
Increased Resection at DSBs in G2-Phase Is a Unique Phenotype Associated with DNA-PKcs Defects That Is Not Shared by Other Factors of c-NHEJ

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Irradiation
2.3. Treatment of Cell Cultures with Inhibitors
2.4. RNA Interference
2.5. Quantitative Image-Based Cytometry (QIBC) Analysis by Indirect Immunofluorescence (IF)
2.6. Flow Cytometry (FC) Analysis of DNA End-Resection by RPA70 Quantification
2.7. Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
2.8. Cytogenetic Analysis of Translocation Formation in Cells Irradiated in G2-Phase
2.9. Statistical Analysis
3. Results
3.1. Specific Analysis of Resection in G2-Phase Cells That Are also Irradiated in G2-Phase
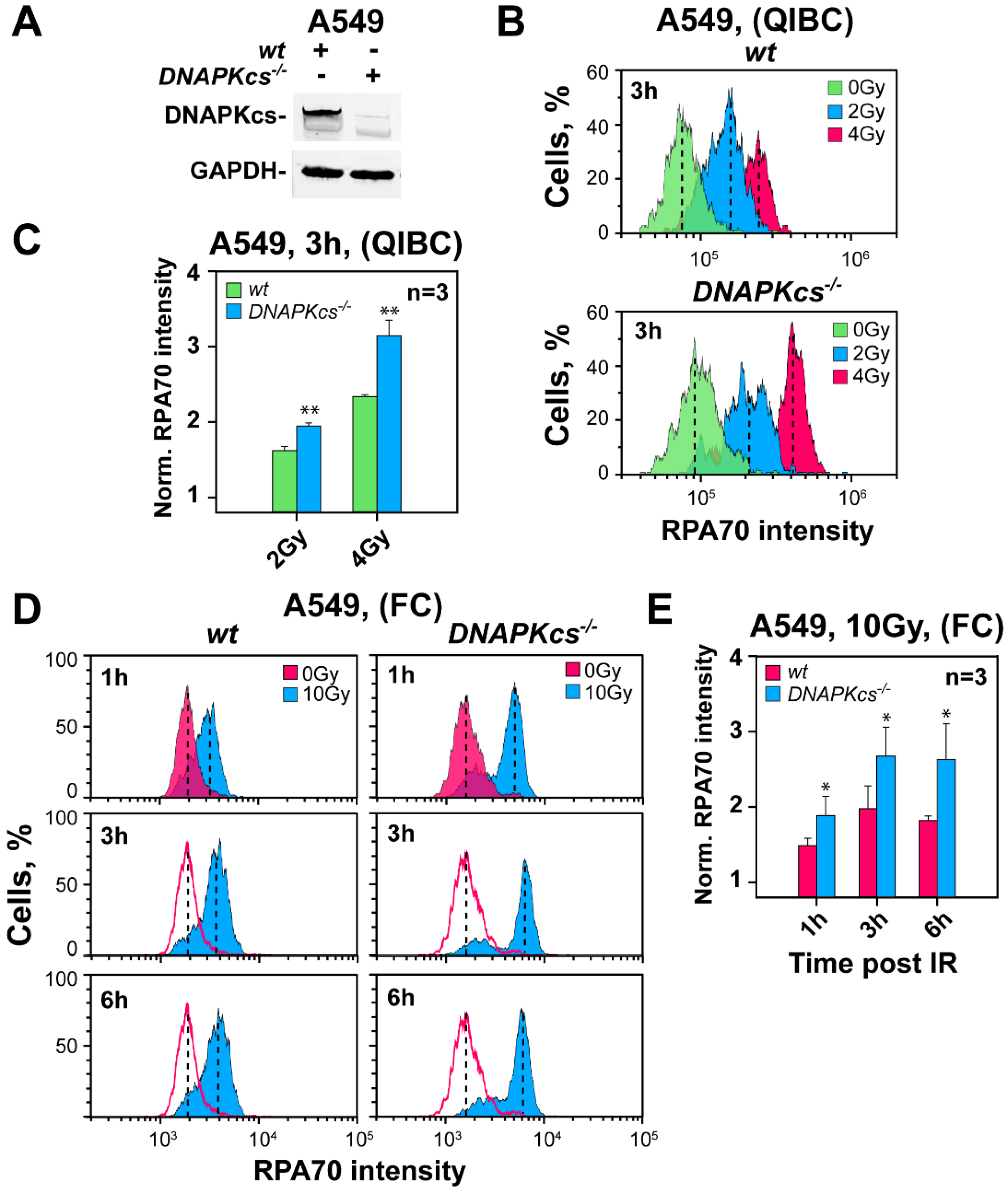
3.2. Genetic Ablation of DNA-PKcs Causes Hyper-Resection in Cells Irradiated in G2-Phase
3.3. Inhibition of DNA-PKcs Using Small Molecule Inhibitors Fails to Cause Hyper-Resection
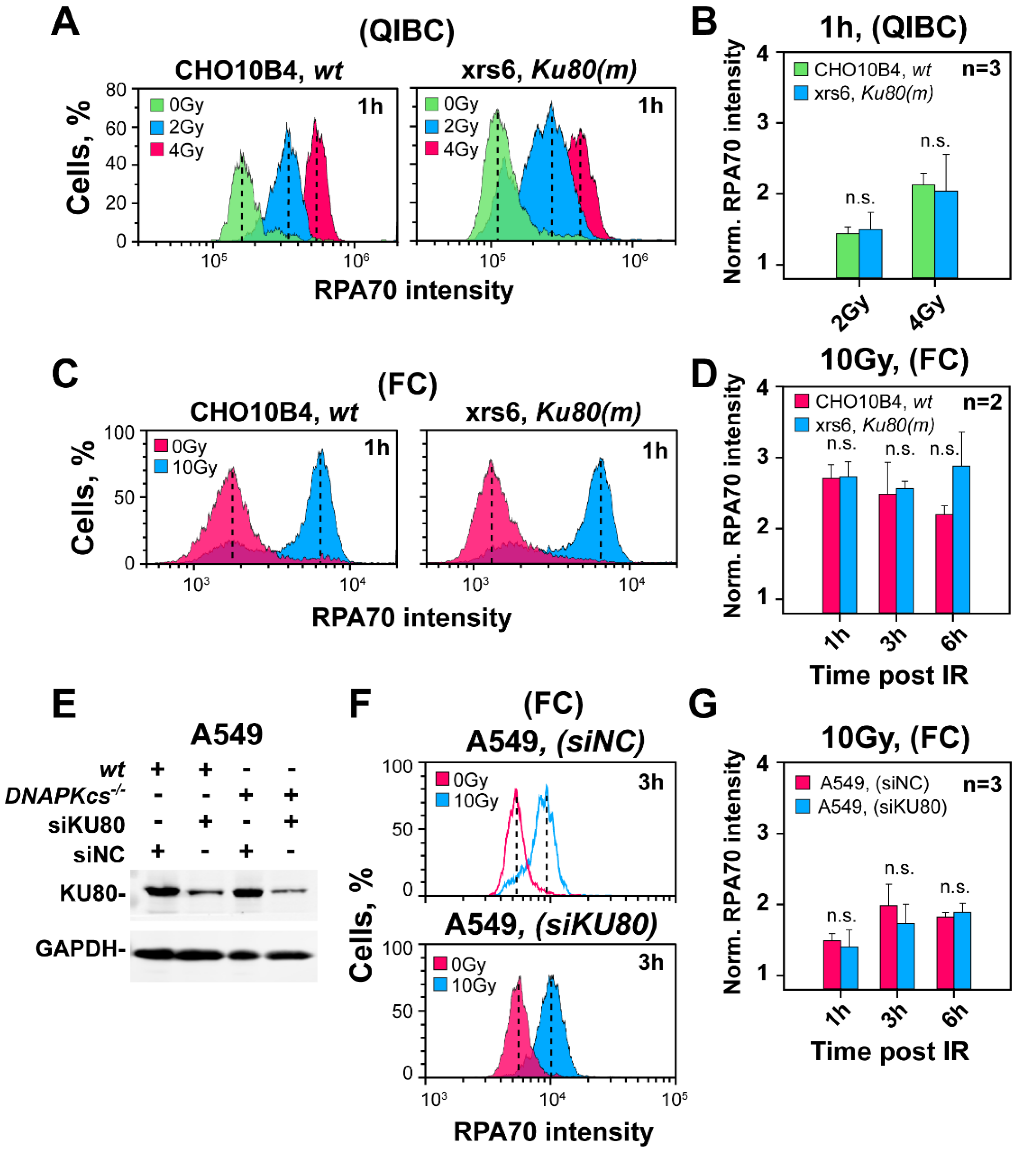
3.4. The Curtailing Function of DNA-PKcs in Resection Is Independent of Ku
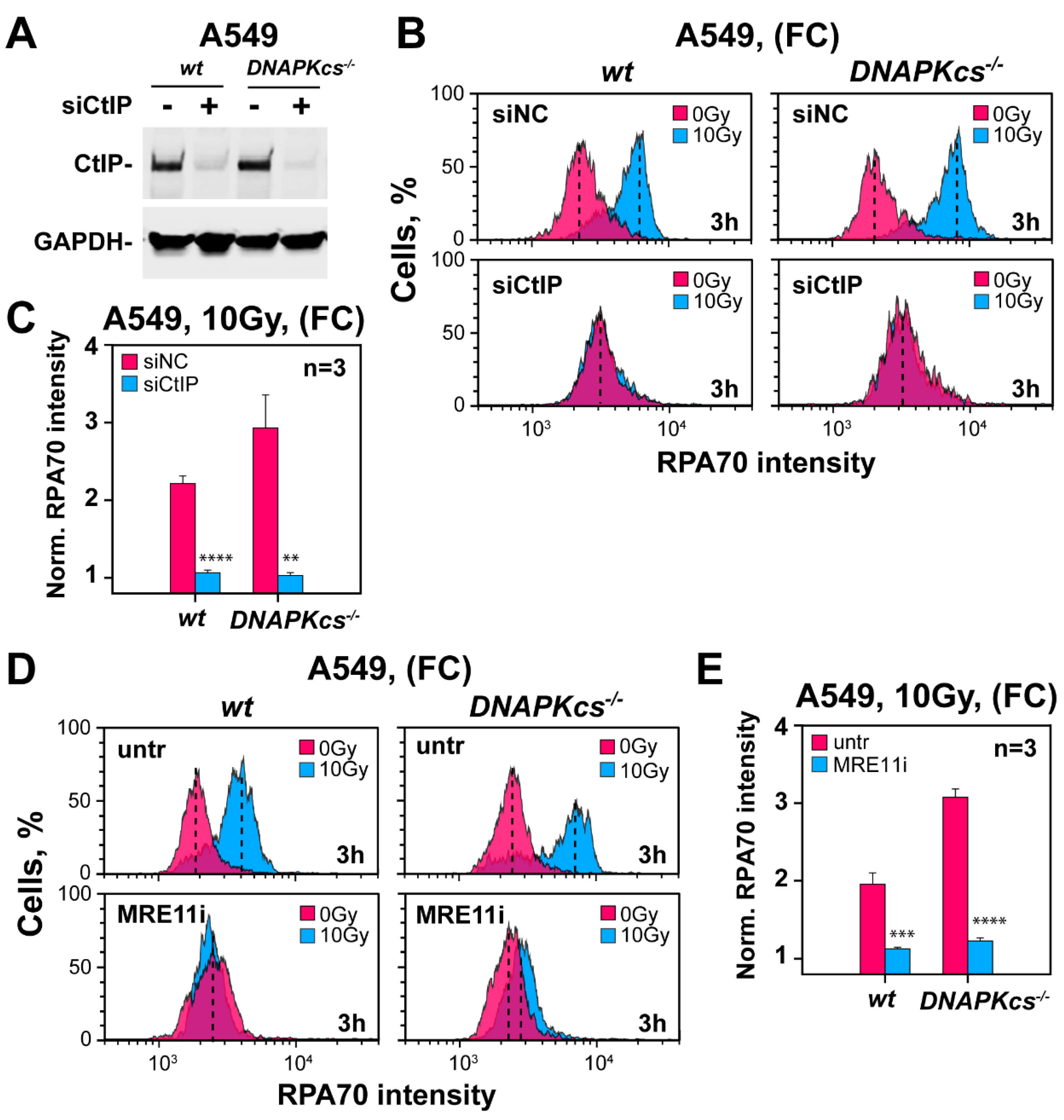
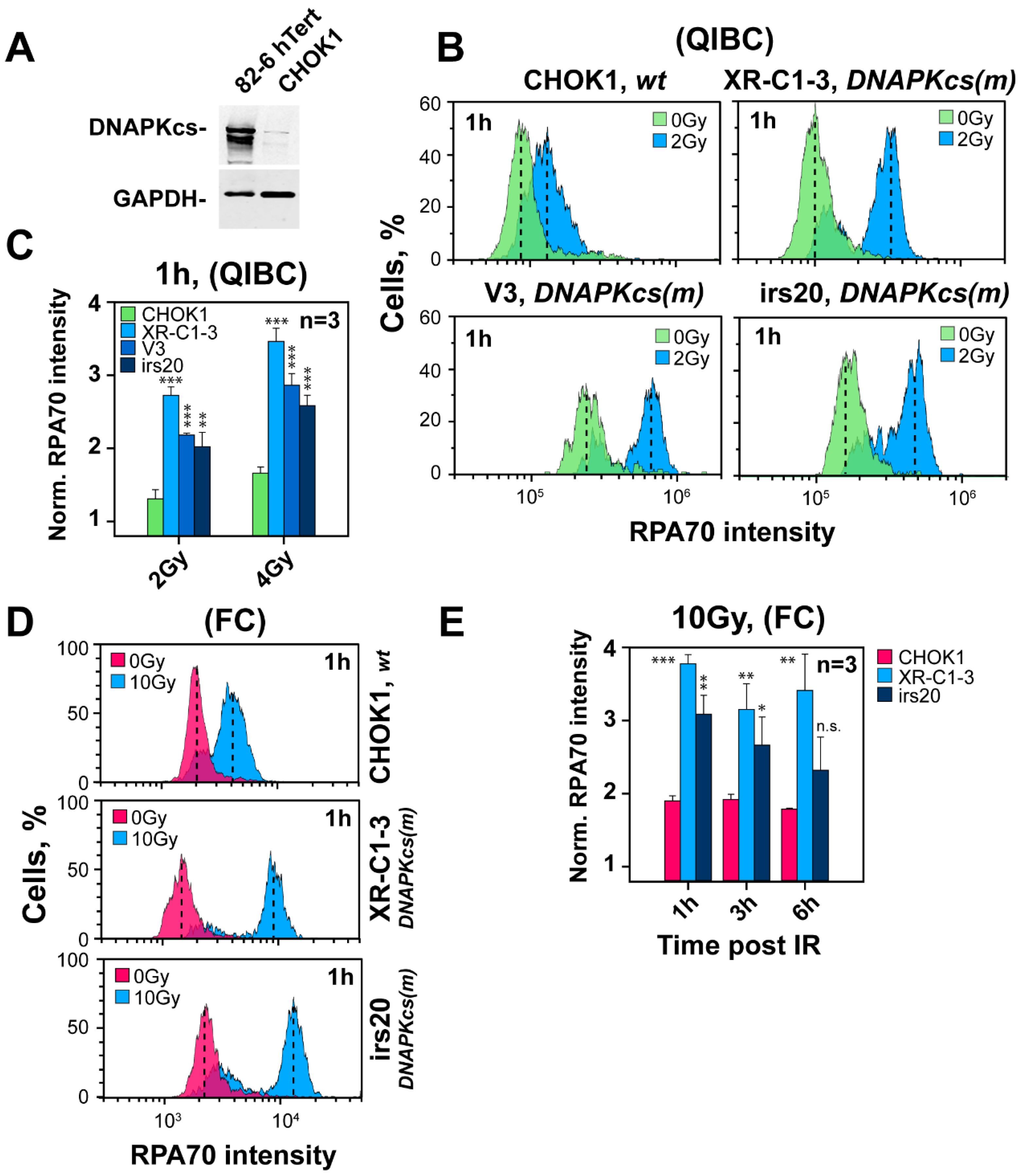
3.5. Defects in Factors of c-NHEJ, Other Than DNA-PKcs, Fail to Cause Hyper-Resection at DSBs
3.6. Hyper-Resection in DNA-PKcs Mutants Causes an Explosion in Chromosomal Translocation Formation
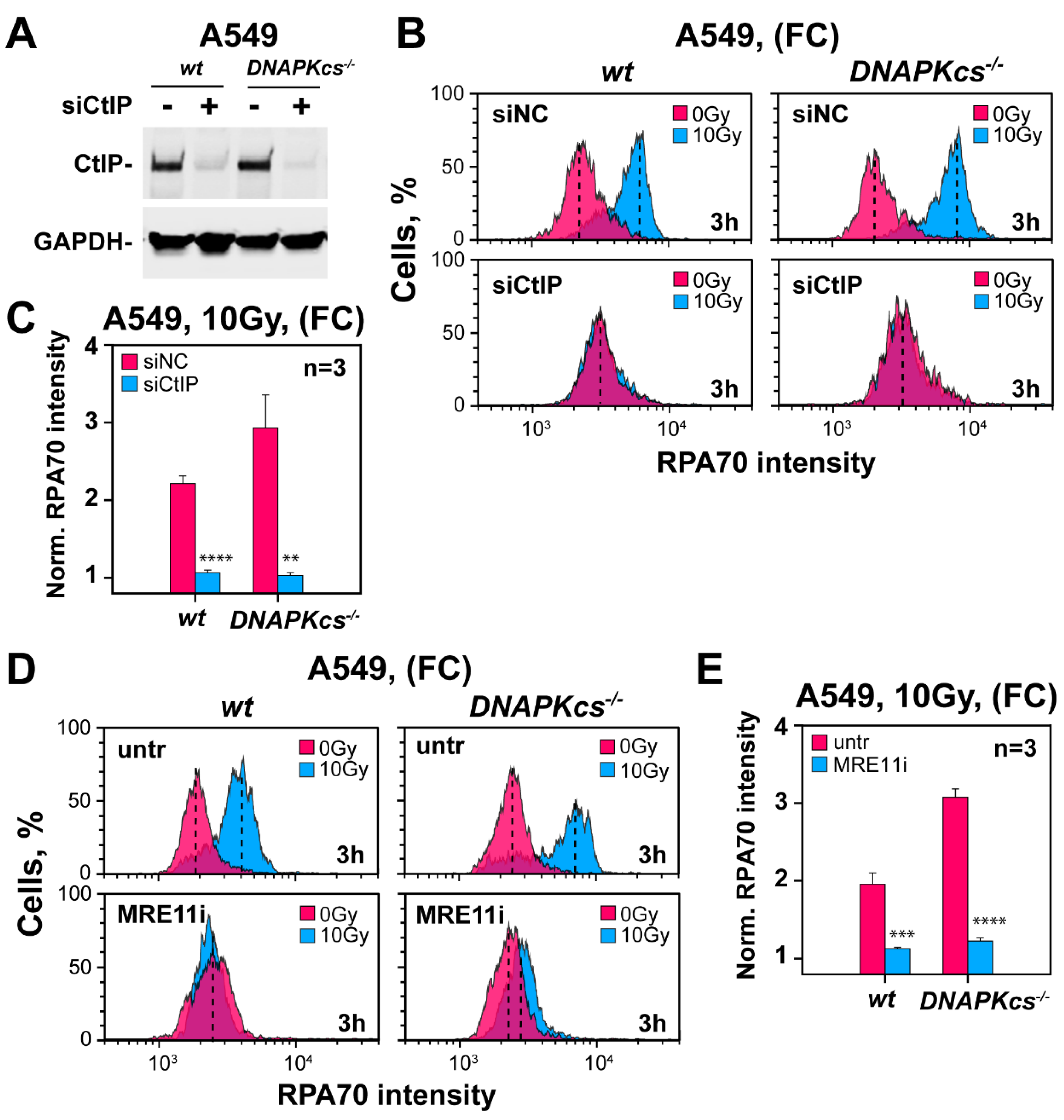
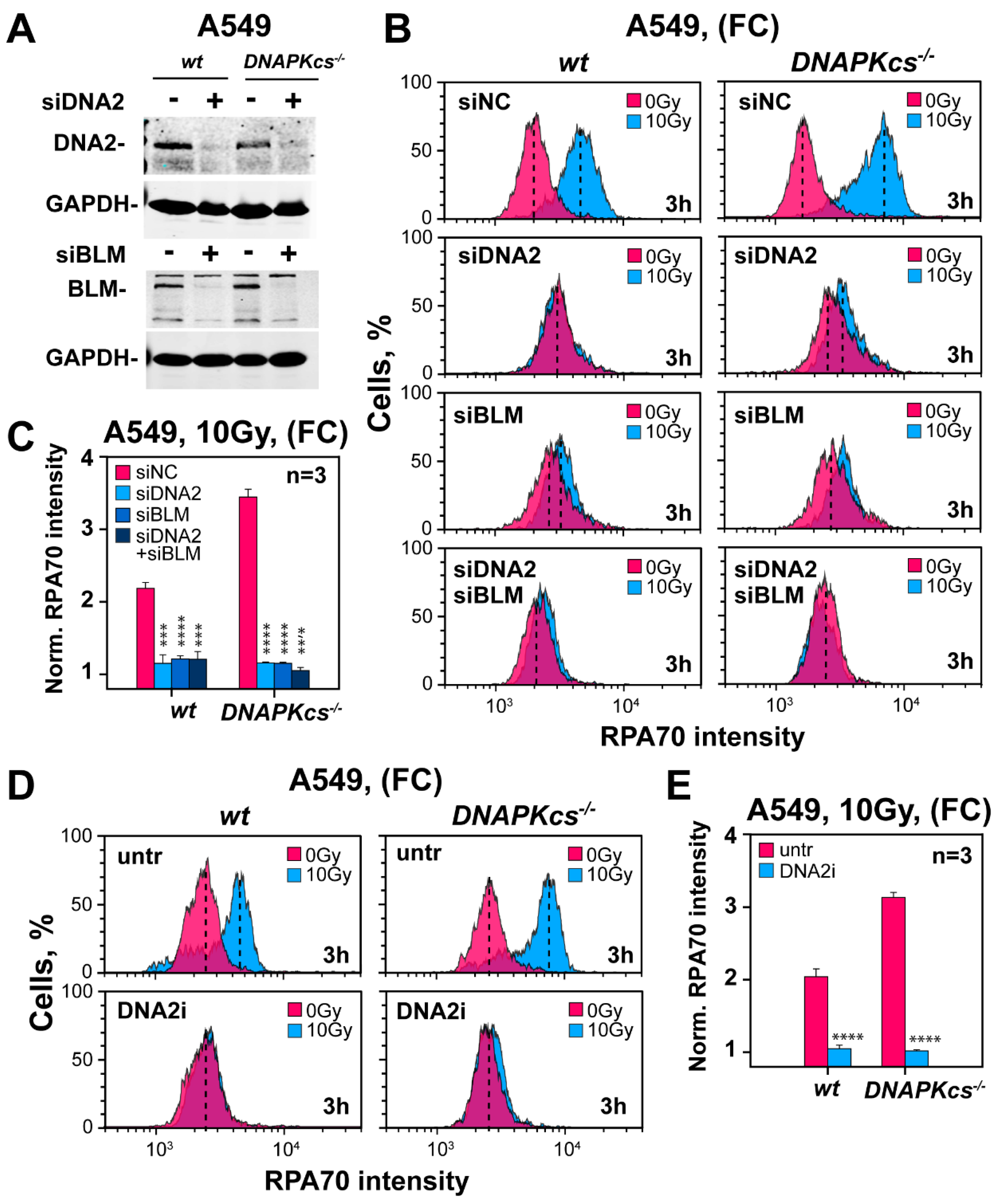
3.7. Factors Involved in DNA End Resection in the Absence of DNA-PKcs
4. Discussion
4.1. Genetic Ablation of DNA-PKcs Causes Hyper-Resection in Cells Irradiated in G2-Phase
4.2. Integration of DNA-PKcs Functions with ATM and ATR Functions
