
cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach

 ,
,
Abstract
1. Introduction
2. The cAMP-PKA Pathway’s Role in the Growth of Various Tumors
3. Involvement of CREB in Tumor Growth
4. Involvement of EPAC in Tumor Growth
5. cAMP and Its Other Effectors Act in Various Signaling Pathways
6. Potential Anticancer Therapeutic Strategies
7. Updated Potential Anticancer Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed]
- Rall, T.W.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [CrossRef]
- Gerlo, S.; Kooijman, R.; Beck, I.M.; Kolmus, K.; Spooren, A.; Haegeman, G. Cyclic AMP: A selective modulator of NF-κB action. Cell. Mol. Life Sci. 2011, 68, 3823–3841. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Zaccolo, M. cAMP signaling in subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Sirotkin, A.V.; Ben, O.A.; Tandlmajerová, A.; Lauková, M.; Vaší Ek, D.; Laurin Ik, J.; Kornhauser, J.; Alwasel, S.; Harrath, A.H. cAMP response element-binding protein 1 controls porcine ovarian cell proliferation, apoptosis, and FSH and insulin-like growth factor 1 response. Reprod. Fertil. Dev. 2018, 30, 1145–1153. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. cAMP, cGMP and Amyloid β: Three Ideal Partners for Memory Formation. Trends Neurosci. 2018, 41, 255–266. [Google Scholar] [CrossRef]
- Numata, T.; Sato-Numata, K.; Okada, Y.; Inoue, R. Cellular mechanism for herbal medicine Junchoto to facilitate intestinal Cl(-)/water secretion that involves cAMP-dependent activation of CFTR. J. Nat. Med. 2018, 72, 694–705. [Google Scholar] [CrossRef]
- Arumugham, V.B.; Baldari, C.T. cAMP: A multifaceted modulator of immune synapse assembly and T cell activation. J. Leukoc. Biol. 2017, 101, 1301–1316. [Google Scholar] [CrossRef]
- Chin, K.V.; Yang, W.L.; Ravatn, R.; Kita, T.; Reitman, E.; Vettori, D.; Cvijic, M.E.; Shin, M.; Iacono, L. Reinventing the wheel of cyclic AMP: Novel mechanisms of cAMP signaling. Ann. N. Y. Acad. Sci. 2002, 968, 49–64. [Google Scholar] [CrossRef]
- Ramms, D.J.; Raimondi, F.; Arang, N.; Herberg, F.W.; Taylor, S.S.; Gutkind, J.S. Gαs-Protein Kinase A (PKA) Pathway Signalopathies: The Emerging Genetic Landscape and Therapeutic Potential of Human Diseases Driven by Aberrant Gαs-PKA Signaling. Pharmacol. Rev. 2021, 73, 155–197. [Google Scholar] [CrossRef]
- Rogue, P.J.; Humbert, J.P.; Meyer, A.; Freyermuth, S.; Krady, M.M.; Malviya, A.N. cAMP-dependent protein kinase phosphorylates and activates nuclear Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1998, 95, 9178–9183. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Petricoin, E., 3rd; Larner, A.C. Activation of protein kinase A inhibits interferon induction of the Jak/Stat pathway in U266 cells. J. Biol. Chem. 1996, 271, 4585–4588. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.J.; McCormick, F. Inhibition by cAMP of Ras-dependent activation of Raf. Science 1993, 262, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Won, S.-Y.; Park, J.-J.; Shin, E.-Y.; Kim, E.-G. PAK4 signaling in health and disease: Defining the PAK4–CREB axis. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Cadd, G.; McKnight, G.S. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron 1989, 3, 71–79. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 16–26. [Google Scholar] [CrossRef]
- Rich, T.C.; Fagan, K.A.; Tse, T.E.; Schaack, J.; Cooper, D.M.; Karpen, J.W. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA 2001, 98, 13049–13054. [Google Scholar] [CrossRef]
- Steinberg, S.F.; Brunton, L.L. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 751–773. [Google Scholar] [CrossRef]
- Smith, F.D.; Langeberg, L.K.; Scott, J.D. The where’s and when’s of kinase anchoring. Trends Biochem. Sci. 2006, 31, 316–323. [Google Scholar] [CrossRef]
- Beene, D.L.; Scott, J.D. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 2007, 19, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.X.; Rehmann, H.; Andres, D.A. A novel cyclic AMP-dependent Epac-Rit signaling pathway contributes to PACAP38-mediated neuronal differentiation. Mol. Cell. Biol. 2006, 26, 9136–9147. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, W.G., 3rd; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef]
- Popovic, M.; Leeuw, M.R.-D.; Rehmann, H. Selectivity of CDC25 homology domain-containing guanine nucleotide exchange factors. J. Mol. Biol. 2013, 425, 2782–2794. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Liu, C.; Takahashi, M.; Li, Y.; Song, S.; Dillon, T.J.; Shinde, U.; Stork, P.J. Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol. Cell. Biol. 2008, 28, 7109–7125. [Google Scholar] [CrossRef]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase A in cancer therapy: An update. Exp. Clin. Sci. J. 2014, 13, 843–855. [Google Scholar]
- Wehbe, N.; Slika, H.; Mesmar, J.; Nasser, S.A.; Pintus, G.; Baydoun, S.; Badran, A.; Kobeissy, F.; Eid, A.H.; Baydoun, E. The Role of Epac in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 6489. [Google Scholar] [CrossRef]
- Lorenowicz, M.J.; Fernandez-Borja, M.; Kooistra, M.R.; Bos, J.L.; Hordijk, P.L. PKA and Epac1 regulate endothelial integrity and migration through parallel and independent pathways. Eur. J. Cell Biol. 2008, 87, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Hochbaum, D.; Hong, K.; Barila, G.; Ribeiro-Neto, F.; Altschuler, D.L. Epac, in synergy with cAMP-dependent protein kinase (PKA), is required for cAMP-mediated mitogenesis. J. Biol. Chem. 2008, 283, 4464–4468. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Boulton, S.; Shao, H.; Akimoto, M.; Natarajan, A.; Cheng, X.; Melacini, G. Recent Advances in EPAC-Targeted Therapies: A Biophysical Perspective. Cells 2019, 8, 1462. [Google Scholar] [CrossRef]
