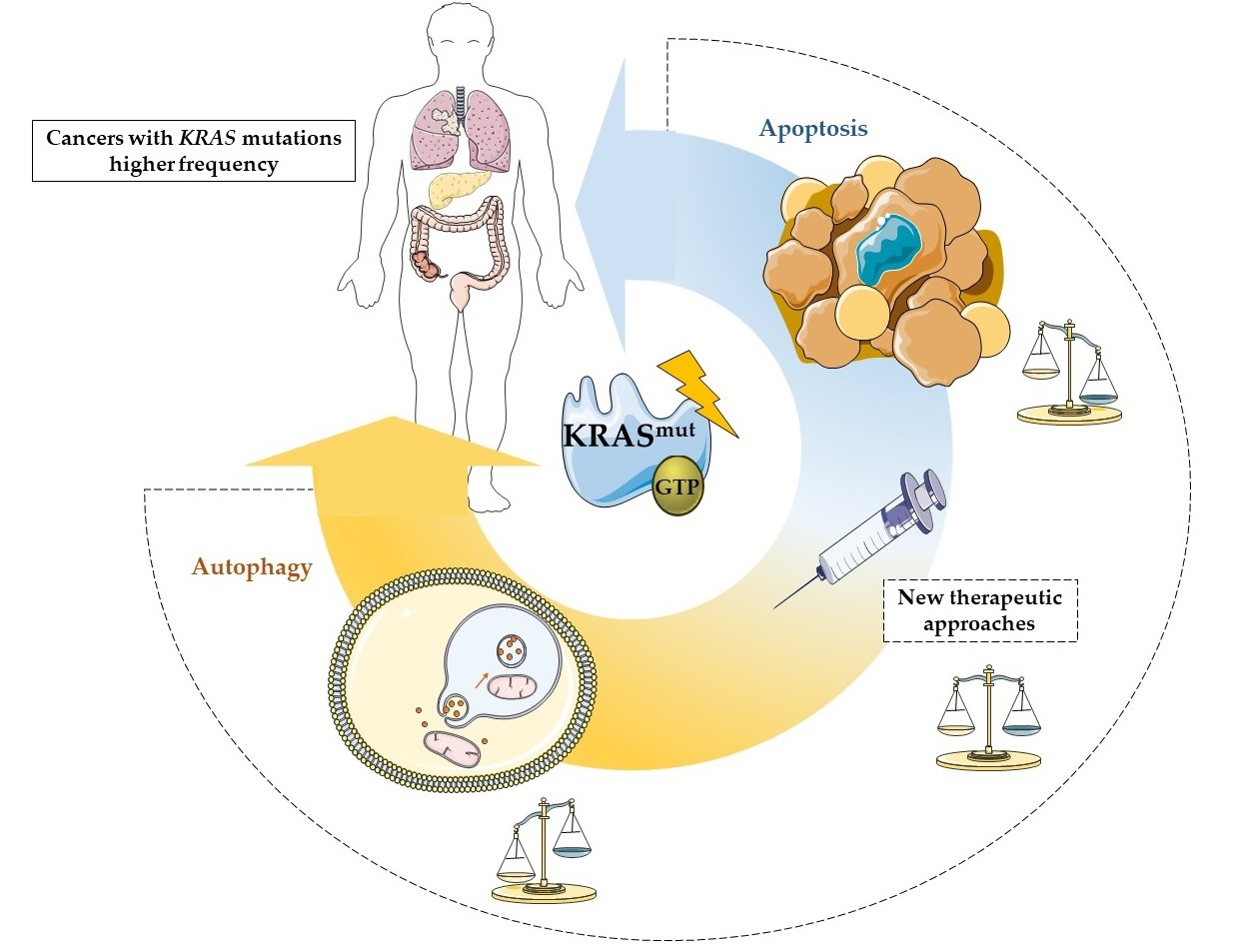
Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications

 , , ,
, , ,  , and
, and 
Abstract

1. Introduction
1.1. The Oncogene KRAS
1.2. KRAS Mutations and Cancer
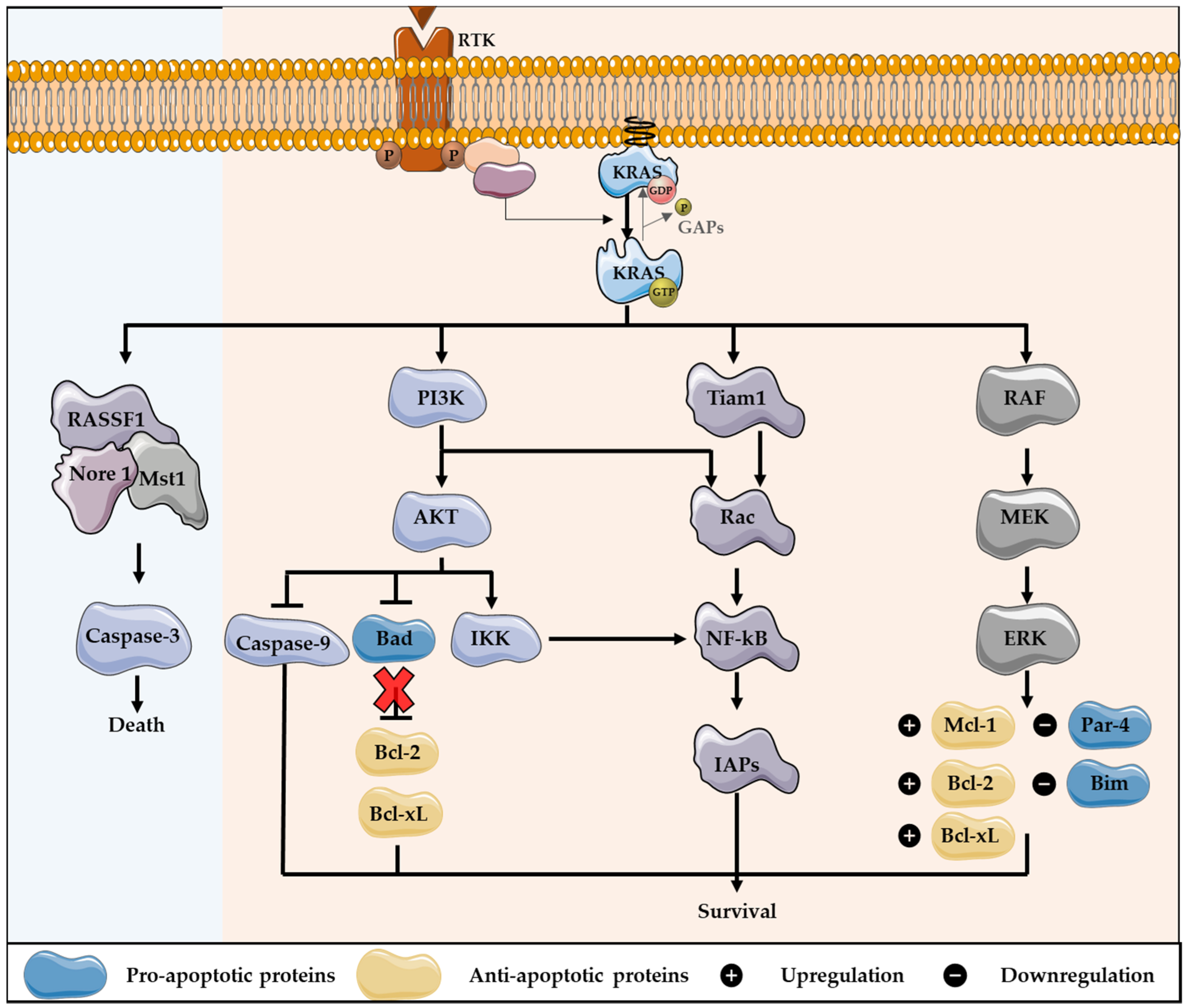
2. KRAS Role on Apoptotic Cell Death
Impact of KRAS Mutations in Resistance to Apoptosis
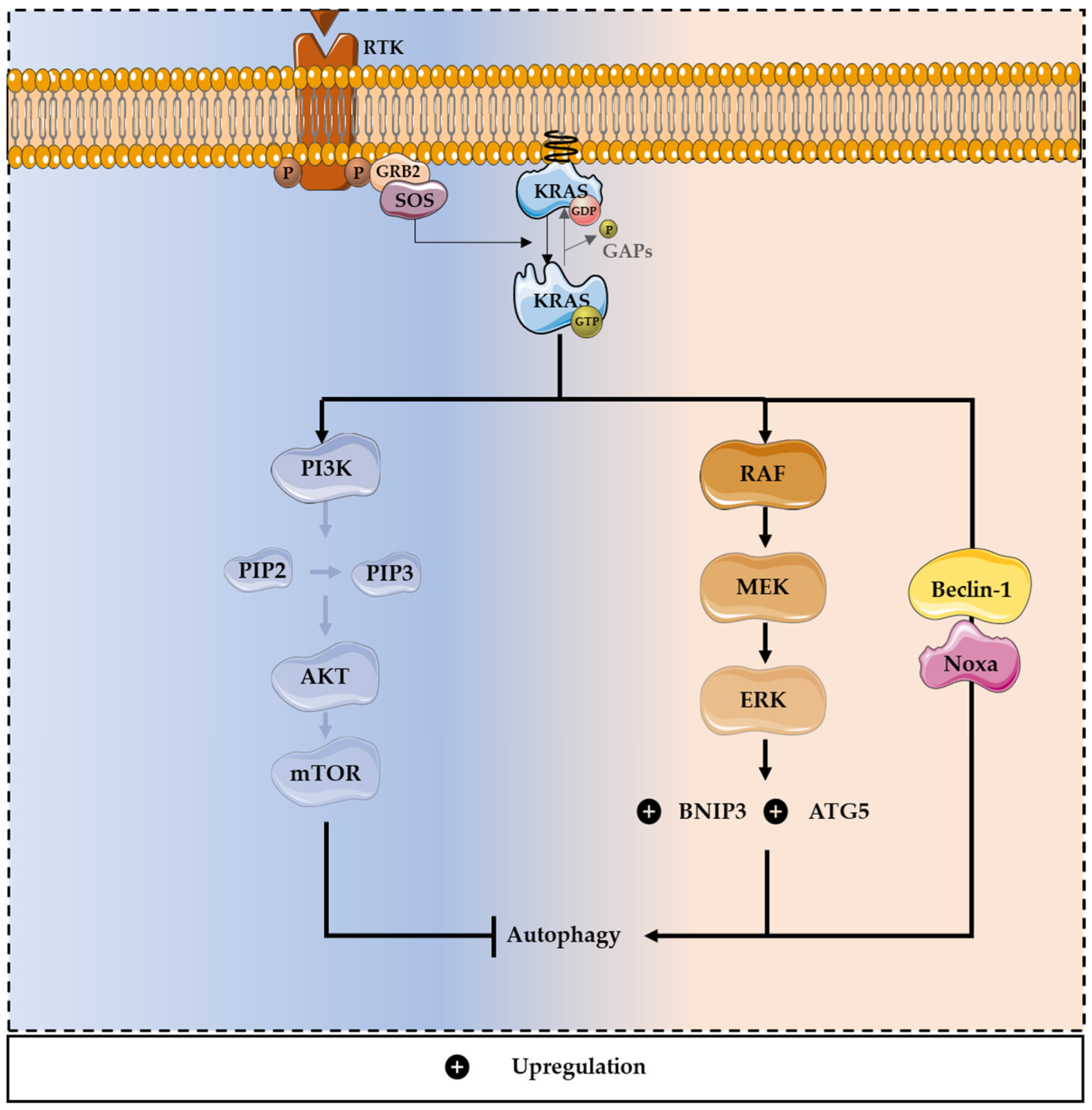
3. Role of Autophagy in Cancer
Modulation of Autophagy by KRAS Mutations
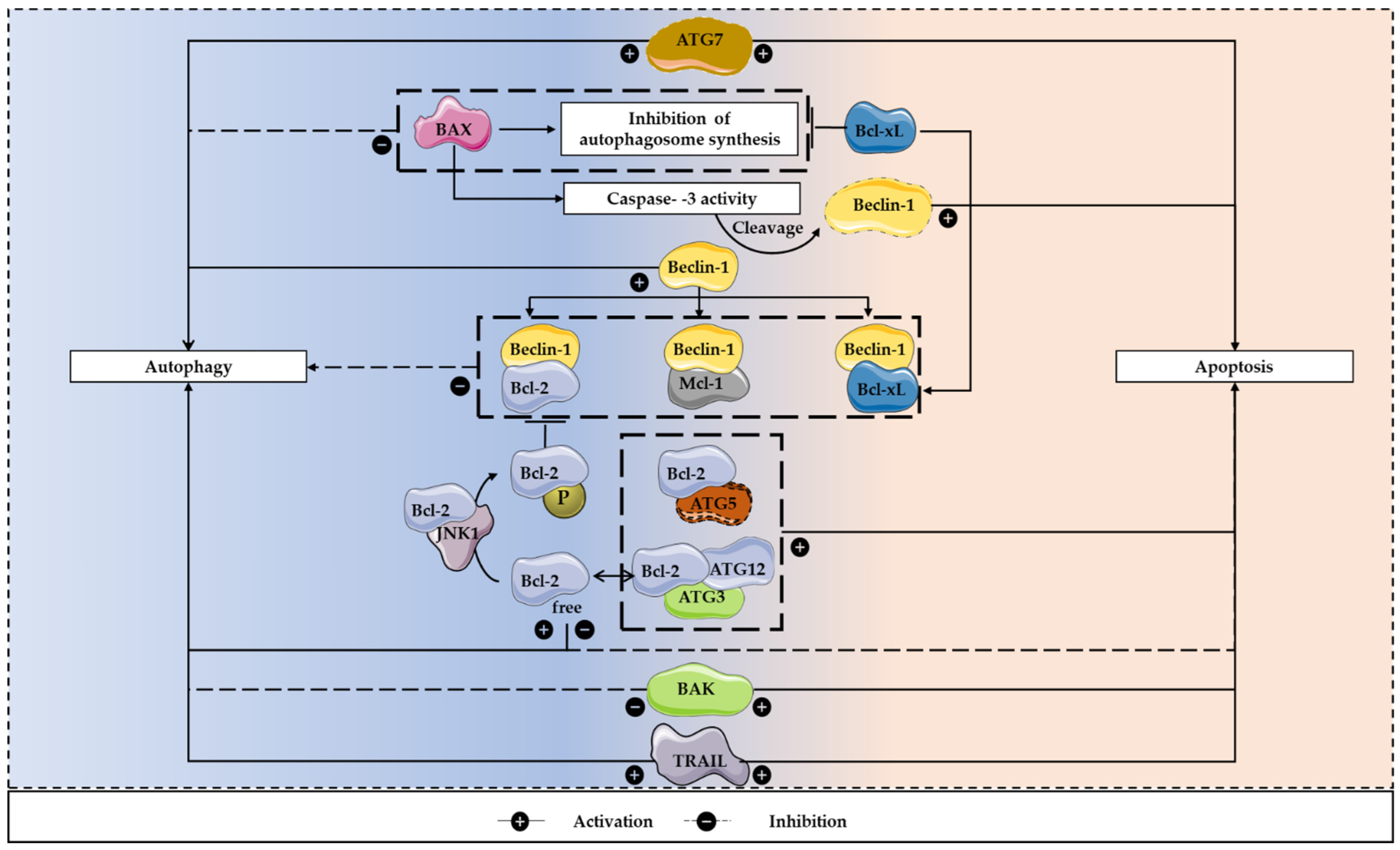
4. The Autophagy/Apoptosis Regulation Loop
5. Therapies Targeting KRAS-Induced Cell Death Resistance
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Bounds, R.; Wolf, H.; Steele, R.; Carey, F.; Wolf, C. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours—implications for personalised cancer medicine. Br. J. Cancer 2010, 102, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.H.; Luo, L.X.; Liu, Z.Q.; Wong, V.K.W.; Lu, L.L.; Xie, Y.; Zhang, N.; Qu, Y.Q.; Fan, X.X.; Li, Y.; et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: Blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and predictive roles of KRAS mutation in colorectal cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168. [Google Scholar] [CrossRef]
- Liu, X.; Jakubowski, M.; Hunt, J.L. KRAS gene mutation in colorectal cancer is correlated with increased proliferation and spontaneous apoptosis. Am. J. Clin. Pathol. 2011, 135, 245–252. [Google Scholar] [CrossRef]
- Overmeyer, J.H.; Maltese, W.A. Death pathways triggered by activated Ras in cancer cells. Front. Biosci. (Landmark Ed.) 2011, 16, 1693–1713. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Cazzanelli, G.; Pereira, F.; Alves, S.; Francisco, R.; Azevedo, L.; Dias Carvalho, P.; Almeida, A.; Côrte-Real, M.; Oliveira, M.J.; Lucas, C.; et al. The Yeast Saccharomyces Cerevisiae as a Model for Understanding RAS Proteins and Their Role in Human Tumorigenesis. Cells 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, M.; Wang, Y. Alternative splicing of the K-ras gene in mouse tissues and cell lines. Exp. Lung Res. 2001, 27, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2468. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Yoo, B.H.; Khan, I.A.; Koomson, A.; Gowda, P.; Sasazuki, T.; Shirasawa, S.; Gujar, S.; Rosen, K.V. Oncogenic RAS-induced downregulation of ATG12 is required for survival of malignant intestinal epithelial cells. Autophagy 2018, 14, 134–151. [Google Scholar] [CrossRef]
- Xu, K.; Park, D.; Magis, A.T.; Zhang, J.; Zhou, W.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Deng, X. Small Molecule KRAS Agonist for Mutant KRAS Cancer Therapy. Mol. Cancer 2019, 18, 85. [Google Scholar] [CrossRef]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef]
