
Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF)

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Idiopathic Pulmonary Fibrosis
1.1. Definition and Epidemiology
1.2. Clinical Manifestation and Diagnosis
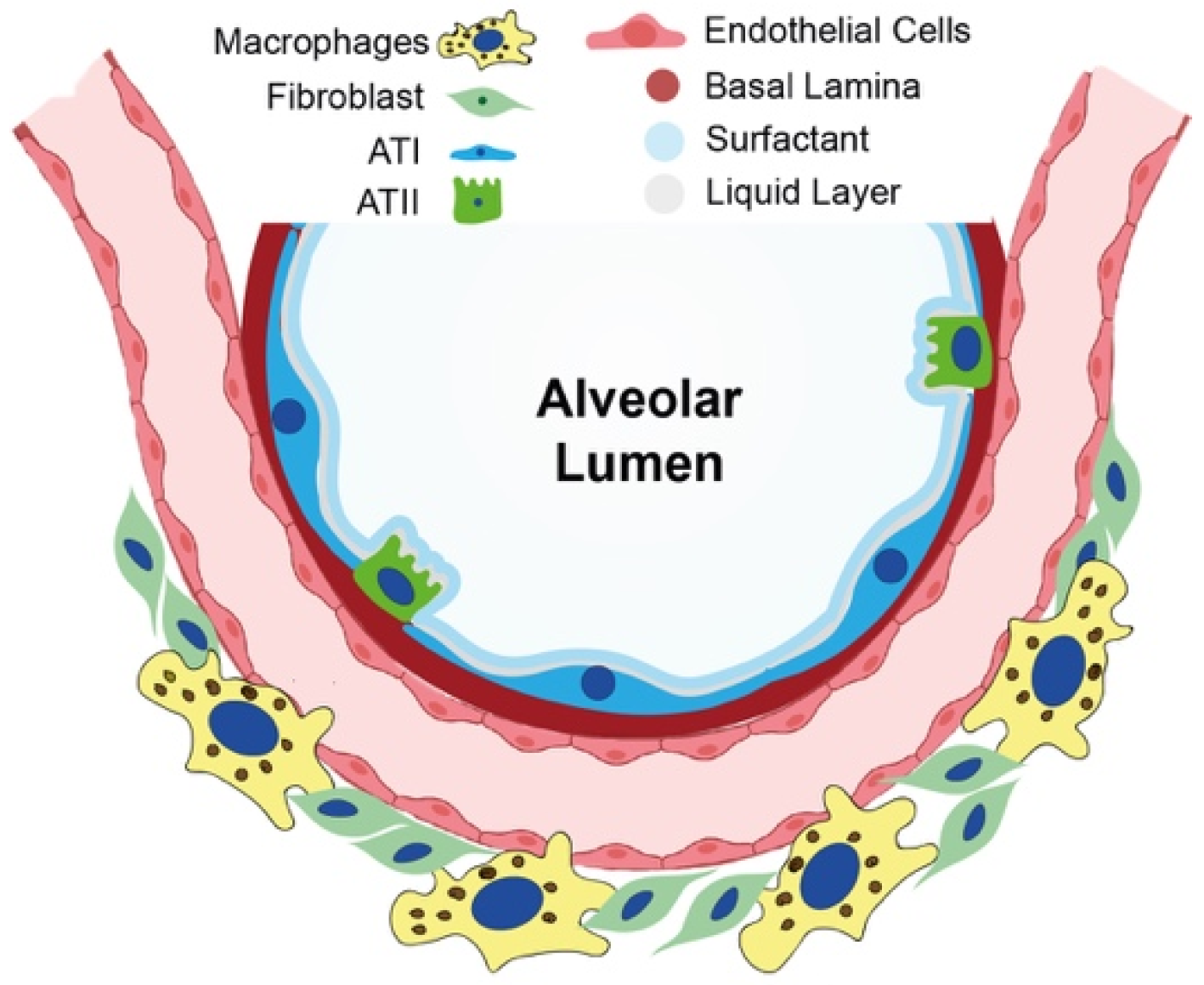
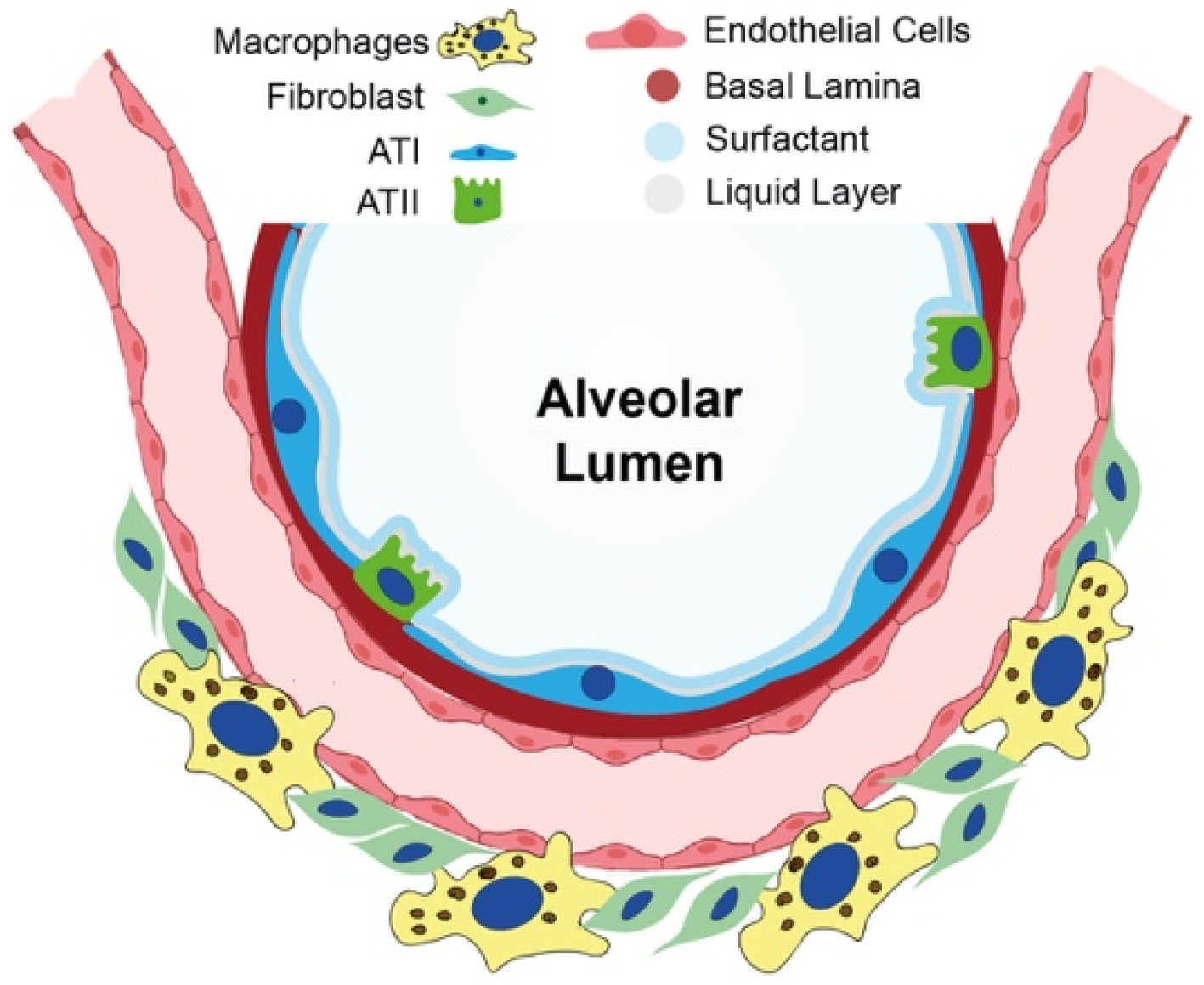
2. Current Knowledge of Cellular and Molecular Mechanisms in IPF Onset
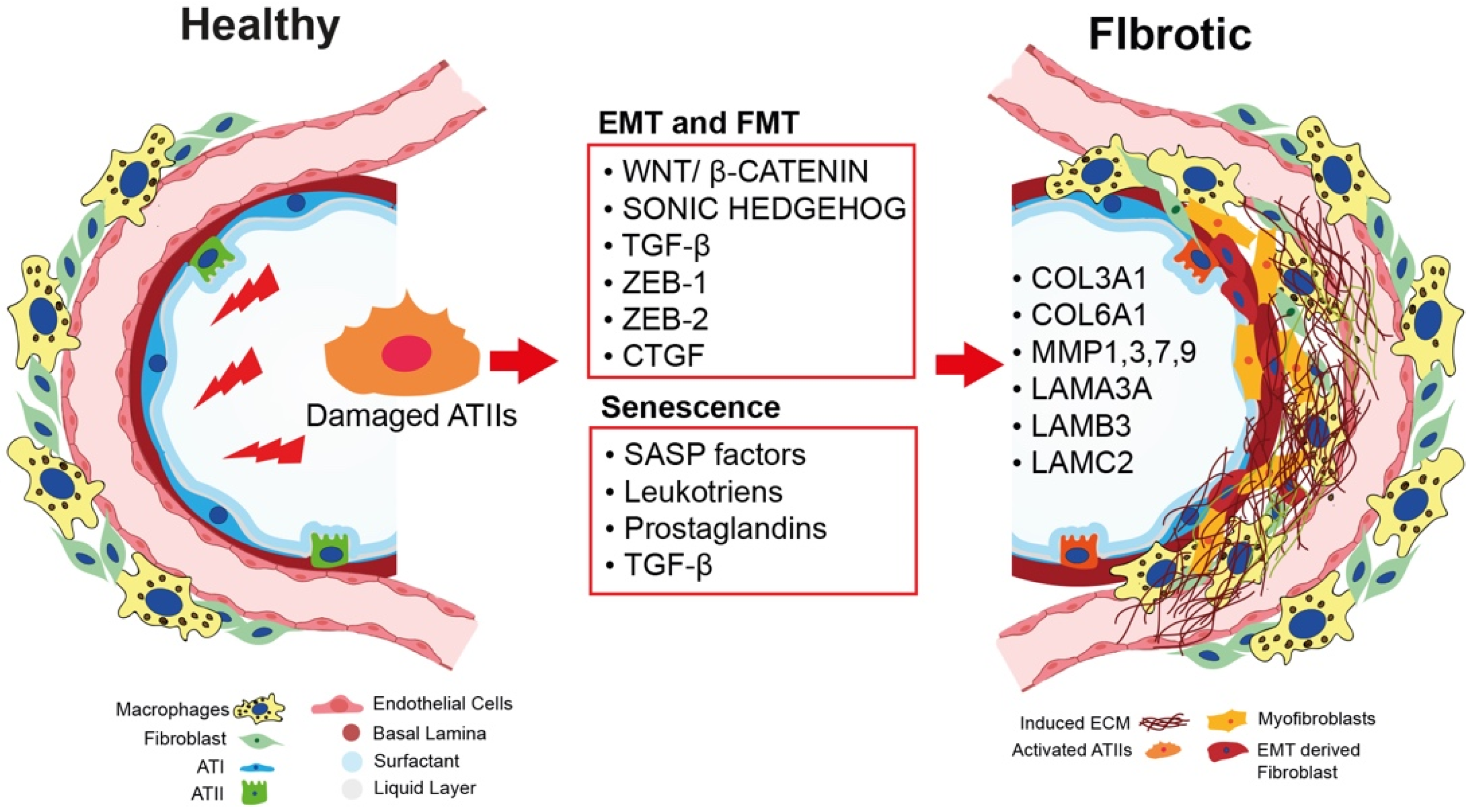