- Elbrashy, A.A.; Kamal, A.; Fahim, M.I. Methods of Treatment and Outcome for Ovarian Germ Cell Tumors. Indian J. Surg. Oncol. 2019, 10, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Chang-Qing, Y.; Jie, L.; Shi-Qi, Z.; Kun, Z.; Zi-Qian, G.; Ran, X.; Hui-Meng, L.; Ren-Bin, Z.; Gang, Z.; Da-Chuan, Y.; et al. Recent treatment progress of triple negative breast cancer. Prog. Biophys. Mol. Biol. 2020, 151, 40–53. [Google Scholar] [CrossRef]
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; Von Hoff, D.D. Tumor-Treating Fields: A Fourth Modality in Cancer Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 266–275. [Google Scholar] [CrossRef]
- Daniel, P.M.; Filiz, G.; Mantamadiotis, T. Sensitivity of GBM cells to cAMP agonist-mediated apoptosis correlates with CD44 expression and agonist resistance with MAPK signaling. Cell Death Dis. 2016, 7, e2494. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, S.; Wang, J.; Jiang, Y. Blockade of AMPK-Mediated cAMP-PKA-CREB/ATF1 Signaling Synergizes with Aspirin to Inhibit Hepatocellular Carcinoma. Cancers 2021, 13, 1738. [Google Scholar] [CrossRef]
- Deng, Z.; Li, X.; Blanca Ramirez, M.; Purtell, K.; Choi, I.; Lu, J.H.; Yu, Q.; Yue, Z. Selective autophagy of AKAP11 activates cAMP/PKA to fuel mitochondrial metabolism and tumor cell growth. Proc. Natl. Acad. Sci. USA 2021, 118, e2020215118. [Google Scholar] [CrossRef]
- Selim, K.A.; Haffner, M.; Burkhardt, M.; Mantovani, O.; Neumann, N.; Albrecht, R.; Seifert, R.; Krüger, L.; Stülke, J.; Hartmann, M.D.; et al. Diurnal metabolic control in cyanobacteria requires perception of second messenger signaling molecule c-di-AMP by the carbon control protein SbtB. Sci. Adv. 2021, 7, eabk0568. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Kwon, Y.; Koo, S.H. Role of CRTC2 in Metabolic Homeostasis: Key Regulator of Whole-Body Energy Metabolism? Diabetes Metab. J. 2020, 44, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Reggi, E.; Diviani, D. The role of A-kinase anchoring proteins in cancer development. Cell. Signal. 2017, 40, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinska, P.; Ptak, A.; Wrobel, A.; Gregoraszczuk, E.L. Hydroxylated estrogens (2-OH-E2 AND 4-OH-E2) do not activate cAMP/PKA and ERK1/2 pathways activation in a breast cancer MCF-7 cell line. Endocr. Regul. 2012, 46, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Chen, Y.; Meng, F.; Zhang, Y.; Wu, H.; Wu, F. Roflumilast restores cAMP/PKA/CREB signaling axis for FtMt-mediated tumor inhibition of ovarian cancer. Oncotarget 2017, 8, 112341–112353. [Google Scholar] [CrossRef] [PubMed]
- Peverelli, E.; Giardino, E.; Mangili, F.; Treppiedi, D.; Catalano, R.; Ferrante, E.; Sala, E.; Locatelli, M.; Lania, A.G.; Arosio, M.; et al. cAMP/PKA-induced filamin A (FLNA) phosphorylation inhibits SST2 signal transduction in GH-secreting pituitary tumor cells. Cancer Lett. 2018, 435, 101–109. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Qian, W.; Ji, D.; Wang, Q.; Ji, B.; Zhang, Y.; Zhang, C.; Sun, Y.; Zhu, C.; et al. Angiogenesis and vasculogenic mimicry are inhibited by 8-Br-cAMP through activation of the cAMP/PKA pathway in colorectal cancer. OncoTargets Ther. 2018, 11, 3765–3774. [Google Scholar] [CrossRef]
- Jiang, K.; Yao, G.; Hu, L.; Yan, Y.; Liu, J.; Shi, J.; Chang, Y.; Zhang, Y.; Liang, D.; Shen, D.; et al. MOB2 suppresses GBM cell migration and invasion via regulation of FAK/Akt and cAMP/PKA signaling. Cell Death Dis. 2020, 11, 230. [Google Scholar] [CrossRef]
- Huang, F.; Ma, G.; Zhou, X.; Zhu, X.; Yu, X.; Ding, F.; Cao, X.; Liu, Z. Depletion of LAMP3 enhances PKA-mediated VASP phosphorylation to suppress invasion and metastasis in esophageal squamous cell carcinoma. Cancer Lett. 2020, 479, 100–111. [Google Scholar] [CrossRef]
- Sapio, L.; Gallo, M.; Illiano, M.; Chiosi, E.; Naviglio, D.; Spina, A.; Naviglio, S. The Natural cAMP Elevating Compound Forskolin in Cancer Therapy: Is It Time? J. Cell. Physiol. 2017, 232, 922–927. [Google Scholar] [CrossRef]
- Tang, C.; Liu, D.; Fan, Y.; Yu, J.; Li, C.; Su, J.; Wang, C. Visualization and bibliometric analysis of cAMP signaling system research trends and hotspots in cancer. J. Cancer 2021, 12, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Richartz, N.; Pietka, W.; Gilljam, K.M.; Skah, S.; Skålhegg, B.S.; Bhagwat, S.; Naderi, E.H.; Ruud, E.; Blomhoff, H.K. cAMP-Mediated Autophagy Promotes Cell Survival via ROS-Induced Activation of PARP1: Implications for Treatment of Acute Lymphoblastic Leukemia. Mol. Cancer Res. 2022, 20, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.E.; Juhnn, Y.S. Cell-type-specific Modulation of Non-homologous End Joining of Gamma Ray-induced DNA Double-strand Breaks by cAMP Signaling in Human Cancer Cells. J. Korean Med. Sci. 2020, 35, e371. [Google Scholar] [CrossRef]
- Patritti Cram, J.; Wu, J.; Coover, R.A.; Rizvi, T.A.; Chaney, K.E.; Ravindran, R.; Cancelas, J.A.; Spinner, R.J.; Ratner, N. P2RY14 cAMP signaling regulates Schwann cell precursor self-renewal, proliferation, and nerve tumor initiation in a mouse model of neurofibromatosis. eLife 2022, 11, e73511. [Google Scholar] [CrossRef] [PubMed]
- Massimi, M.; Ragusa, F.; Cardarelli, S.; Giorgi, M. Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma. Cells 2019, 8, 1511. [Google Scholar] [CrossRef]
- Massimi, M.; Cardarelli, S.; Galli, F.; Giardi, M.F.; Ragusa, F.; Panera, N.; Cinque, B.; Cifone, M.G.; Biagioni, S.; Giorgi, M. Increase of Intracellular Cyclic AMP by PDE4 Inhibitors Affects HepG2 Cell Cycle Progression and Survival. J. Cell. Biochem. 2017, 118, 1401–1411. [Google Scholar] [CrossRef]
- Peng, T.; Gong, J.; Jin, Y.; Zhou, Y.; Tong, R.; Wei, X.; Bai, L.; Shi, J. Inhibitors of phosphodiesterase as cancer therapeutics. Eur. J. Med. Chem. 2018, 150, 742–756. [Google Scholar] [CrossRef]
- Tonucci, F.M.; Almada, E.; Borini-Etichetti, C.; Pariani, A.; Hidalgo, F.; Rico, M.J.; Girardini, J.; Favre, C.; Goldenring, J.R.; Menacho-Marquez, M.; et al. Identification of a CIP4 PKA phosphorylation site involved in the regulation of cancer cell invasiveness and metastasis. Cancer Lett. 2019, 461, 65–77. [Google Scholar] [CrossRef]