4.3. DNA-PKcs Is a Candidate Regulator of HR with Decreasing IR Dose
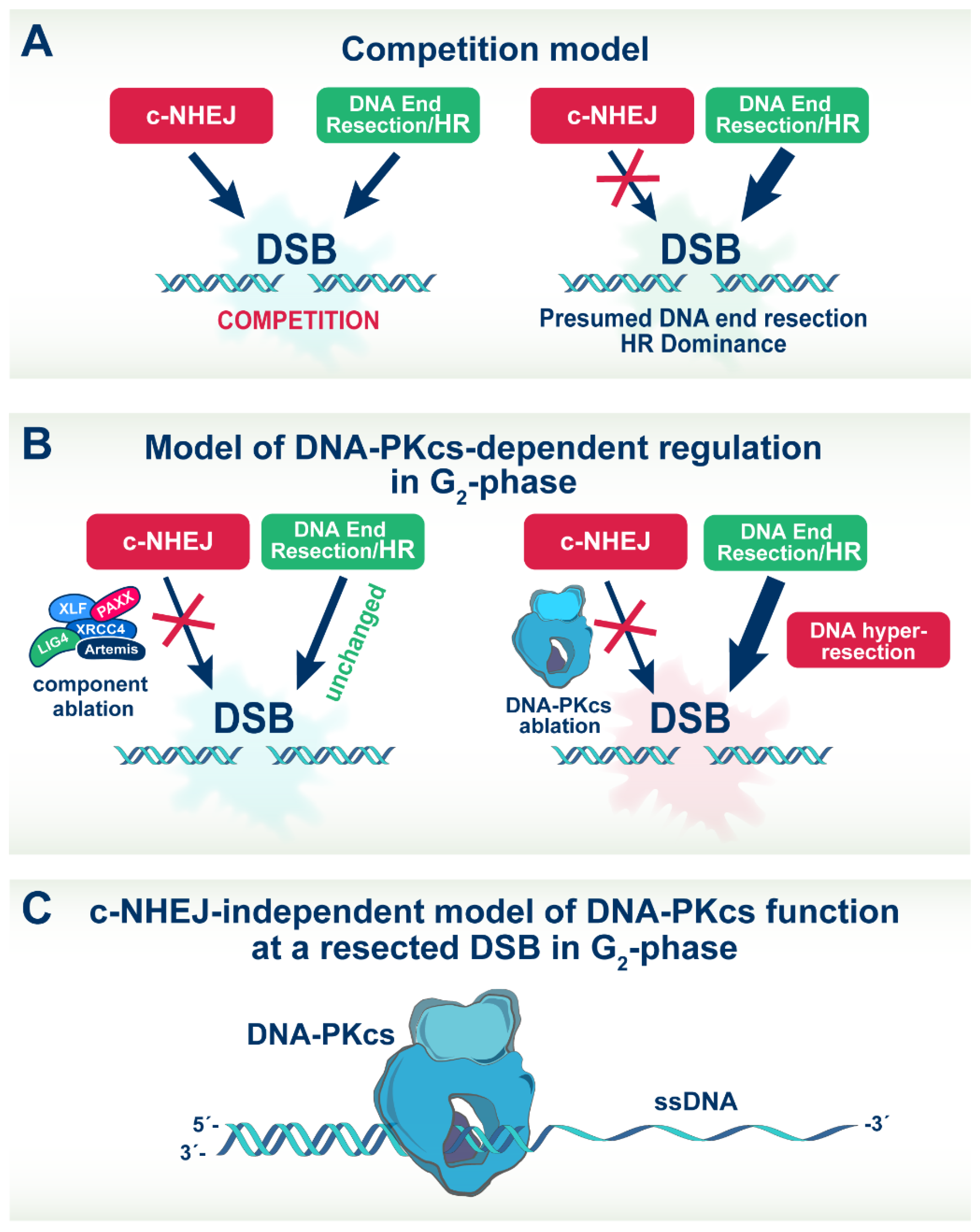
4.4. A model of DNA-PKcs Function in DSB Repair Pathway Choice at Low DSB Loads
5. Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Aylon, Y.; Kupiec, M. New insights into the mechanism of homologous recombination in yeast. Mutat. Res. 2004, 566, 231–248. [Google Scholar] [CrossRef]
- Shrivastav, M.; de Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [Green Version]
- Lees-Miller, J.P.; Cobban, A.; Katsonis, P.; Bacolla, A.; Tsutakawa, S.E.; Hammel, M.; Meek, K.; Anderson, D.W.; Lichtarge, O.; Tainer, J.A.; et al. Uncovering DNA-PKcs ancient phylogeny, unique sequence motifs and insights for human disease. Prog. Biophys. Mol. Biol. 2021, 163, 87–108. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.S.; Cortez, D. Rapid Activation of ATR by Ionizing Radiation Requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, K.E.; Medhurst, A.L.; Dart, D.A.; Lakin, N.D. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 2006, 25, 3894–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, M.; Martinez-Pastor, B.; Murga, M.; Toledo, L.I.; Gutierrez-Martinez, P.; Lopez, E.; Fernandez-Capetillo, O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006, 203, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Löbrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. γH2AX foci analysis for monitoring DNA double-strand break repair-Strengths, limitations and optimization. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [Green Version]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Pantelias, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Dahm-Daphi, J.; Dikomey, E. DNA Repair and Cell Cycle Regulation After Ionizing Irradiation. In The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies, Section: DNA Repair/Reviews-General ed.; Molls, M., Vaupel, P., Nieder, C., Anscher, M.S., Eds.; Medical Radiology-Radiation Oncology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 251–272. [Google Scholar]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Reginato, G.; Cejka, P. The MRE11 complex: A versatile toolkit for the repair of broken DNA. DNA Repair (Amst) 2020, 91–92, 102869. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Hammel, M.; Radhakrishnan, S.K.; Ramsden, D.; Lees-Miller, S.P.; Tainer, J.A. Structural insights into NHEJ: Building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair 2014, 17, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Soni, A.; Mladenov, E.; Iliakis, G. Proficiency in homologous recombination repair is prerequisite for activation of G2-checkpoint at low radiation doses. DNA Repair 2021, 101, 103076. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.A.; Dang, V.; Douglas, P.; Wold, M.S.; Lees-Miller, S.P.; Meek, K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation. Mol. Cell. Biol. 2011, 31, 1719–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.A.; Meek, K. Choosing the right path: Does DNA-PK help make the decision? Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2011, 711, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.W.; Chen, B.P.-C.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Merkle, D.; Douglas, P.; Moorhead, G.B.; Leonenko, Z.; Yu, Y.; Cramb, D.; Bazett-Jones, D.P.; Lees-Miller, S.P. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry 2002, 41, 12706–12714. [Google Scholar] [CrossRef] [PubMed]