- Fröjdö, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta—Mol. Basis Dis. 2009, 1792, 83–92. [Google Scholar] [CrossRef]
- Ihle, N. Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention. Ph.D. Thesis, The University of Texas Graduate School of Biomedical Sciences, Houston, TX, USA, 2012. [Google Scholar]
- Sancho, E.; Batlle, E.; Clevers, H. Signaling Pathways in Intestinal Development and Cancer. Annu. Rev. Cell Dev. Biol. 2004, 20, 695–723. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Biran, A.; Poirier, F.; Raz, A.; Kloog, Y. Galectin-3 mediates cross-talk between K-ras and let-7c tumor suppressor microRNA. PLoS ONE 2011, 6, e27490. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, K.; Zhang, L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015, 2, 4–12. [Google Scholar] [CrossRef]
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Okudela, K.; Hayashi, H.; Ito, T.; Yazawa, T.; Suzuki, T.; Nakane, Y.; Sato, H.; Ishi, H.; KeQin, X.; Masuda, A.; et al. K-ras Gene Mutation Enhances Motility of Immortalized Airway Cells and Lung Adenocarcinoma Cells via Akt Activation: Possible Contribution to Non-Invasive Expansion of Lung Adenocarcinoma. Am. J. Pathol. 2004, 164, 91–100. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Efferth, T. Signal Transduction Pathways of the Epidermal Growth Factor Receptor in Colorectal Cancer and their Inhibition by Small Molecules. Curr. Med. Chem. 2012, 19, 5735–5744. [Google Scholar] [CrossRef]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, Š. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Lautwein, A.; Kabsch, W.; Ahmadian, M.R.; Wittinghofer, A. Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature 1996, 384, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Fan, G.; Lou, L.; Song, Z.; Zhang, X.; Xiong, X.F. Targeting mutated GTPase KRAS in tumor therapies. Eur. J. Med. Chem. 2021, 226, 113816. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Worthley, D.-L.; Leggett, B.-A. Colorectal cancer: Molecular features and clinical opportunities. Clin. Biochem. Rev. 2010, 31, 31–38. [Google Scholar]
- Krasinskas, A.M. EGFR Signaling in Colorectal Carcinoma. Patholog. Res. Int. 2011, 2011, 932932. [Google Scholar] [CrossRef] [PubMed]
- Bruera, G.; Cannita, K.; Di Giacomo, D.; Lamy, A.; Frébourg, T.; Sabourin, J.C.; Tosi, M.; Alesse, E.; Ficorella, C.; Ricevuto, E. Worse prognosis of KRAS c.35 G > A mutant metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx). BMC Med. 2013, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Hung, K.E. Molecular Mechanisms of Colorectal Carcinogenesis. In Molecular Pathogenesis of Colorectal Cancer; Haigis, K.M., Ed.; Springer: New York, NY, USA, 2013; pp. 25–66. ISBN 978-1-4614-8411-0. [Google Scholar]