- Hara, M.; Takeba, Y.; Iiri, T.; Ohta, Y.; Ootaki, M.; Watanabe, M.; Watanabe, D.; Koizumi, S.; Otsubo, T.; Matsumoto, N. Vasoactive intestinal peptide increases apoptosis of hepatocellular carcinoma by inhibiting the cAMP/Bcl-xL pathway. Cancer Sci. 2019, 110, 235–244. [Google Scholar] [CrossRef]
- Honeyman, J.N.; Simon, E.P.; Robine, N.; Chiaroni-Clarke, R.; Darcy, D.G.; Lim, I.I.P.; Gleason, C.E.; Murphy, J.M.; Rosenberg, B.R.; Teegan, L.J.S. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 2014, 343, 1010–1014. [Google Scholar] [CrossRef]
- Riggle, K.M.; Riehle, K.J.; Kenerson, H.L.; Turnham, R.; Homma, M.K.; Kazami, M.; Samelson, B.; Bauer, R.; McKnight, G.S.; Scott, J.D.J.P.r. Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr. Res. 2016, 80, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lalazar, G.; Houlihan, S.L.; Tschaharganeh, D.F.; Baslan, T.; Chen, C.-C.; Requena, D.; Tian, S.; Bosbach, B.; Wilkinson, J.E. DNAJB1–PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 13076–13084. [Google Scholar] [CrossRef] [PubMed]
- Cornella, H.; Alsinet, C.; Sayols, S.; Zhang, Z.; Hao, K.; Cabellos, L.; Hoshida, Y.; Villanueva, A.; Thung, S.; Ward, S.C.; et al. Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology 2015, 148, 806–818.e10. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Lackner, C.; Terracciano, L.; González-Cantú, Y.; Maleszewski, J.J.; Greipp, P.T.; Simon, S.M.; Torbenson, M.S.J.H. Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology 2018, 68, 1441–1447. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Singhi, A.D.; Wood, L.D.; Parks, E.; Torbenson, M.S.; Felsenstein, M.; Hruban, R.H.; Nikiforova, M.N.; Wald, A.I.; Kaya, C.; Nikiforov, Y.E.J.G. Recurrent rearrangements in PRKACA and PRKACB in intraductal oncocytic papillary neoplasms of the pancreas and bile duct. Gastroenterology 2020, 158, 573–582.e572. [Google Scholar] [CrossRef]
- Vyas, M.; Hechtman, J.F.; Zhang, Y.; Benayed, R.; Yavas, A.; Askan, G.; Shia, J.; Klimstra, D.S.; Basturk, O.J.M.P. DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod. Pathol. 2020, 33, 648–656. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein–coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef]
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 2020, 594, 4201–4232. [Google Scholar] [CrossRef]
- Innamorati, G.; Wilkie, T.M.; Kantheti, H.S.; Valenti, M.T.; Dalle Carbonare, L.; Giacomello, L.; Parenti, M.; Melisi, D.; Bassi, C. The curious case of Gαs gain-of-function in neoplasia. BMC Cancer 2018, 18, 293. [Google Scholar] [CrossRef]
- Deeble, P.D.; Cox, M.E.; Frierson, H.F.; Sikes, R.A.; Palmer, J.B.; Davidson, R.J.; Casarez, E.V.; Amorino, G.P.; Parsons, S.J.J.C.r. Androgen-independent growth and tumorigenesis of prostate cancer cells are enhanced by the presence of PKA-differentiated neuroendocrine cells. Cancer Res. 2007, 67, 3663–3672. [Google Scholar] [CrossRef] [PubMed]
- Boora, G.K.; Kanwar, R.; Kulkarni, A.A.; Pleticha, J.; Ames, M.; Schroth, G.; Beutler, A.S.; Banck, M.S. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015, 208, 374–381. [Google Scholar] [CrossRef]
- Coles, G.L.; Cristea, S.; Webber, J.T.; Levin, R.S.; Moss, S.M.; He, A.; Sangodkar, J.; Hwang, Y.C.; Arand, J.; Drainas, A.P.J.C.C. Unbiased proteomic profiling uncovers a targetable GNAS/PKA/PP2A axis in small cell lung cancer stem cells. Cancer Cell 2020, 38, 129–143.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Hinton, D.R.; Zidovetzki, R.; Hofman, F.M. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab. Investig. 1998, 78, 165–174. [Google Scholar] [PubMed]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.S.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 2005, 7, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Odreman, F.; Vindigni, M.; Gonzales, M.L.; Niccolini, B.; Candiano, G.; Zanotti, B.; Skrap, M.; Pizzolitto, S.; Stanta, G.; Vindigni, A. Proteomic studies on low- and high-grade human brain astrocytomas. J. Proteome Res. 2005, 4, 698–708. [Google Scholar] [CrossRef]
- Hanson, A.J.; Nahreini, P.; Andreatta, C.; Yan, X.D.; Prasad, K.N. Role of the adenosine 3′,5′-cyclic monophosphate (cAMP) in enhancing the efficacy of siRNA-mediated gene silencing in neuroblastoma cells. Oncogene 2005, 24, 4149–4154. [Google Scholar] [CrossRef]
- Døskeland, S.O.; Maronde, E.; Gjertsen, B.T. The genetic subtypes of cAMP-dependent protein kinase—functionally different or redundant? Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1993, 1178, 249–258. [Google Scholar] [CrossRef]
- Moreno, M.J.; Ball, M.; Andrade, M.F.; McDermid, A.; Stanimirovic, D.B. Insulin-like growth factor binding protein-4 (IGFBP-4) is a novel anti-angiogenic and anti-tumorigenic mediator secreted by dibutyryl cyclic AMP (dB-cAMP)-differentiated glioblastoma cells. Glia 2006, 53, 845–857. [Google Scholar] [CrossRef]
- Chen, T.C.; Wadsten, P.; Su, S.; Rawlinson, N.; Hofman, F.M.; Hill, C.K.; Schönthal, A.H. The type IV phosphodiesterase inhibitor rolipram induces expression of the cell cycle inhibitors p21(Cip1) and p27(Kip1), resulting in growth inhibition, increased differentiation, and subsequent apoptosis of malignant A-172 glioma cells. Cancer Biol. Ther. 2002, 1, 268–276. [Google Scholar] [CrossRef]
- Li, Y.; Yin, W.; Wang, X.; Zhu, W.; Huang, Y.; Yan, G. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Onuma, T.; Birukawa, N.; Abe, M.; Ito, E.; Chen, Z.; Urano, A. Change of morphology and cytoskeletal protein gene expression during dibutyryl cAMP-induced differentiation in C6 glioma cells. Cell. Mol. Neurobiol. 2008, 28, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Choi, M.R.; Song, D.K.; Huh, S.O.; Kim, Y.H.; Suh, H.W. Activation of adenylate cyclase results in down-regulation of c-jun mRNA expression in rat C6 glioma cells. Neurosci. Lett. 1999, 276, 53–56. [Google Scholar] [CrossRef]