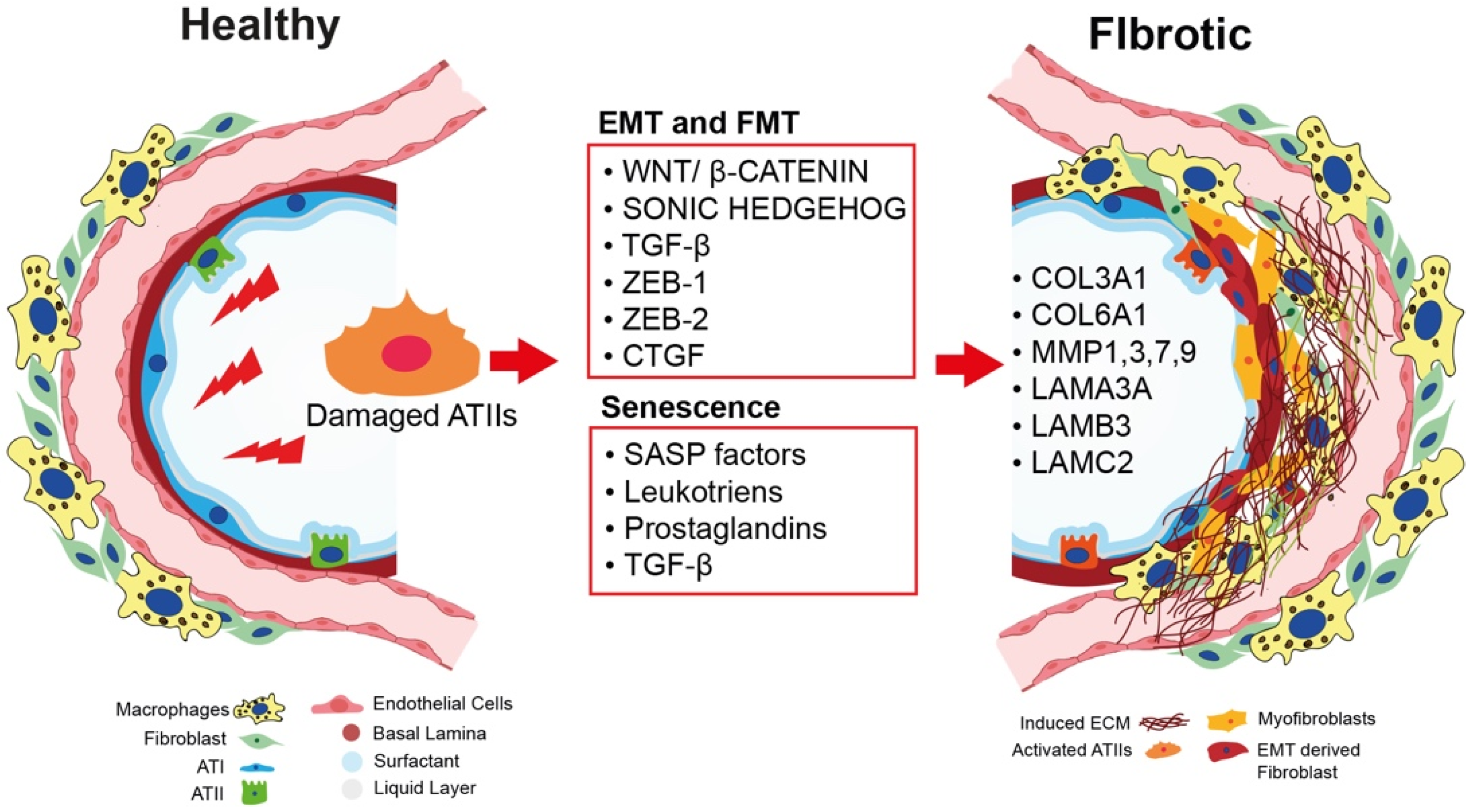
2.1. Epithelial-to-Mesenchymal Transition as a Nonmesenchymal Source of Fibrosis
2.2. The Role of Bidirectional Epithelial–Mesenchymal Crosstalk in Fibroblast-to-Myofibroblast Activation
2.3. Cellular Senescence and IPF: A Causative Link
2.4. The Role of ECM in IPF Onset and Progression
3. Current Knowledge of Cellular and Molecular Mechanisms in Lung Regeneration