- Block, W.D.; Merkle, D.; Meek, K.; Lees-Miller, S.P. Selective inhibition of the DNA-dependent protein kinase (DNA-PK) by the radiosensitizing agent caffeine. Nucleic Acids Res. 2004, 32, 1967–1972. [Google Scholar] [CrossRef] [Green Version]
- Hammel, M.; Yu, Y.; Mahaney, B.L.; Cai, B.; Ye, R.; Phipps, B.M.; Rambo, R.P.; Hura, G.L.; Pelikan, M.; So, S.; et al. Ku and DNA-dependent Protein Kinase Dynamic Conformations and Assembly Regulate DNA Binding and the Initial Non-homologous End Joining Complex. J. Biol. Chem. 2010, 285, 1414–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Reconsidering pathway choice: A sequential model of mammalian DNA double-strand break pathway decisions. Curr. Opin. Genet. Dev. 2021, 71, 55–62. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [Green Version]
- Kass, E.M.; Jasin, M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010, 584, 3703–3708. [Google Scholar] [CrossRef] [Green Version]
- Dynan, W.S.; Yoo, S. Interaction of ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blier, P.R.; Griffith, A.J.; Craft, J.; Hardin, J.A. Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J. Biol. Chem. 1993, 268, 7594–7601. [Google Scholar] [CrossRef]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001, 15, 3237–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delacote, F.; Han, M.; Stamato, T.D.; Jasin, M.; Lopez, B.S. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002, 30, 3454–3463. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.; Kurimasa, A.; Brenneman, M.A.; Chen, D.J.; Nickoloff, J.A. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc. Natl. Acad. Sci. USA 2002, 99, 3758–3763. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Siemann, M.; Pantelias, G.E.; Iliakis, G. Marked cell cycle-dependent contribution of alternative end joining to formation of chromosome translocations by stochastically induced DNA double strand breaks in human cells. Mutat. Res. 2015, 793, 2–8. [Google Scholar] [CrossRef]
- Soni, A.; Siemann, M.; Grabos, M.; Murmann, T.; Pantelias, G.E.; Iliakis, G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014, 42, 6380–6392. [Google Scholar] [CrossRef] [Green Version]
- Forment, J.V.; Walker, R.V.; Jackson, S.P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytom. Part A 2012, 81A, 922–928. [Google Scholar] [CrossRef] [Green Version]
- Dueva, R.; Iliakis, G. Replication protein A: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [CrossRef]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshous, D.; Callebaut, I.; de Chasseval, R.; Corneo, B.; Cavazzana-Calvo, M.; Le Deist, F.; Tezcan, I.; Sanal, O.; Bertrand, Y.; Philippe, N.; et al. Artemis, a novel dna double-strand break repair/v(d)j recombination protein, is mutated in human severe combined immune deficiency. Cell 2001, 105, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.M.; Recio, M.-J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, artemis, and proteins locating to g-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef]