- Anciaux, K.; Van Dommelen, K.; Nicolai, S.; Van Mechelen, E.; Slegers, H. Cyclic AMP-mediated induction of the glial fibrillary acidic protein is independent of protein kinase A activation in rat C6 glioma. J. Neurosci. Res. 1997, 48, 324–333. [Google Scholar] [CrossRef]
- Oh-hashi, K.; Hirata, Y.; Koga, H.; Kiuchi, K. GRP78-binding protein regulates cAMP-induced glial fibrillary acidic protein expression in rat C6 glioblastoma cells. FEBS Lett. 2006, 580, 3943–3947. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Moriuchi, S.; Shimizu, K.; Miyao, Y.; Kishima, H.; Okawa, M.; Hayakawa, T. Decreased N-myc expression in human medulloblastoma cell lines during differentiation. Anticancer Res. 1997, 17, 301–306. [Google Scholar]
- Kobsar, A.; Heeg, S.; Krohne, K.; Opitz, A.; Walter, U.; Böck, M.; Gambaryan, S.; Eigenthaler, M. Cyclic nucleotide-regulated proliferation and differentiation vary in human hematopoietic progenitor cells derived from healthy persons, tumor patients, and chronic myelocytic leukemia patients. Stem Cells Dev. 2008, 17, 81–91. [Google Scholar] [CrossRef]
- Yang, L.; Jackson, E.; Woerner, B.M.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007, 67, 651–658. [Google Scholar] [CrossRef]
- Huang, W.C.; Xie, Z.; Konaka, H.; Sodek, J.; Zhau, H.E.; Chung, L.W. Human osteocalcin and bone sialoprotein mediating osteomimicry of prostate cancer cells: Role of cAMP-dependent protein kinase A signaling pathway. Cancer Res. 2005, 65, 2303–2313. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, H.S.; Lee, C.S.; You, S.T.; Kim, D.J.; Park, B.H.; Kang, M.J.; Heo, W.D.; Shin, E.Y.; Schwartz, M.A.; et al. p21-Activated kinase 4 promotes prostate cancer progression through CREB. Oncogene 2013, 32, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Gao, X.H.; Li, X.J.; Cao, Q.H.; Zhao, D.D.; Zhou, J.R.; Wu, H.X.; Wang, Y.; You, L.J.; Yang, H.B.; et al. Depression promotes prostate cancer invasion and metastasis via a sympathetic-cAMP-FAK signaling pathway. Oncogene 2018, 37, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R., Jr.; Danial, N.; et al. Behavioral stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, V.O.; Dobroff, A.S.; Zigler, M.; Villares, G.J.; Braeuer, R.R.; Wang, H.; Huang, L.; Bar-Eli, M. CREB inhibits AP-2alpha expression to regulate the malignant phenotype of melanoma. PLoS ONE 2010, 5, e12452. [Google Scholar] [CrossRef]
- Tan, Y.; Rouse, J.; Zhang, A.; Cariati, S.; Cohen, P.; Comb, M.J. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996, 15, 4629–4642. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef]
- Abramovitch, R.; Tavor, E.; Jacob-Hirsch, J.; Zeira, E.; Amariglio, N.; Pappo, O.; Rechavi, G.; Galun, E.; Honigman, A. A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer Res. 2004, 64, 1338–1346. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Nafa, K.; Segal, N.H.; Dal Cin, P.; Ladanyi, M. EWS-CREB1: A recurrent variant fusion in clear cell sarcoma—Association with gastrointestinal location and absence of melanocytic differentiation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 5356–5362. [Google Scholar] [CrossRef]
- Braeuer, R.R.; Zigler, M.; Villares, G.J.; Dobroff, A.S.; Bar-Eli, M. Transcriptional control of melanoma metastasis: The importance of the tumor microenvironment. Semin. Cancer Biol. 2011, 21, 83–88. [Google Scholar] [CrossRef]
- Chhabra, A.; Fernando, H.; Watkins, G.; Mansel, R.E.; Jiang, W.G. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol. Rep. 2007, 18, 953–958. [Google Scholar] [CrossRef]
- Fan, C.F.; Mao, X.Y.; Wang, E.H. Elevated p-CREB-2 (ser 245) expression is potentially associated with carcinogenesis and development of breast carcinoma. Mol. Med. Rep. 2012, 5, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Jean, D.; Bar-Eli, M. Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Mol. Cell. Biochem. 2000, 212, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pigazzi, M.; Manara, E.; Bresolin, S.; Tregnago, C.; Beghin, A.; Baron, E.; Giarin, E.; Cho, E.C.; Masetti, R.; Rao, D.S.; et al. MicroRNA-34b promoter hypermethylation induces CREB overexpression and contributes to myeloid transformation. Haematologica 2013, 98, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Wang, S.; Yang, B.; Zhu, L.; Yin, B.; Chao, T.; Zhao, J.; Yuan, J.; Qiang, B.; Peng, X. The CREB-miR-9 negative feedback minicircuitry coordinates the migration and proliferation of glioma cells. PLoS ONE 2012, 7, e49570. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Amann, J.M.; Fukuda, K.; Takeuchi, S.; Fujita, N.; Uehara, H.; Iwakiri, S.; Itoi, K.; Shilo, K.; Yano, S.; et al. Akt Kinase-Interacting Protein 1 Signals through CREB to Drive Diffuse Malignant Mesothelioma. Cancer Res. 2015, 75, 4188–4197. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Kinjo, K.; Judelson, D.R.; Chang, J.; Wu, W.S.; Schmid, I.; Shankar, D.B.; Kasahara, N.; Stripecke, R.; Bhatia, R.; et al. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood 2008, 111, 1182–1192. [Google Scholar] [CrossRef]
- Cho, E.C.; Mitton, B.; Sakamoto, K.M. CREB and leukemogenesis. Crit. Rev. Oncog. 2011, 16, 37–46. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Zimmer, S.N.; Lemieux, M.E.; Karia, B.P.; Day, C.; Zhou, T.; Zhou, Q.; Kung, A.L.; Suresh, U.; Chen, Y.; Kinney, M.C.; et al. Mice heterozygous for CREB binding protein are hypersensitive to γ-radiation and invariably develop myelodysplastic/myeloproliferative neoplasm. Exp. Hematol. 2012, 40, 295–306.e5. [Google Scholar] [CrossRef]
- Suarez, C.D.; Deng, X.; Hu, C.D. Targeting CREB inhibits radiation-induced neuroendocrine differentiation and increases radiation-induced cell death in prostate cancer cells. Am. J. Cancer Res. 2014, 4, 850–861. [Google Scholar]