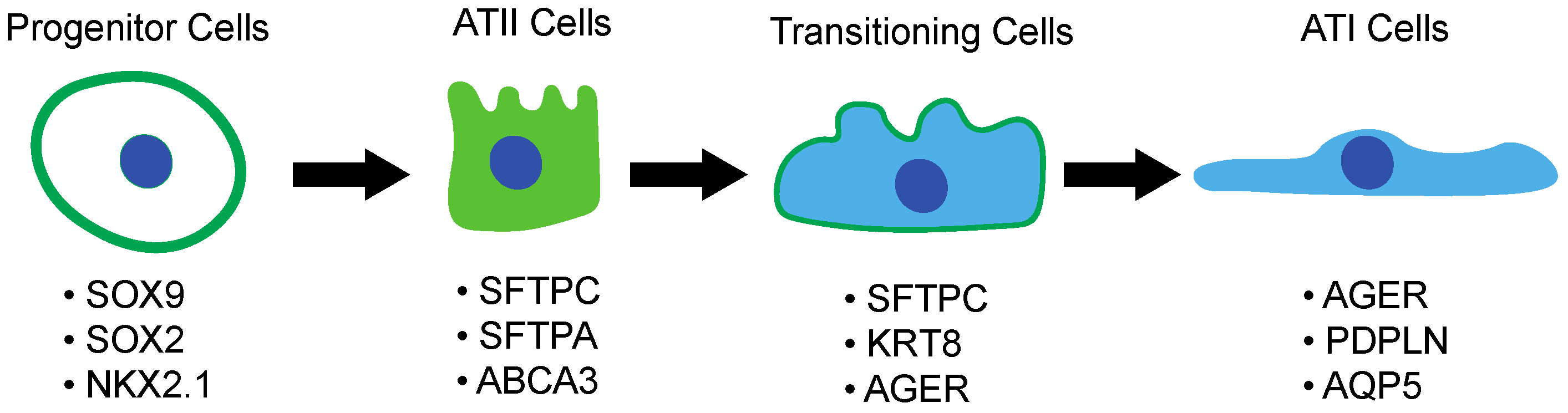
3.1. Lung Embryogenesis and Development
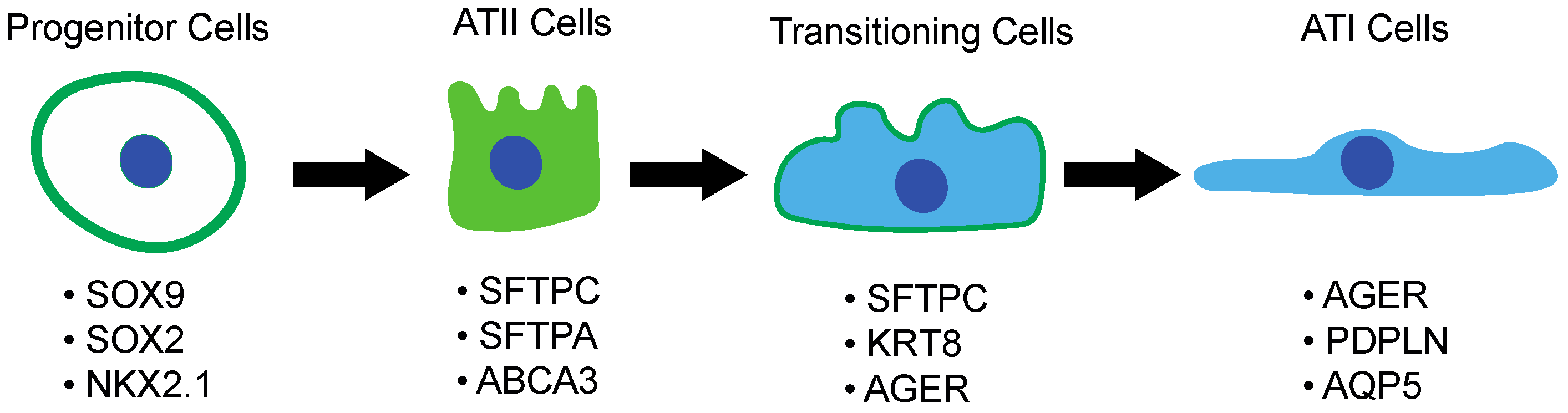
3.2. Alveolar Type II Pneumocytes and Their Response to Lung Injury
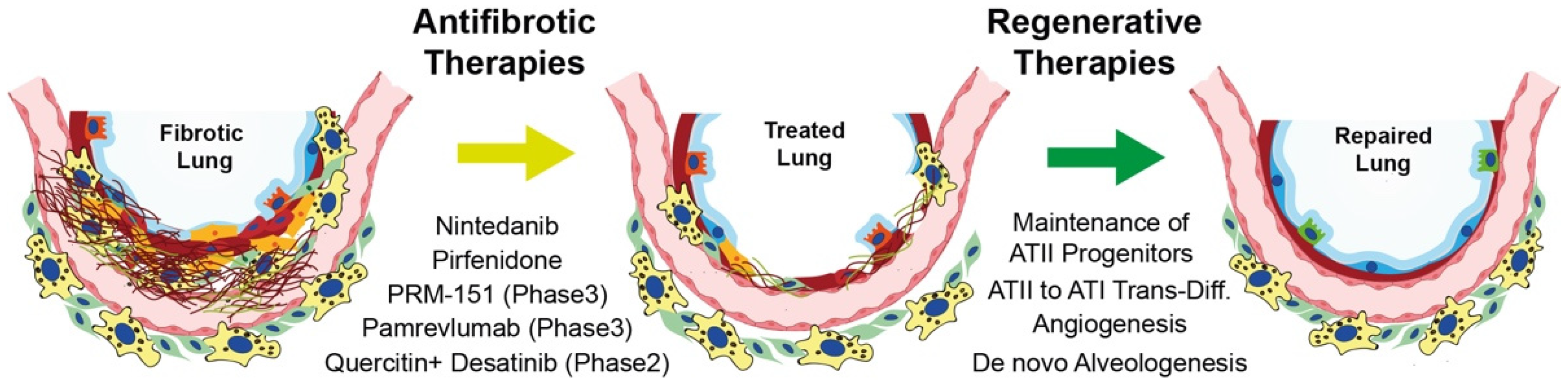
4. Current Therapies for IPF
- (1)
- Obtain better mechanistic insights into the interplay between ATII cells and fibroblasts during fibrosis progression, with the aim of identifying novel ATII-related druggable targets for the development of the most effective antifibrotic therapies;
- (2)
- Systematically dissect the molecular mechanisms of ATII-driven physiological lung regeneration in order to find new druggable targets for the development of effective regenerative therapies;
- (3)
- Tackle this disease by simultaneously blocking fibrosis progression and promoting alveolar repair and organ regeneration.