- Hammond, D.E.; Mageean, C.J.; Rusilowicz, E.V.; Wickenden, J.A.; Clague, M.J.; Prior, I.A. Differential Reprogramming of Isogenic Colorectal Cancer Cells by Distinct Activating KRAS Mutations. J. Proteome Res. 2015, 14, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Grafi-Cohen, M.; Kraiem, Z.; Kloog, Y. Galectin-3 Promotes Chronic Activation of K-Ras and Differentiation Block in Malignant Thyroid Carcinomas. Am. Assoc. Cancer Res. 2010, 9, 2208–2219. [Google Scholar] [CrossRef]
- Sideris, M.; Emin, E.I.; Abdullah, Z.; Hanrahan, J.; Stefatou, K.M.; Sevas, V.; Emin, E.; Hollingworth, T.; Odejinmi, F.; Papagrigoriadis, S.; et al. The role of KRAS in endometrial cancer: A mini-review. Anticancer Res. 2019, 39, 533–540. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Sakanashi, F.; Shintani, M.; Tsuneyoshi, M.; Ohsaki, H.; Kamoshida, S. Apoptosis, necroptosis and autophagy in colorectal cancer: Associations with tumor aggressiveness and p53 status. Pathol. Res. Pract. 2019, 215, 152425. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Brit 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Renault, T.T.; Teijido, O.; Missire, F.; Ganesan, Y.T.; Velours, G.; Arokium, H.; Beaumatin, F.; Llanos, R.; Athané, A.; Camougrand, N.; et al. Bcl-xL stimulates Bax relocation to mitochondria and primes cells to ABT-737. Int. J. Biochem. Cell Biol. 2015, 64, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Lalier, L.; Vallette, F.; Manon, S. Bcl-2 Family Members and the Mitochondrial Import Machineries: The Roads to Death. Biomolecules 2022, 12, 162. [Google Scholar] [CrossRef]
- Okamoto, K.; Zaanan, A.; Kawakami, H.; Huang, S.; Sinicrope, F.A. Reversal of mutant KRAS-mediated apoptosis resistance by concurrent Noxa/Bik induction and Bcl-2/Bcl-xL antagonism in colon cancer cells. Mol. Cancer Res. 2015, 13, 659–669. [Google Scholar] [CrossRef]
- Hata, A.N.; Yeo, A.; Faber, A.C.; Lifshits, E.; Chen, Z.; Cheng, K.A.; Walton, Z.; Sarosiek, K.A.; Letai, A.; Heist, R.S.; et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS mutant lung cancers. Cancer Res. 2014, 74, 3146–3156. [Google Scholar]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Mayo, M.W.; Baldwin, A.S. The transcription factor NF-kB: Control of oncogenesis and cancer therapy resistance. Biochim. Biophys. Acta 2000, 1470, 55–62. [Google Scholar]
- Sulciner, D.J.; Irani, K.; Yu, Z.X.; Ferrans, V.J.; Goldschmidt-Clermont, P.; Finkel, T. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kB activation. Mol. Cell. Biol. 1996, 16, 7115–7121. [Google Scholar] [CrossRef]