- Cheng, J.C.; Esparza, S.; Sandoval, S.; Shankar, D.; Fu, C.; Sakamoto, K.M. Potential role of CREB as a prognostic marker in acute myeloid leukemia. Future Oncol. 2007, 3, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Liu, H.; Huang, J.; Cheng, L.; Keller, E.T.; Parsons, S.J.; Hu, C.D. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: Implications for disease progression. Cancer Res. 2008, 68, 9663–9670. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Frank, D.A. CREB in the pathophysiology of cancer: Implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Liu, D.D.; Bekele, B.N.; Kim, M.K.; Pisters, K.; Lippman, S.M.; Wistuba, I.I.; Koo, J.S. Cyclic AMP response element-binding protein overexpression: A feature associated with negative prognosis in never smokers with non-small cell lung cancer. Cancer Res. 2008, 68, 6065–6073. [Google Scholar] [CrossRef]
- Impey, S.; McCorkle, S.R.; Cha-Molstad, H.; Dwyer, J.M.; Yochum, G.S.; Boss, J.M.; McWeeney, S.; Dunn, J.J.; Mandel, G.; Goodman, R.H. Defining the CREB regulon: A genome-wide analysis of transcription factor regulatory regions. Cell 2004, 119, 1041–1054. [Google Scholar] [CrossRef]
- Pigazzi, M.; Ricotti, E.; Germano, G.; Faggian, D.; Aricò, M.; Basso, G. cAMP response element binding protein (CREB) overexpression CREB has been described as critical for leukemia progression. Haematologica 2007, 92, 1435–1437. [Google Scholar] [CrossRef]
- Shankar, D.B.; Cheng, J.C.; Sakamoto, K.M. Role of cyclic AMP response element binding protein in human leukemias. Cancer 2005, 104, 1819–1824. [Google Scholar] [CrossRef]
- Zhang, X.; Odom, D.T.; Koo, S.H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef]
- Phuong, N.T.; Lim, S.C.; Kim, Y.M.; Kang, K.W. Aromatase induction in tamoxifen-resistant breast cancer: Role of phosphoinositide 3-kinase-dependent CREB activation. Cancer Lett. 2014, 351, 91–99. [Google Scholar] [CrossRef]
- Donnelly, S.M.; Paplomata, E.; Peake, B.M.; Sanabria, E.; Chen, Z.; Nahta, R. P38 MAPK contributes to resistance and invasiveness of HER2-overexpressing breast cancer. Curr. Med. Chem. 2014, 21, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Juhnn, Y.S. Cyclic AMP signaling reduces sirtuin 6 expression in non-small cell lung cancer cells by promoting ubiquitin-proteasomal degradation via inhibition of the Raf-MEK-ERK (Raf/mitogen-activated extracellular signal-regulated kinase/extracellular signal-regulated kinase) pathway. J. Biol. Chem. 2015, 290, 9604–9613. [Google Scholar] [CrossRef] [PubMed]
- James, M.A.; Lu, Y.; Liu, Y.; Vikis, H.G.; You, M. RGS17, an overexpressed gene in human lung and prostate cancer, induces tumor cell proliferation through the cyclic AMP-PKA-CREB pathway. Cancer Res. 2009, 69, 2108–2116. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Banat, G.A.; Schmall, A.; Szibor, M.; Pomagruk, D.; Hänze, J.; Kolosionek, E.; Wilhelm, J.; Braun, T.; Grimminger, F.; et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene 2013, 32, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.A.; Kim, E.J.; Kwak, S.J.; Juhnn, Y.S. cAMP signaling inhibits radiation-induced ATM phosphorylation leading to the augmentation of apoptosis in human lung cancer cells. Mol. Cancer 2014, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Sola-Penna, M.; Paixão, L.P.; Branco, J.R.; Ochioni, A.C.; Albanese, J.M.; Mundim, D.M.; Baptista-de-Souza, D.; Figueiredo, C.P.; Coelho, W.S.; Marcondes, M.C.; et al. Serotonin activates glycolysis and mitochondria biogenesis in human breast cancer cells through activation of the Jak1/STAT3/ERK1/2 and adenylate cyclase/PKA, respectively. Br. J. Cancer 2020, 122, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.E.; et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Grandoch, M.; López de Jesús, M.; Oude Weernink, P.A.; Weber, A.A.; Jakobs, K.H.; Schmidt, M. B cell receptor-induced growth arrest and apoptosis in WEHI-231 immature B lymphoma cells involve cyclic AMP and Epac proteins. Cell. Signal. 2009, 21, 609–621. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Johnson, B.H.; Thompson, E.B.; Cheng, X. Protein kinase A, not Epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J. Biol. Chem. 2007, 282, 37370–37377. [Google Scholar] [CrossRef]
- Vitali, E.; Cambiaghi, V.; Spada, A.; Tresoldi, A.; Zerbi, A.; Peverelli, E.; Carnaghi, C.; Mantovani, G.; Lania, A.G. cAMP effects in neuroendocrine tumors: The role of Epac and PKA in cell proliferation and adhesion. Exp. Cell Res. 2015, 339, 241–251. [Google Scholar] [CrossRef]
- Cho, E.A.; Juhnn, Y.S. The cAMP signaling system inhibits the repair of γ-ray-induced DNA damage by promoting Epac1-mediated proteasomal degradation of XRCC1 protein in human lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 422, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Soares-Silva, C.; Brandão, D.; Marino, F.; Cosentino, M.; Ribeiro, L. β-Adrenergic modulation of cancer cell proliferation: Available evidence and clinical perspectives. J. Cancer Res. Clin. Oncol. 2017, 143, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Granholm, S.; Lundberg, P.; Lerner, U.H. Calcitonin inhibits osteoclast formation in mouse haematopoetic cells independently of transcriptional regulation by receptor activator of NF-{kappa}B and c-Fms. J. Endocrinol. 2007, 195, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Miller, A.L.; Thompson, E.B.; Cheng, X. Protein kinase A (PKA) isoform RIIbeta mediates the synergistic killing effect of cAMP and glucocorticoid in acute lymphoblastic leukemia cells. J. Biol. Chem. 2008, 283, 21920–21925. [Google Scholar] [CrossRef]
- Moon, E.Y.; Lee, G.H.; Lee, M.S.; Kim, H.M.; Lee, J.W. Phosphodiesterase inhibitors control A172 human glioblastoma cell death through cAMP-mediated activation of protein kinase A and Epac1/Rap1 pathways. Life Sci. 2012, 90, 373–380. [Google Scholar] [CrossRef]
- Tiwari, S.; Felekkis, K.; Moon, E.Y.; Flies, A.; Sherr, D.H.; Lerner, A. Among circulating hematopoietic cells, B-CLL uniquely expresses functional EPAC1, but EPAC1-mediated Rap1 activation does not account for PDE4 inhibitor-induced apoptosis. Blood 2004, 103, 2661–2667. [Google Scholar] [CrossRef]