- Zhang, Y.; Jasin, M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Brunet, E.; Jasin, M. Induction of Chromosomal Translocations with CRISPR-Cas9 and Other Nucleases: Understanding the Repair Mechanisms That Give Rise to Translocations. Adv. Exp. Med. Biol. 2018, 1044, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Mozaffari, N.L.; Pagliarulo, F.; Sartori, A.A. Human CtIP: A ‘double agent’ in DNA repair and tumorigenesis. Semin. Cell Dev. Biol. 2021, 113, 47–56. [Google Scholar] [CrossRef]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Soniat, M.M.; Myler, L.R.; Kuo, H.-C.; Paull, T.T.; Finkelstein, I.J. RPA Phosphorylation Inhibits DNA Resection. Mol. Cell 2019, 75, 145–153.e145. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Karanja, K.K.; Popuri, V.; et al. A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huston, E.; Lynch, M.J.; Mohamed, A.; Collins, D.M.; Hill, E.V.; MacLeod, R.; Krause, E.; Baillie, G.S.; Houslay, M.D. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc. Natl. Acad. Sci. USA 2008, 105, 12791–12796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.H. The role of DNA-PK in aging and energy metabolism. FEBS J. 2018, 285, 1959–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.H.F.; Chang, I.; Hudak, C.S.S.; Hyun, S.; Kwan, H.-Y.; Sul, H.S. A Role of DNA-PK for the Metabolic Gene Regulation in Response to Insulin. Cell 2009, 136, 1056–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Gavrilova, O.; Brown, A.L.; Soto, J.E.; Bremner, S.; Kim, J.; Xu, X.; Yang, S.; Um, J.H.; Koch, L.G.; et al. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017, 26, 447. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Kothari, V.; Shafi, A.A.; Semenova, G.; Gallagher, P.T.; Guan, Y.F.; Pang, A.; Goodwin, J.F.; Irani, S.; McCann, J.J.; et al. A Novel Role for DNA-PK in Metabolism by Regulating Glycolysis in Castration Resistant Prostate Cancer. Clin. Cancer Res. 2022, 28, 1446–1459. [Google Scholar] [CrossRef]
- Chen, X.; Xu, X.; Chen, Y.; Cheung, J.C.; Wang, H.; Jiang, J.; de Val, N.; Fox, T.; Gellert, M.; Yang, W. Structure of an activated DNA-PK and its implications for NHEJ. Mol. Cell 2021, 81, 801–810.e3. [Google Scholar] [CrossRef]
- Chaplin, A.K.; Hardwick, S.W.; Liang, S.; Kefala Stavridi, A.; Hnizda, A.; Cooper, L.R.; De Oliveira, T.M.; Chirgadze, D.Y.; Blundell, T.L. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nat. Struct. Mol. Biol. 2021, 28, 13–19. [Google Scholar] [CrossRef]
- Cui, X.; Yu, Y.; Gupta, S.; Cho, Y.M.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell Biol. 2005, 25, 10842–10852. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-S.; Wang, R.W.; Chang, H.-H.; Capurso, D.; Segal, M.R.; Haber, J.E. Chromosome position determines the success of double-strand break repair. Proc. Natl. Acad. Sci. USA 2016, 113, E146–E154. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, A.K.; Hardwick, S.W.; Stavridi, A.K.; Buehl, C.J.; Goff, N.J.; Ropars, V.; Liang, S.; De Oliveira, T.M.; Chirgadze, D.Y.; Meek, K.; et al. Cryo-EM of NHEJ supercomplexes provides insights into DNA repair. Mol. Cell 2021, 81, 3400–3409.e3403. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, A.; Johnson, F.B.; Oliver, A.; Tresini, M.; Smith, J.S.; Hdeib, M.; Sell, C.; Cristofalo, V.J.; Stamato, T.D. Significant correlation of species longevity with DNA double strand break recognition but not with telomere length. Mech. Ageing Dev. 2009, 130, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, H.; Li, F.; Mladenov, E.; Soni, A.; Mladenova, V.; Pan, B.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Increased Resection at DSBs in G2-Phase Is a Unique Phenotype Associated with DNA-PKcs Defects That Is Not Shared by Other Factors of c-NHEJ. Cells 2022, 11, 2099. https://doi.org/10.3390/cells11132099
Xiao H, Li F, Mladenov E, Soni A, Mladenova V, Pan B, Dueva R, Stuschke M, Timmermann B, Iliakis G. Increased Resection at DSBs in G2-Phase Is a Unique Phenotype Associated with DNA-PKcs Defects That Is Not Shared by Other Factors of c-NHEJ. Cells. 2022; 11(13):2099. https://doi.org/10.3390/cells11132099
Chicago/Turabian StyleXiao, Huaping, Fanghua Li, Emil Mladenov, Aashish Soni, Veronika Mladenova, Bing Pan, Rositsa Dueva, Martin Stuschke, Beate Timmermann, and George Iliakis. 2022. "Increased Resection at DSBs in G2-Phase Is a Unique Phenotype Associated with DNA-PKcs Defects That Is Not Shared by Other Factors of c-NHEJ" Cells 11, no. 13: 2099. https://doi.org/10.3390/cells11132099
APA StyleXiao, H., Li, F., Mladenov, E., Soni, A., Mladenova, V., Pan, B., Dueva, R., Stuschke, M., Timmermann, B., & Iliakis, G. (2022). Increased Resection at DSBs in G2-Phase Is a Unique Phenotype Associated with DNA-PKcs Defects That Is Not Shared by Other Factors of c-NHEJ. Cells, 11(13), 2099. https://doi.org/10.3390/cells11132099