Author Contributions
Funding
Conflicts of Interest
References
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Ley, B.; Collard, H.R. Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Chen, S.Y.; Yeh, W.S.; Li, Q.; Lee, Y.C.; Wang, A.; Raghu, G. Health care utilization and costs of idiopathic pulmonary fibrosis in U.S. Medicare beneficiaries aged 65 years and older. Ann. Am. Thorac. Soc. 2015, 12, 981–987. [Google Scholar] [CrossRef]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, L.W.; Angampalli, S.; Guttentag, S.H.; Beers, M.F.; Feinstein, S.I.; Matlapudi, A.; Ballard, P.L. Maintenance of differentiated function of the surfactant system in human fetal lung type II epithelial cells cultured on plastic. Pediatric Pathol. Mol. Med. 2001, 20, 387–412. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; Coultas, D.B.; Stidley, C.A.; Hunt, W.C.; Colby, T.V.; Waldron, J.A. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: A multicenter case-control study. Collaborating Centers. Am. J. Epidemiol. 2000, 152, 307–315. [Google Scholar] [CrossRef]
- Paolocci, G.; Folletti, I.; Toren, K.; Ekstrom, M.; Dell’Omo, M.; Muzi, G.; Murgia, N. Occupational risk factors for idiopathic pulmonary fibrosis in Southern Europe: A case-control study. BMC Pulm. Med. 2018, 18, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Campo, I.; Zorzetto, M.; Mariani, F.; Kadija, Z.; Morbini, P.; Dore, R.; Kaltenborn, E.; Frixel, S.; Zarbock, R.; Liebisch, G.; et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir. Res. 2014, 15, 43. [Google Scholar] [CrossRef] [Green Version]
- Mulugeta, S.; Nureki, S.; Beers, M.F. Lost after translation: Insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L507–L525. [Google Scholar] [CrossRef] [Green Version]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Guenther, A.; Krauss, E.; Tello, S.; Wagner, J.; Paul, B.; Kuhn, S.; Maurer, O.; Heinemann, S.; Costabel, U.; Barbero, M.A.N.; et al. The European IPF registry (eurIPFreg): Baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 141. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Krauss, E.; Gehrken, G.; Drakopanagiotakis, F.; Tello, S.; Dartsch, R.C.; Maurer, O.; Windhorst, A.; von der Beck, D.; Griese, M.; Seeger, W.; et al. Clinical characteristics of patients with familial idiopathic pulmonary fibrosis (f-IPF). BMC Pulm. Med. 2019, 19, 130. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Pastre, J.; Barnett, S.; Ksovreli, I.; Taylor, J.; Brown, A.W.; Shlobin, O.A.; Ahmad, K.; Khangoora, V.; Aryal, S.; King, C.S.; et al. Idiopathic pulmonary fibrosis patients with severe physiologic impairment: Characteristics and outcomes. Respir. Res. 2021, 22, 5. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Moimas, S.; Salton, F.; Kosmider, B.; Ring, N.; Volpe, M.C.; Bahmed, K.; Braga, L.; Rehman, M.; Vodret, S.; Graziani, M.L.; et al. miR-200 family members reduce senescence and restore idiopathic pulmonary fibrosis type II alveolar epithelial cell transdifferentiation. ERJ Open Res. 2019, 5, 00138–2019. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial-Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef]
- Bartis, D.; Mise, N.; Mahida, R.Y.; Eickelberg, O.; Thickett, D.R. Epithelial-mesenchymal transition in lung development and disease: Does it exist and is it important? Thorax 2014, 69, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tian, X.J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-beta, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef]
- Rock, J.R.; Hogan, B.L. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 493–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G. Idiopathic pulmonary fibrosis: Lessons from clinical trials over the past 25 years. Eur. Respir. J. 2017, 50, 1701209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.M.; Liu, G. Cell senescence and fibrotic lung diseases. Exp. Gerontol. 2020, 132, 110836. [Google Scholar] [CrossRef] [PubMed]
- Muthuramalingam, K.; Cho, M.; Kim, Y. Cellular senescence and EMT crosstalk in bleomycin-induced pathogenesis of pulmonary fibrosis-an in vitro analysis. Cell Biol. Int. 2020, 44, 477–487. [Google Scholar] [CrossRef]