- Almahariq, M.; Mei, F.C.; Cheng, X. The pleiotropic role of exchange protein directly activated by cAMP 1 (EPAC1) in cancer: Implications for therapeutic intervention. Acta Biochim. Biophys. Sin. 2016, 48, 75–81. [Google Scholar] [CrossRef]
- Li, K.; Liang, J.; Lin, Y.; Zhang, H.; Xiao, X.; Tan, Y.; Cai, J.; Zhu, W.; Xing, F.; Hu, J.; et al. A classical PKA inhibitor increases the oncolytic effect of M1 virus via activation of exchange protein directly activated by cAMP 1. Oncotarget 2016, 7, 48443–48455. [Google Scholar] [CrossRef]
- Li, K.; Zhang, H.; Qiu, J.; Lin, Y.; Liang, J.; Xiao, X.; Fu, L.; Wang, F.; Cai, J.; Tan, Y.; et al. Activation of Cyclic Adenosine Monophosphate Pathway Increases the Sensitivity of Cancer Cells to the Oncolytic Virus M1. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 156–165. [Google Scholar] [CrossRef]
- Vacas, E.; Fernández-Martínez, A.B.; Bajo, A.M.; Sánchez-Chapado, M.; Schally, A.V.; Prieto, J.C.; Carmena, M.J. Vasoactive intestinal peptide (VIP) inhibits human renal cell carcinoma proliferation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1676–1685. [Google Scholar] [CrossRef]
- Fernández-Martínez, A.B.; Carmena, M.J.; Bajo, A.M.; Vacas, E.; Sánchez-Chapado, M.; Prieto, J.C. VIP induces NF-κB1-nuclear localisation through different signalling pathways in human tumour and non-tumour prostate cells. Cell. Signal. 2015, 27, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Flacke, J.P.; Flacke, H.; Appukuttan, A.; Palisaar, R.J.; Noldus, J.; Robinson, B.D.; Reusch, H.P.; Zippin, J.H.; Ladilov, Y. Type 10 soluble adenylyl cyclase is overexpressed in prostate carcinoma and controls proliferation of prostate cancer cells. J. Biol. Chem. 2013, 288, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Frevert, U.; Engelmann, S.; Zougbédé, S.; Stange, J.; Ng, B.; Matuschewski, K.; Liebes, L.; Yee, H. Intravital observation of Plasmodium berghei sporozoite infection of the liver. PLoS Biol. 2005, 3, e192. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Ma, Y.; Bast, R.C., Jr.; Li, Y.; Wan, L.; Liu, Y.; Sun, Y.; Fang, Z.; Zhang, L.; Wang, X.; et al. Epac1 knockdown inhibits the proliferation of ovarian cancer cells by inactivating AKT/Cyclin D1/CDK4 pathway in vitro and in vivo. Med. Oncol. 2016, 33, 73. [Google Scholar] [CrossRef] [PubMed]
- Onodera, Y.; Nam, J.M.; Bissell, M.J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 2014, 124, 367–384. [Google Scholar] [CrossRef]
- Baljinnyam, E.; Umemura, M.; Chuang, C.; De Lorenzo, M.S.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Ishikawa, Y.; Whitelock, J.M.; Iwatsubo, K. Epac1 increases migration of endothelial cells and melanoma cells via FGF2-mediated paracrine signaling. Pigment Cell Melanoma Res. 2014, 27, 611–620. [Google Scholar] [CrossRef]
- Baljinnyam, E.; Umemura, M.; De Lorenzo, M.S.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Iwatsubo, K. Epac1 promotes melanoma metastasis via modification of heparan sulfate. Pigment Cell Melanoma Res. 2011, 24, 680–687. [Google Scholar] [CrossRef]
- Menon, J.; Doebele, R.C.; Gomes, S.; Bevilacqua, E.; Reindl, K.M.; Rosner, M.R. A novel interplay between Rap1 and PKA regulates induction of angiogenesis in prostate cancer. PLoS ONE 2012, 7, e49893. [Google Scholar] [CrossRef]
- Rangarajan, S.; Enserink, J.M.; Kuiperij, H.B.; de Rooij, J.; Price, L.S.; Schwede, F.; Bos, J.L. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J. Cell Biol. 2003, 160, 487–493. [Google Scholar] [CrossRef]
- Almahariq, M.; Chao, C.; Mei, F.C.; Hellmich, M.R.; Patrikeev, I.; Motamedi, M.; Cheng, X. Pharmacological inhibition and genetic knockdown of exchange protein directly activated by cAMP 1 reduce pancreatic cancer metastasis in vivo. Mol. Pharmacol. 2015, 87, 142–149. [Google Scholar] [CrossRef]
- Wang, X.; Luo, C.; Cheng, X.; Lu, M. Lithium and an EPAC-specific inhibitor ESI-09 synergistically suppress pancreatic cancer cell proliferation and survival. Acta Biochim. Biophys. Sin. 2017, 49, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, J.; Moon, E.Y. HeLa human cervical cancer cell migration is inhibited by treatment with dibutyryl-cAMP. Anticancer Res. 2014, 34, 3447–3455. [Google Scholar] [PubMed]
- Harper, K.; Arsenault, D.; Boulay-Jean, S.; Lauzier, A.; Lucien, F.; Dubois, C.M. Autotaxin promotes cancer invasion via the lysophosphatidic acid receptor 4: Participation of the cyclic AMP/EPAC/Rac1 signaling pathway in invadopodia formation. Cancer Res. 2010, 70, 4634–4643. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.R.; Poppinga, W.J.; de Jager, W.; Lezoualc’h, F.; Cheng, X.; Wieland, T.; Yarwood, S.J.; Gosens, R.; Schmidt, M. Epac1 links prostaglandin E2 to β-catenin-dependent transcription during epithelial-to-mesenchymal transition. Oncotarget 2016, 7, 46354–46370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lim, J.A.; Juhnn, Y.S. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp. Mol. Med. 2016, 48, e204. [Google Scholar] [CrossRef]
- Grandoch, M.; Rose, A.; ter Braak, M.; Jendrossek, V.; Rübben, H.; Fischer, J.W.; Schmidt, M.; Weber, A.A. Epac inhibits migration and proliferation of human prostate carcinoma cells. Br. J. Cancer 2009, 101, 2038–2042. [Google Scholar] [CrossRef]
- Baljinnyam, E.; Iwatsubo, K.; Kurotani, R.; Wang, X.; Ulucan, C.; Iwatsubo, M.; Lagunoff, D.; Ishikawa, Y. Epac increases melanoma cell migration by a heparan sulfate-related mechanism. Am. J. Physiol. Cell Physiol. 2009, 297, C802–C813. [Google Scholar] [CrossRef]
- Baljinnyam, E.; De Lorenzo, M.S.; Xie, L.H.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Nowycky, M.C.; Iwatsubo, K. Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2+-dependent mechanism. Cancer Res. 2010, 70, 5607–5617. [Google Scholar] [CrossRef]