- Nalca, A.; Qiu, S.G.; El-Guendy, N.; Krishnan, S.; Rangnekar, V.M. Oncogenic Ras sensitizes cells to apoptosis by Par-4. J. Biol. Chem. 1999, 274, 29976–29983. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Kijima, H.; Hori, S.; Oshika, Y.; Tokunaga, T.; Kawai, K.; Yamazaki, H.; Ueyama, Y.; Scanlon, K.J.; Tamaoki, N.; et al. Adenovirus-mediated anti-K-ras ribozyme induces apoptosis and growth suppression of human pancreatic carcinoma. Cancer Gene Ther. 2000, 7, 373–383. [Google Scholar] [CrossRef][Green Version]
- Navarro, P.; Valverde, A.M.; Benito, M.; Lorenzo, M. Activated Ha-ras induces apoptosis by association with phosphorylated Bcl-2 in a mitogen-activated protein kinase-independent manner. J. Biol. Chem. 1999, 274, 18857–18863. [Google Scholar] [CrossRef]
- Boucher, M.J.; Morisset, J.; Vachon, P.H.; Reed, J.C.; Lainé, J.; Rivard, N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J. Cell. Biochem. 2000, 79, 355–369. [Google Scholar] [CrossRef]
- Rebollo, A.; Pérez-Sala, D.; Martínez-A, C. Bcl-2 differentially targets K-, N-, and H-Ras to mitochondria in IL-2 supplemented or deprived cells: Implications in prevention of apoptosis. Oncogene 1999, 18, 4930–4939. [Google Scholar] [CrossRef] [PubMed]
- Eckfeld, K.; Hesson, L.; Vos, M.D.; Bieche, I.; Latif, F.; Clark, G.J. RASSF4/AD037 is a potential Ras effector/tumor suppressor of the RASSF family. Cancer Res. 2004, 64, 8688–8693. [Google Scholar] [CrossRef]
- Lee, K.K.; Ohyama, T.; Yajima, N.; Tsubuki, S.; Yonehara, S. MST, a Physiological Caspase Substrate, Highly Sensitizes Apoptosis Both Upstream and Downstream of Caspase Activation. J. Biol. Chem. 2001, 276, 19276–19285. [Google Scholar] [CrossRef]
- Downward, J. Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 1998, 8, 49–54. [Google Scholar] [CrossRef]
- Zaanan, A.; Okamoto, K.; Kawakami, H.; Khazaie, K.; Huang, S.; Sinicrope, F.A. The Mutant KRAS Gene Up-regulates BCL-XL Protein via STAT3 to Confer Apoptosis Resistance That Is Reversed by BIM Protein Induction and BCL-XL Antagonism. J. Biol. Chem. 2015, 290, 23838–23849. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.P.; Batra, S.; Kandala, P.K.; Brown, T.L.; Srivastava, S.K. The role of K-ras gene mutation in TRAIL-induced apoptosis in pancreatic and lung cancer cell lines. Cancer Chemoterapy Pharmacol. 2011, 67, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Sung, B.J.; Cho, Y.S.; Kim, H.J.; Ha, N.C.; Hwang, J.I.; Chung, C.W.; Jung, Y.K.; Oh, B.H. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry 2001, 40, 1117–1123. [Google Scholar] [CrossRef]
- Tecleab, A.; Sebti, S.M. Depletion of K-Ras promotes proteasome degradation of survivin. Cell Cycle 2013, 12, 522–532. [Google Scholar] [CrossRef][Green Version]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Sun, J.; Shen, Q.; Lu, H.; Jiang, Z.; Xu, W.; Feng, L.; Li, L.; Wang, X.; Cai, X.; Jin, H. Oncogenic Ras suppresses ING4-TDG-Fas axis to promote apoptosis resistance. Oncotarget 2015, 6, 41997–42007. [Google Scholar] [CrossRef]