- Faheem, M.M.; Seligson, N.D.; Ahmad, S.M.; Rasool, R.U.; Gandhi, S.G.; Bhagat, M.; Goswami, A. Convergence of therapy-induced senescence (TIS) and EMT in multistep carcinogenesis: Current opinions and emerging perspectives. Cell Death Discov. 2020, 6, 51. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial(-)Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMaio, L.; Buckley, S.T.; Krishnaveni, M.S.; Flodby, P.; Dubourd, M.; Banfalvi, A.; Xing, Y.; Ehrhardt, C.; Minoo, P.; Zhou, B.; et al. Ligand-independent transforming growth factor-beta type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J. Pathol. 2012, 226, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degryse, A.L.; Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Jones, B.R.; Boomershine, C.S.; Ortiz, C.; Sherrill, T.P.; McMahon, F.B.; Gleaves, L.A.; et al. TGFbeta signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L887–L897. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zhou, Y.; Li, J.; Wickens, L.; Conforti, F.; Rattu, A.; Ibrahim, F.M.; Alzetani, A.; Marshall, B.G.; Fletcher, S.V.; et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 2021, 297, 101096. [Google Scholar] [CrossRef]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Gyetko, M.R.; Idell, S.; Gharaee-Kermani, M.; Shetty, R.S.; Starcher, B.C.; Shetty, S. Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L463–L473. [Google Scholar] [CrossRef] [Green Version]
- Uhal, B.D.; Gidea, C.; Bargout, R.; Bifero, A.; Ibarra-Sunga, O.; Papp, M.; Flynn, K.; Filippatos, G. Captopril inhibits apoptosis in human lung epithelial cells: A potential antifibrotic mechanism. Am. J. Physiol. 1998, 275, L1013–L1017. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Aoshiba, K.; Tsuji, T.; Kameyama, S.; Itoh, M.; Semba, S.; Yamaguchi, K.; Nakamura, H. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Exp. Toxicol. Pathol. 2013, 65, 1053–1062. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Hansel, C.; Jendrossek, V.; Klein, D. Cellular Senescence in the Lung: The Central Role of Senescent Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3279. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 2019, 4, e130056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesemann, A.; Ketteler, J.; Slama, A.; Wirsdorfer, F.; Hager, T.; Rock, K.; Engel, D.R.; Fischer, J.W.; Aigner, C.; Jendrossek, V.; et al. Inhibition of Radiation-Induced Ccl2 Signaling Protects Lungs from Vascular Dysfunction and Endothelial Cell Loss. Antioxid Redox Signal 2019, 30, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [Green Version]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [Green Version]
- Amsellem, V.; Gary-Bobo, G.; Marcos, E.; Maitre, B.; Chaar, V.; Validire, P.; Stern, J.B.; Noureddine, H.; Sapin, E.; Rideau, D.; et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1358–1366. [Google Scholar] [CrossRef]
- Moliva, J.I.; Rajaram, M.V.; Sidiki, S.; Sasindran, S.J.; Guirado, E.; Pan, X.J.; Wang, S.H.; Ross, P., Jr.; Lafuse, W.P.; Schlesinger, L.S.; et al. Molecular composition of the alveolar lining fluid in the aging lung. Age 2014, 36, 9633. [Google Scholar] [CrossRef] [Green Version]
- Dickens, J.A.; Rutherford, E.N.; Abreu, S.; Chambers, J.E.; Ellis, M.O.; van Schadewijk, A.; Hiemstra, P.S.; Marciniak, S.J. Novel insights into surfactant protein C trafficking revealed through the study of a pathogenic mutant. Eur. Respir. J. 2022, 59, 2100267. [Google Scholar] [CrossRef] [PubMed]
- Savani, R.C.; Godinez, R.I.; Godinez, M.H.; Wentz, E.; Zaman, A.; Cui, Z.; Pooler, P.M.; Guttentag, S.H.; Beers, M.F.; Gonzales, L.W.; et al. Respiratory distress after intratracheal bleomycin: Selective deficiency of surfactant proteins B and C. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L685–L696. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, E.; Boden, C.; Echaide, M.; Perez-Gil, J.; Kolb, M.; Gauldie, J.; Maus, U.A.; Ochs, M.; Knudsen, L. Surfactant dysfunction during overexpression of TGF-beta1 precedes profibrotic lung remodeling in vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1260–L1271. [Google Scholar] [CrossRef] [PubMed]
- Yazicioglu, T.; Muhlfeld, C.; Autilio, C.; Huang, C.K.; Bar, C.; Dittrich-Breiholz, O.; Thum, T.; Perez-Gil, J.; Schmiedl, A.; Brandenberger, C. Aging impairs alveolar epithelial type II cell function in acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L755–L769. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers. 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Hashimoto, M.; Asai, A.; Kawagishi, H.; Mikawa, R.; Iwashita, Y.; Kanayama, K.; Sugimoto, K.; Sato, T.; Maruyama, M.; Sugimoto, M. Elimination of p19(ARF)-expressing cells enhances pulmonary function in mice. JCI Insight 2016, 1, e87732. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Todd, J.L.; Vinisko, R.; Liu, Y.; Neely, M.L.; Overton, R.; Flaherty, K.R.; Noth, I.; Newby, L.K.; Lasky, J.A.; Olman, M.A.; et al. Circulating matrix metalloproteinases and tissue metalloproteinase inhibitors in patients with idiopathic pulmonary fibrosis in the multicenter IPF-PRO Registry cohort. BMC Pulm. Med. 2020, 20, 64. [Google Scholar] [CrossRef] [Green Version]