- Lorenz, R.; Aleksic, T.; Wagner, M.; Adler, G.; Weber, C.K. The cAMP/Epac1/Rap1 pathway in pancreatic carcinoma. Pancreas 2008, 37, 102–103. [Google Scholar] [CrossRef]
- Sun, D.P.; Fang, C.L.; Chen, H.K.; Wen, K.S.; Hseu, Y.C.; Hung, S.T.; Uen, Y.H.; Lin, K.Y. EPAC1 overexpression is a prognostic marker and its inhibition shows promising therapeutic potential for gastric cancer. Oncol. Rep. 2017, 37, 1953–1960. [Google Scholar] [CrossRef]
- Guan, Z.; Zhuang, W.; Lei, H.; Wang, D.; Yao, Y.; Guo, D.; Sun, Q.; Chen, Y.; Chen, X.; Lin, H.; et al. Epac1, PDE4, and PKC protein expression and their correlation with AKAP95 and Cx43 in esophagus cancer tissues. Thorac. Cancer 2017, 8, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Gupta, S.; Dabral, S.; Singh, S.; Sehrawat, S. Role of exchange protein directly activated by cAMP (EPAC1) in breast cancer cell migration and apoptosis. Mol. Cell. Biochem. 2017, 430, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Sun, Q.; Zhuang, W.; Peng, K.; Wang, D.; Yao, Y.; Guo, D.; Zhang, L.; Shen, C.; Sun, M.; et al. Epac1, PDE4, and PKC protein expression and their association with AKAP95, Cx43, and cyclinD2/E1 in breast cancer tissues. Thorac. Cancer 2017, 8, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Tcherkezian, J.; Roux, P.P. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis 2015, 30, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Juhnn, Y.S. cAMP signaling increases histone deacetylase 8 expression via the Epac2-Rap1A-Akt pathway in H1299 lung cancer cells. Exp. Mol. Med. 2017, 49, e297. [Google Scholar] [CrossRef]
- Misra, U.K.; Pizzo, S.V. Epac1-induced cellular proliferation in prostate cancer cells is mediated by B-Raf/ERK and mTOR signaling cascades. J. Cell. Biochem. 2009, 108, 998–1011. [Google Scholar] [CrossRef]
- Misra, U.K.; Pizzo, S.V. Evidence for a pro-proliferative feedback loop in prostate cancer: The role of Epac1 and COX-2-dependent pathways. PLoS ONE 2013, 8, e63150. [Google Scholar] [CrossRef]
- Vossler, M.R.; Yao, H.; York, R.D.; Pan, M.G.; Rim, C.S.; Stork, P.J. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 1997, 89, 73–82. [Google Scholar] [CrossRef]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Fitchman, B.; Ronai, Z. Understanding signaling cascades in melanoma. Photochem. Photobiol. 2008, 84, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Pollock, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Kolch, W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol. Cell. Biol. 2002, 22, 3237–3246. [Google Scholar] [CrossRef]
- Laroche-Joubert, N.; Marsy, S.; Michelet, S.; Imbert-Teboul, M.; Doucet, A. Protein kinase A-independent activation of ERK and H,K-ATPase by cAMP in native kidney cells: Role of Epac I. J. Biol. Chem. 2002, 277, 18598–18604. [Google Scholar] [CrossRef]
- Li, Y.; Dillon, T.J.; Takahashi, M.; Earley, K.T.; Stork, P.J. Protein Kinase A-independent Ras Protein Activation Cooperates with Rap1 Protein to Mediate Activation of the Extracellular Signal-regulated Kinases (ERK) by cAMP. J. Biol. Chem. 2016, 291, 21584–21595. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, O.; Qin, J.; Liu, S.; Sun, S.; Liu, H.; Kuang, J.; Jiang, G.; Zhang, W. cis-Acting elements and trans-acting factors in the transcriptional regulation of raf kinase inhibitory protein expression. PLoS ONE 2013, 8, e83097. [Google Scholar] [CrossRef]
- Lee, H.C.; Tian, B.; Sedivy, J.M.; Wands, J.R.; Kim, M. Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology 2006, 131, 1208–1217. [Google Scholar] [CrossRef]
- Park, S.; Yeung, M.L.; Beach, S.; Shields, J.M.; Yeung, K.C. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene 2005, 24, 3535–3540. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Mooi, W.J.; Peeper, D.S. BRAF(E600) in benign and malignant human tumours. Oncogene 2008, 27, 877–895. [Google Scholar] [CrossRef]
- Schindler, R.F.; Brand, T. The Popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Amunjela, J.N.; Swan, A.H.; Brand, T.J.C. The role of the Popeye domain containing gene family in organ homeostasis. Cells 2019, 8, 1594. [Google Scholar] [CrossRef] [PubMed]
- Parang, B.; Thompson, J.J.; Williams, C.S. Blood Vessel Epicardial Substance (BVES) in junctional signaling and cancer. Tissue Barriers 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.K.; Hager, H.A.; Francis, R.; Kilkenny, D.M.; Lo, C.W.; Bader, D.M. Bves directly interacts with GEFT, and controls cell shape and movement through regulation of Rac1/Cdc42 activity. Proc. Natl. Acad. Sci. USA 2008, 105, 8298–8303. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Lei, Y.; Li, D.; Liu, J.; Yan, W.; Tian, D. Ten years of research on the role of BVES/POPDC1 in human disease: A review. OncoTargets Ther. 2019, 12, 1279–1291. [Google Scholar] [CrossRef]
- Brand, T. POPDC proteins and cardiac function. Biochem. Soc. Trans. 2019, 47, 1393–1404. [Google Scholar] [CrossRef]
- Lee, H.; Paik, S.G. Regulation of BNIP3 in normal and cancer cells. Mol. Cells 2006, 21, 1–6. [Google Scholar]
- Wang, S.C.; Lin, X.L.; Li, J.; Zhang, T.T.; Wang, H.Y.; Shi, J.W.; Yang, S.; Zhao, W.T.; Xie, R.Y.; Wei, F.; et al. MicroRNA-122 triggers mesenchymal-epithelial transition and suppresses hepatocellular carcinoma cell motility and invasion by targeting RhoA. PLoS ONE 2014, 9, e101330. [Google Scholar] [CrossRef]
- Han, P.; Fu, Y.; Liu, J.; Wang, Y.; He, J.; Gong, J.; Li, M.; Tan, Q.; Li, D.; Luo, Y.; et al. Netrin-1 promotes cell migration and invasion by down-regulation of BVES expression in human hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 1396–1409. [Google Scholar]
- Amunjela, J.N.; Tucker, S.J. POPDC1 is suppressed in human breast cancer tissues and is negatively regulated by EGFR in breast cancer cell lines. Cancer Lett. 2017, 406, 81–92. [Google Scholar] [CrossRef]
- Amunjela, J.N.; Tucker, S.J. POPDC proteins as potential novel therapeutic targets in cancer. Drug Discov. Today 2016, 21, 1920–1927. [Google Scholar] [CrossRef]