- Zhu, J.K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef]
- Schickel, R.; Park, S.M.; Murmann, A.E.; Peter, M.E. miR-200c Regulates Induction of Apoptosis through CD95 by Targeting FAP-1. Mol. Cell 2010, 38, 908–915. [Google Scholar] [CrossRef]
- Zhong, X.; Zheng, L.; Shen, J.; Zhang, D.; Xiong, M.; Zhang, Y.; He, X.; Tanyi, J.L.; Yang, F.; Montone, K.T.; et al. Suppression of MicroRNA 200 Family Expression by Oncogenic KRAS Activation Promotes Cell Survival and Epithelial-Mesenchymal Transition in KRAS-Driven Cancer. Mol. Cell. Biol. 2016, 36, 2742–2754. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef]
- Priault, M.; Hue, E.; Marhuenda, F.; Pilet, P.; Oliver, L.; Vallette, F.M. Differential dependence on Beclin 1 for the regulation of pro-survival autophagy by Bcl-2 and Bcl-xL in HCT116 colorectal cancer cells. PLoS ONE 2010, 5, e8755. [Google Scholar] [CrossRef]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation by energy sensing. Curr. Biol. 2011, 21, R227–R229. [Google Scholar] [CrossRef]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Kim, M.J.; Woo, S.J.; Yoon, C.H.; Lee, J.S.; An, S.; Choi, Y.H.; Hwang, S.G.; Yoon, G.; Lee, S.J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of Autophagy in Cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Frankel, L.B.; Lund, A.H. MicroRNA regulation of autophagy. Carcinogenesis 2012, 33, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-M.; Huang, J.-J.; Du, J.-J.; Zhang, N.; Long, Z.; Yang, Y.; Zhong, F.-F.; Zheng, B.-W.; Shen, Y.-F.; Huang, Z.; et al. Autophagy inhibitors increase the susceptibility of KRAS-mutant human colorectal cancer cells to a combined treatment of 2-deoxy-D-glucose and lovastatin. Acta Pharmacol. Sin. 2021, 42, 1875–1887. [Google Scholar] [CrossRef]
- Devenport, S.N.; Singhal, R.; Radyk, M.D.; Taranto, J.G.; Kerk, S.A.; Chen, B.; Goyert, J.W.; Jain, C.; Das, N.K.; Oravecz-Wilson, K.; et al. Colorectal cancer cells utilize autophagy to maintain mitochondrial metabolism for cell proliferation under nutrient stress. JCI Insight 2021, 6, e138835. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.; Chiang, H.; Kaklamani, V.G.; et al. Tamoxifen resistance in Breast Cancer is regulated by the EZH2–ERα–GREB1 Transcriptional Axis. Cancer Res. 2018, 78, 671–684. [Google Scholar] [CrossRef]