- Okuda, R.; Aoshiba, K.; Matsushima, H.; Ogura, T.; Okudela, K.; Ohashi, K. Cellular senescence and senescence-associated secretory phenotype: Comparison of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung disease, and chronic obstructive pulmonary disease. J. Thorac.Dis. 2019, 11, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H.; et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 2012, 186, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePianto, D.J.; Chandriani, S.; Abbas, A.R.; Jia, G.; N’Diaye, E.N.; Caplazi, P.; Kauder, S.E.; Biswas, S.; Karnik, S.K.; Ha, C.; et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2015, 70, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Placido, L.; Romero, Y.; Maldonado, M.; Toscano-Marquez, F.; Ramirez, R.; Calyeca, J.; Mora, A.L.; Selman, M.; Pardo, A. Loss of MT1-MMP in Alveolar Epithelial Cells Exacerbates Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 2923. [Google Scholar] [CrossRef]
- Gutierrez-Fernandez, A.; Soria-Valles, C.; Osorio, F.G.; Gutierrez-Abril, J.; Garabaya, C.; Aguirre, A.; Fueyo, A.; Fernandez-Garcia, M.S.; Puente, X.S.; Lopez-Otin, C. Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. EMBO J. 2015, 34, 1875–1888. [Google Scholar] [CrossRef] [Green Version]
- Kotton, D.N.; Morrisey, E.E. Lung regeneration: Mechanisms, applications and emerging stem cell populations. Nat. Med. 2014, 20, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, W.V.; Lu, J. Regulation of early lung morphogenesis: Questions, facts and controversies. Development 2006, 133, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Pepicelli, C.V.; Lewis, P.M.; McMahon, A.P. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol. 1998, 8, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Massaro, G.D.; Massaro, D. Postnatal treatment with retinoic acid increases the number of pulmonary alveoli in rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 1996, 270, L305–L310. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Ohuchi, H.; Fujiwara, M.; Yamasaki, M.; Yoshizawa, T.; Sato, T.; Yagishita, N.; Matsui, D.; Koga, Y.; Itoh, N. Fgf10 is essential for limb and lung formation. Nat. Genet. 1999, 21, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Volckaert, T.; De Langhe, S.P. Wnt and FGF mediated epithelial-mesenchymal crosstalk during lung development. Dev. Dyn. 2015, 244, 342–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrycaj, S.M.; Dye, B.R.; Baker, N.C.; Larsen, B.M.; Burke, A.C.; Spence, J.R.; Wellik, D.M. Hox5 genes regulate the Wnt2/2b-Bmp4-signaling axis during lung development. Cell Rep. 2015, 12, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Yin, Y. Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb. Perspect. Biol. 2012, 4, a008318. [Google Scholar] [CrossRef]
- Frank, D.B.; Penkala, I.J.; Zepp, J.A.; Sivakumar, A.; Linares-Saldana, R.; Zacharias, W.J.; Stolz, K.G.; Pankin, J.; Lu, M.; Wang, Q.; et al. Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc. Natl. Acad. Sci. USA 2019, 116, 4362–4371. [Google Scholar] [CrossRef] [Green Version]
- Morrisey, E.E.; Cardoso, W.V.; Lane, R.H.; Rabinovitch, M.; Abman, S.H.; Ai, X.; Albertine, K.H.; Bland, R.D.; Chapman, H.A.; Checkley, W. Molecular determinants of lung development. Ann. Am. Thorac. Soc. 2013, 10, S12–S16. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.-E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.-K.; Han, N.; Lee, J.-H. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 2020, 27, 366–382.e367. [Google Scholar] [CrossRef]
- Danto, S.I.; Shannon, J.M.; Borok, Z.; Zabski, S.M.; Crandall, E.D. Reversible transdifferentiation of alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 1995, 12, 497–502. [Google Scholar] [CrossRef]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488.e410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, F.; Kramer, P.; Jacob, A.; Driver, I.; Thomas, D.C.; McCauley, K.B.; Skvir, N.; Crane, A.M.; Kurmann, A.A.; Hollenberg, A.N.; et al. Prospective isolation of NKX2-1-expressing human lung progenitors derived from pluripotent stem cells. J. Clin. Investig. 2017, 127, 2277–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoo, P.; Su, G.; Drum, H.; Bringas, P.; Kimura, S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(-/-) mouse embryos. Dev. Biol. 1999, 209, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, J.; Leitz, D.H.W.; Szczygiel, M.; Dvornikov, D.; Fraumann, S.G.; Kreutz, C.; Zadora, P.K.; Seyhan Agircan, A.; Konietzke, P.; Engelmann, T.A.; et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 2020, 11, 2012. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Mollar, A.; Nacher, M.; Gay-Jordi, G.; Closa, D.; Xaubet, A.; Bulbena, O. Intratracheal transplantation of alveolar type II cells reverses bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Guillamat-Prats, R.; Gay-Jordi, G.; Xaubet, A.; Peinado, V.I.; Serrano-Mollar, A. Alveolar type II cell transplantation restores pulmonary surfactant protein levels in lung fibrosis. J. Heart Lung Transplant. 