- Pattingre, S.; Bauvy, C.; Codogno, P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 2003, 278, 16667–16674. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.D.; Lapi, E.; Sullivan, A.; Jia, W.; He, Y.-W.; Ratnayaka, I.; Zhong, S.; Goldin, R.D.; Goemans, C.G.; et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc. Natl. Acad. Sci. USA 2012, 109, 13325–13330. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Zhou, H.; Yuan, M.; Yu, Q.; Zhou, X.; Min, W.; Gao, D. Autophagy regulation and its role in gastric cancer and colorectal cancer. Cancer Biomark. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Manent, J.; Banerjee, S.; De Matos Simoes, R.; Zoranovic, T.; Mitsiades, C.; Penninger, J.M.; Simpson, K.J.; Humbert, P.O.; Richardson, H.E. Autophagy suppresses Ras-driven epithelial tumourigenesis by limiting the accumulation of reactive oxygen species. Oncogene 2017, 36, 5576–5592. [Google Scholar] [CrossRef] [PubMed]
- Swiderek, E.; Kalas, W.; Wysokinska, E.; Pawlak, A.; Rak, J.; Strzadala, L. Biochemical and Biophysical Research Communications The interplay between epigenetic silencing, oncogenic KRas and HIF-1 regulatory pathways in control of BNIP3 expression in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2013, 441, 707–712. [Google Scholar] [CrossRef]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-Induced Expression of Noxa and Beclin-1 Promotes Autophagic Cell Death and Limits Clonogenic Survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 2016, 7, 9188–9221. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, D.S.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Lee, C.S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Zeng, X.; Overmeyer, J.H.; Maltese, W.A. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell Sci. 2006, 119, 259–270. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Wei, P.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Heinlein, M.; Huang, D.C.S.; Vaux, D.L. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 2019, 136, 27–41. [Google Scholar] [CrossRef]
- Scott, R.C.; Juhász, G.; Neufeld, T.P. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 2007, 17, 1–11. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Souquère, S.; Pierron, G.; Codogno, P. NF-κB activation represses tumor necrosis factor-α-induced autophagy. J. Biol. Chem. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting kras in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R. Therapeutic Approach to KRAS Mutated Colorectal Cancer. In Cancer Therapy; MedDocs Publishers: Reno, NV, USA, 2021. [Google Scholar]
- Bellio, H.; Fumet, J.D.; Ghiringhelli, F. Targeting BRAF and RAS in colorectal cancer. Cancers 2021, 13, 2201. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Karamouzis, M.V.; Mihailidou, C.; Schizas, D.; Papavassiliou, A.G. Co-targeting of EGFR and autophagy signaling is an emerging treatment strategy in metastatic colorectal cancer. Cancer Lett. 2017, 396, 94–102. [Google Scholar] [CrossRef]
- Cui, J.; Hu, Y.F.; Feng, X.M.; Tian, T.; Guo, Y.H.; Ma, J.W.; Nan, K.J.; Zhang, H.Y. EGFR inhibitors and autophagy in cancer treatment. Tumor Biol. 2014, 35, 11701–11709. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ji, Q.; Li, Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: Underlying mechanisms and reversal strategies. J. Exp. Clin. Cancer Res. 2021, 40, 328. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef]
- Uras, I.Z.; Moll, H.P.; Casanova, E. Targeting KRAS mutant non-small-cell lung cancer: Past, present and future. Int. J. Mol. Sci. 2020, 21, 4325. [Google Scholar] [CrossRef] [PubMed]
- Ghimessy, A.; Radeczky, P.; Laszlo, V.; Hegedus, B.; Renyi-Vamos, F.; Fillinger, J.; Klepetko, W.; Lang, C.; Dome, B.; Megyesfalvi, Z. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020, 39, 1159–1177. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS mutant pancreatic cancer: No lone path to an effective treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Korzeniecki, C.; Priefer, R. Targeting KRAS mutant cancers by preventing signaling transduction in the MAPK pathway. Eur. J. Med. Chem. 2021, 211, 113006. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef]
- Awasthi, N.; Monahan, S.; Stefaniak, A.; Schwarz, M.A.; Schwarz, R.E. Inhibition of the MEK/ERK pathway augments nab-paclitaxel-based chemotherapy effects in preclinical models of pancreatic cancer. Oncotarget 2017, 9, 5274–5286. [Google Scholar] [CrossRef]
- Kim, A.; Lee, J.-E.; Lee, S.-S.; Kim, C.; Lee, S.-J.; Jang, W.-S.; Park, S. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int. J. Cancer 2013, 133, 984–996. [Google Scholar] [CrossRef]
- Burmi, R.S.; Maginn, E.N.; Gabra, H.; Stronach, E.A.; Wasan, H.S. Combined inhibition of the PI3K/mTOR/MEK pathway induces Bim/Mcl-2-regulated apoptosis in pancreatic cancer cells. Cancer Biol. Ther. 2019, 20, 21–30. [Google Scholar] [CrossRef]
- Ozkan-Dagliyan, I.; Diehl, J.N.; George, S.D.; Schaefer, A.; Papke, B.; Klotz-Noack, K.; Waters, A.M.; Goodwin, C.M.; Gautam, P.; Pierobon, M.; et al. Low-Dose Vertical Inhibition of the RAF-MEK-ERK Cascade Causes Apoptotic Death of KRAS Mutant Cancers. Cell Rep. 2020, 31, 107764. [Google Scholar] [CrossRef]
- Faber, A.C.; Coffee, E.M.; Costa, C.; Dastur, A.; Ebi, H.; Hata, A.N.; Yeo, A.T.; Edelman, E.J.; Song, Y.; Tam, A.T.; et al. mTOR Inhibition Specifically Sensitizes Colorectal Cancers with KRAS or BRAF Mutations to BCL-2/BCL-XL Inhibition by Suppressing MCL-1. Cancer Discov. 2014, 4, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; O’Reilly, E.M. New Treatment Strategies for Metastatic Pancreatic Ductal Adenocarcinoma. Drugs 2020, 80, 647–669. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Heiden, M.G.V.; Mccormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The developing story of predictive biomarkers in colorectal cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef]