2014, 33, 758–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Mollar, A.; Gay-Jordi, G.; Guillamat-Prats, R.; Closa, D.; Hernandez-Gonzalez, F.; Marin, P.; Burgos, F.; Martorell, J.; Sanchez, M.; Arguis, P.; et al. Safety and Tolerability of Alveolar Type II Cell Transplantation in Idiopathic Pulmonary Fibrosis. Chest 2016, 150, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Palomo, B.; Sanchez-Lopez, L.I.; Moodley, Y.; Edel, M.J.; Serrano-Mollar, A. Induced pluripotent stem cell-derived lung alveolar epithelial type II cells reduce damage in bleomycin-induced lung fibrosis. Stem Cell Res. Ther. 2020, 11, 213. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir Crit Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Vancheri, C.; Kreuter, M.; Richeldi, L.; Ryerson, C.J.; Valeyre, D.; Grutters, J.C.; Wiebe, S.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Nintedanib with Add-on Pirfenidone in Idiopathic Pulmonary Fibrosis. Results of the INJOURNEY Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 356–363. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Fell, C.D.; Huggins, J.T.; Nunes, H.; Sussman, R.; Valenzuela, C.; Petzinger, U.; Stauffer, J.L.; Gilberg, F.; Bengus, M.; et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur. Respir. J. 2018, 52, 1800230. [Google Scholar] [CrossRef] [Green Version]
- Doubkova, M.; Kriegova, E.; Littnerova, S.; Schneiderova, P.; Sterclova, M.; Bartos, V.; Plackova, M.; Zurkova, M.; Bittenglova, R.; Lostakova, V.; et al. DSP rs2076295 variants influence nintedanib and pirfenidone outcomes in idiopathic pulmonary fibrosis: A pilot study. Ther. Adv. Respir. Dis. 2021, 15, 17534666211042529. [Google Scholar] [CrossRef]
- Pilling, D.; Buckley, C.D.; Salmon, M.; Gomer, R.H. Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunol. 2003, 171, 5537–5546. [Google Scholar] [CrossRef] [Green Version]
- Dillingh, M.R.; van den Blink, B.; Moerland, M.; van Dongen, M.G.; Levi, M.; Kleinjan, A.; Wijsenbeek, M.S.; Lupher, M.L., Jr.; Harper, D.M.; Getsy, J.A.; et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm. Pharmacol. Ther. 2013, 26, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, M.; Herzog, E.L.; et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients With Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Martin, G.; Margetts, P.J.; Ask, K.; Robertson, J.; Gauldie, J.; Kolb, M. Connective tissue growth factor is crucial to inducing a profibrotic environment in "fibrosis-resistant" BALB/c mouse lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 510–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, M.; Nakamura, Y.; Suda, T.; Kato, M.; Kaida, Y.; Hashimoto, D.; Inui, N.; Hamada, E.; Miyazaki, O.; Kurashita, S.; et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin. Chim. Acta 2011, 412, 2211–2215. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Estimated Enrolled Patients | Type | Clinical Trial ID |
|---|---|---|---|
| Pamrevlumab (FG-3019) | 340 | Phase 3 | NCT03955146 |
| PRM-151 | 700 | Phase 3 | NCT04594707–NCT04552899 |
| Treprostinil | 396 | Phase 3 | NCT04708782 |
| ENV-101 | 60 | Phase 2 | NCT04968574 |
| PLN_74809 | 84 | Phase 2 | NCT04396756 |
| GKT137831 | 60 | Phase 2 | NCT03865927 |
| GB0139 | 500 | Phase 2 | NCT03832946 |
| C21 | 60 | Phase 2 | NCT04533022 |
| Jaktinib Dihydrochloride Monohydrate | 90 | Phase 2 | NCT04312594 |
| Saracatinab | 100 | Phase 1 | NCT04598919 |
| ZSP1603 | 36 | Phase 1 | NCT05119972 |
| INS018_055 | 80 | Phase 1 | NCT05154240 |
| AZD5055 | 104 | Phase 1 | NCT05134727 |
| SHR-1906 | 60 | Phase 1 | NCT04986540 |
| NIP292 | 72 | Phase 1 | NCT04720443 |
| Inhaled Nitric Oxide (iNO) | 40 | Early Phase 1 | NCT05052229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. https://doi.org/10.3390/cells11132095
Confalonieri P, Volpe MC, Jacob J, Maiocchi S, Salton F, Ruaro B, Confalonieri M, Braga L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells. 2022; 11(13):2095. https://doi.org/10.3390/cells11132095
Chicago/Turabian StyleConfalonieri, Paola, Maria Concetta Volpe, Justin Jacob, Serena Maiocchi, Francesco Salton, Barbara Ruaro, Marco Confalonieri, and Luca Braga. 2022. "Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF)" Cells 11, no. 13: 2095. https://doi.org/10.3390/cells11132095
APA StyleConfalonieri, P., Volpe, M. C., Jacob, J., Maiocchi, S., Salton, F., Ruaro, B., Confalonieri, M., & Braga, L. (2022). Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells, 11(13), 2095. https://doi.org/10.3390/cells11132095