- Liu, S.; Mithat, G.; Stadler, Z.K.; Weiser, M.R.; Hechtman, J.F.; Vakiani, E.; Wang, T.; Vyas, M.; Joneja, U.; Al-bayati, M.; et al. Cellular localization of PD-L1 expression in mismatch-repair-deficient and proficient colorectal carcinomas. Mod. Pathol. 2019, 32, 110–121. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
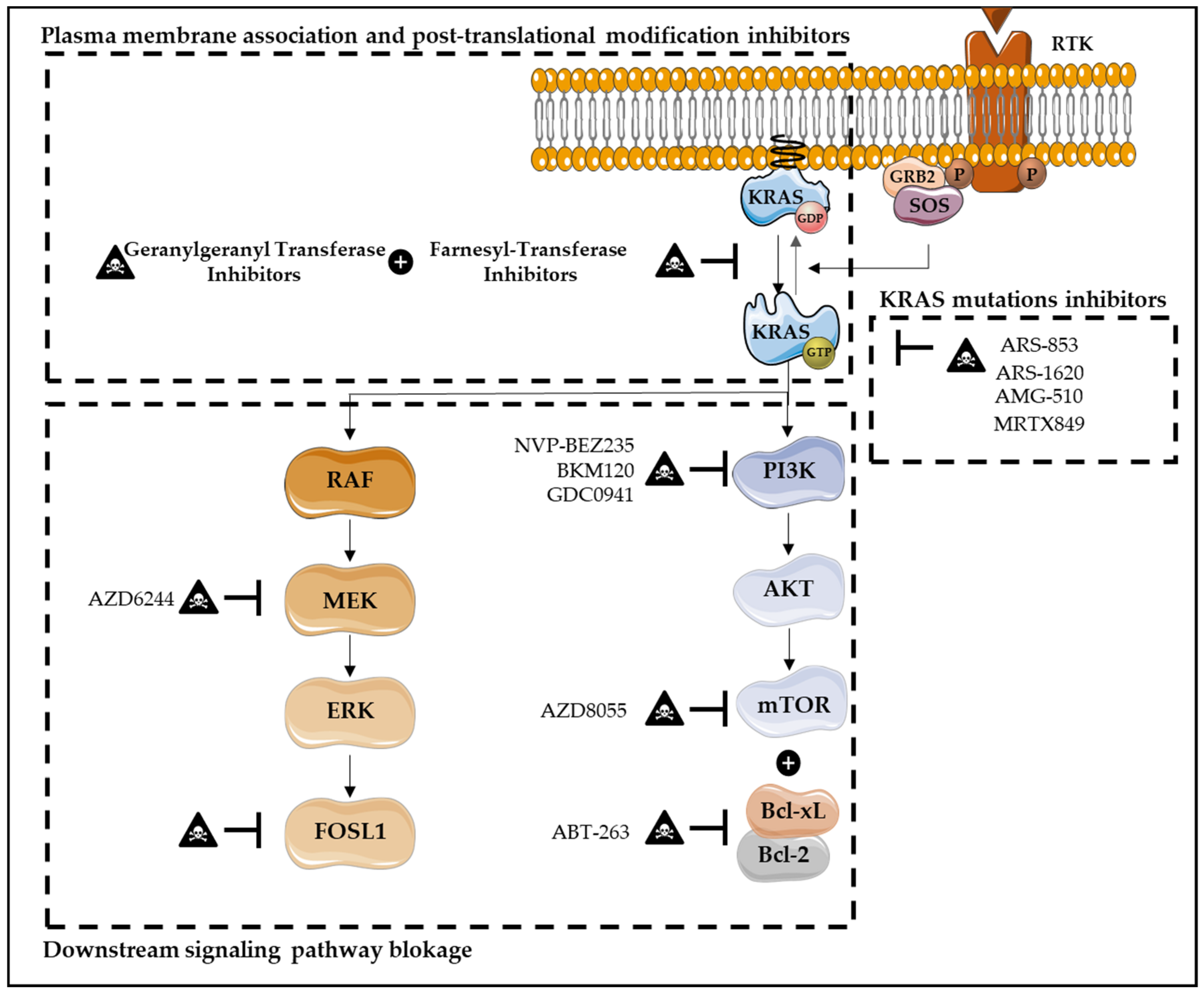{kind=link}
| Target/Biomarkers | Therapies | Impact on Cell Death Processes |
|---|---|---|
| KRASG12C mutation | KRASG12C inhibitors (ARS-853, ARS-1620, AMG-510 and MRTX849) [32,123,125,137] | Apoptosis Induction |
| MEK and PI3K | MEK inhibitor (AZD6244) with PI3K inhibitor (BKM120 or GDC0941) [139] | Apoptosis Induction |
| MEK | MEK inhibitor (trametinib) with Chemotherapy (nab-paclitaxel) [140] | Apoptosis Induction |
| PI3K/mTOR | Dual PI3K-mTOR inhibitor (NVP-BEZ235) [141,142] | Apoptosis Induction |
| RAF/ERK | pan-RAFi with an ERK-selective inhibitor [143] | Apoptosis Induction |
| mTOR and BCL-XL/BCL-2 | mTOR inhibitor (AZD8055) with the dual BCL-XL/BCL-2 inhibitor, (ABT-263) [144] | Apoptosis Induction |
| MEK | Autophagy inhibitor (hydroxychloroquine) with MEK inhibitor (trametinib) [138,145] | Autophagy Inhibition |
| MEK | Autophagy inhibitor (hydroxychloroquine) with MEK inhibitor (binimetinib) [145,146] | Autophagy Inhibition |
| ERK | Autophagy inhibitor (hydroxychloroquine) with ERK inhibitor (ulixertinib) [145,146] | Autophagy Inhibition |
| Mutated KRAS | Autophagy inhibitor (hydroxychloroquine) with KRAS inhibitor (gemcitabine) [145,146] | Autophagy Inhibition |
| PD-1/PD-L1 | Anti-PD-1/PD-L1 monoclonal antibodies [125,133,150,151,152] | Autophagy Induction |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, A.; Pereira, F.; Reis, C.; Oliveira, M.J.; Sousa, M.J.; Preto, A. Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications. Cells 2022, 11, 2183. https://doi.org/10.3390/cells11142183
Ferreira A, Pereira F, Reis C, Oliveira MJ, Sousa MJ, Preto A. Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications. Cells. 2022; 11(14):2183. https://doi.org/10.3390/cells11142183
Chicago/Turabian StyleFerreira, Anabela, Flávia Pereira, Celso Reis, Maria José Oliveira, Maria João Sousa, and Ana Preto. 2022. "Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications" Cells 11, no. 14: 2183. https://doi.org/10.3390/cells11142183
APA StyleFerreira, A., Pereira, F., Reis, C., Oliveira, M. J., Sousa, M. J., & Preto, A. (2022). Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications. Cells, 11(14), 2183. https://doi.org/10.3390/cells11142183