- Kim, M.; Jang, H.R.; Haam, K.; Kang, T.W.; Kim, J.H.; Kim, S.Y.; Noh, S.M.; Song, K.S.; Cho, J.S.; Jeong, H.Y.; et al. Frequent silencing of popeye domain-containing genes, BVES and POPDC3, is associated with promoter hypermethylation in gastric cancer. Carcinogenesis 2010, 31, 1685–1693. [Google Scholar] [CrossRef]
- Parang, B.; Kaz, A.M.; Barrett, C.W.; Short, S.P.; Ning, W.; Keating, C.E.; Mittal, M.K.; Naik, R.D.; Washington, M.K.; Revetta, F.L.; et al. BVES regulates c-Myc stability via PP2A and suppresses colitis-induced tumourigenesis. Gut 2017, 66, 852–862. [Google Scholar] [CrossRef]
- Williams, C.S.; Zhang, B.; Smith, J.J.; Jayagopal, A.; Barrett, C.W.; Pino, C.; Russ, P.; Presley, S.H.; Peng, D.; Rosenblatt, D.O.; et al. BVES regulates EMT in human corneal and colon cancer cells and is silenced via promoter methylation in human colorectal carcinoma. J. Clin. Investig. 2011, 121, 4056–4069. [Google Scholar] [CrossRef]
- Russ, P.K.; Pino, C.J.; Williams, C.S.; Bader, D.M.; Haselton, F.R.; Chang, M.S. Bves modulates tight junction associated signaling. PLoS ONE 2011, 6, e14563. [Google Scholar] [CrossRef]
- Thompson, J.J.; Short, S.P.; Parang, B.; Brown, R.E.; Li, C.; Ng, V.H.; Saito-Diaz, K.; Choksi, Y.A.; Washington, M.K.; Smith, J.J.; et al. Blood vessel epicardial substance (BVES) reduces LRP6 receptor and cytoplasmic β-catenin levels to modulate Wnt signaling and intestinal homeostasis. Carcinogenesis 2019, 40, 1086–1098. [Google Scholar] [CrossRef]
- He, X.; Xu, H.; Zhao, W.; Zhan, M.; Li, Y.; Liu, H.; Tan, L.; Lu, L. POPDC3 is a potential biomarker for prognosis and radioresistance in patients with head and neck squamous cell carcinoma. Oncol. Lett. 2019, 18, 5468–5480. [Google Scholar] [CrossRef]
- Gupta, S.; Li, J.; Kemeny, G.; Bitting, R.L.; Beaver, J.; Somarelli, J.A.; Ware, K.E.; Gregory, S.; Armstrong, A.J. Whole genomic copy number alterations in circulating tumor cells from men with abiraterone or enzalutamide-resistant metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2017, 23, 1346–1357. [Google Scholar] [CrossRef]
- Er, T.-K.; Su, Y.-F.; Wu, C.-C.; Chen, C.-C.; Wang, J.; Hsieh, T.-H.; Herreros-Villanueva, M.; Chen, W.-T.; Chen, Y.-T.; Liu, T.-C. Targeted next-generation sequencing for molecular diagnosis of endometriosis-associated ovarian cancer. Klin. Wochenschr. 2016, 94, 835–847. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Ma, Z.; Ma, Y.; Zhao, J.; Peng, B.; Qiao, Z. Sequencing study on familial lung squamous cancer. Oncol. Lett. 2015, 10, 2634–2638. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Liu, B.; Connolly, I.D.; Kakusa, B.W.; Pan, W.; Nagpal, S.; Montgomery, S.B.; Gephart, M.H. Recurrently mutated genes differ between leptomeningeal and solid lung cancer brain metastases. J. Thorac. Oncol. 2018, 13, 1022–1027. [Google Scholar] [CrossRef]
- Yao, H.; Wu, C.; Chen, Y.; Guo, L.; Chen, W.; Pan, Y.; Fu, X.; Wang, G.; Ding, Y. Spectrum of gene mutations identified by targeted next-generation sequencing in Chinese leukemia patients. Mol. Genet. Genom. Med. 2020, 8, e1369. [Google Scholar] [CrossRef]
- Bouguenina, H.; Salaun, D.; Mangon, A.; Muller, L.; Baudelet, E.; Camoin, L.; Tachibana, T.; Cianférani, S.; Audebert, S.; Verdier-Pinard, P. EB1-binding–myomegalin protein complex promotes centrosomal microtubules functions. Proc. Natl. Acad. Sci. USA 2017, 114, E10687–E10696. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, J.; Ya, A.; Ma, W.; Villa, S.; Sukenik, S.; Ge, X. Myomegalin regulates Hedgehog pathway by controlling PDE4D at the centrosome. Mol. Biol. Cell 2021, 32, 1807–1817. [Google Scholar] [CrossRef]
- Shimada, H.; Kuboshima, M.; Shiratori, T.; Nabeya, Y.; Takeuchi, A.; Takagi, H.; Nomura, F.; Takiguchi, M.; Ochiai, T.; Hiwasa, T. Serum anti-myomegalin antibodies in patients with esophageal squamous cell carcinoma. Int. J. Oncol. 2007, 30, 97–103. [Google Scholar] [CrossRef]
- O’Bleness, M.; Searles, V.B.; Dickens, C.M.; Astling, D.; Albracht, D.; Mak, A.C.; Lai, Y.Y.; Lin, C.; Chu, C.; Graves, T. Finished sequence and assembly of the DUF1220-rich 1q21 region using a haploid human genome. BMC Genom. 2014, 15, 387. [Google Scholar] [CrossRef]
- Snuderl, M.; Kannan, K.; Pfaff, E.; Wang, S.; Stafford, J.M.; Serrano, J.; Heguy, A.; Ray, K.; Faustin, A.; Aminova, O. Recurrent homozygous deletion of DROSHA and microduplication of PDE4DIP in pineoblastoma. Nat. Commun. 2018, 9, 2868. [Google Scholar] [CrossRef]
- Dumas, L.J.; O’Bleness, M.S.; Davis, J.M.; Dickens, C.M.; Anderson, N.; Keeney, J.; Jackson, J.; Sikela, M.; Raznahan, A.; Giedd, J. DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am. J. Hum. Genet. 2012, 91, 444–454. [Google Scholar] [CrossRef]
- Neary, C.L.; Nesterova, M.; Cho, Y.S.; Cheadle, C.; Becker, K.G.; Cho-Chung, Y.S. Protein kinase A isozyme switching: Eliciting differential cAMP signaling and tumor reversion. Oncogene 2004, 23, 8847–8856. [Google Scholar] [CrossRef]
- Tortora, G.; Ciardiello, F.; Pepe, S.; Tagliaferri, P.; Ruggiero, A.; Bianco, C.; Guarrasi, R.; Miki, K.; Bianco, A.R. Phase I clinical study with 8-chloro-cAMP and evaluation of immunological effects in cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1995, 1, 377–384. [Google Scholar]
- Tortora, G.; Ciardiello, F. Protein kinase A as target for novel integrated strategies of cancer therapy. Ann. N. Y. Acad. Sci. 2002, 968, 139–147. [Google Scholar] [CrossRef]
- Tortora, G.; Caputo, R.; Damiano, V.; Melisi, D.; Bianco, R.; Fontanini, G.; Veneziani, B.M.; De Placido, S.; Bianco, A.R.; Ciardiello, F. Combination of a selective cyclooxygenase-2 inhibitor with epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 and protein kinase A antisense causes cooperative antitumor and antiangiogenic effect. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1566–1572. [Google Scholar]
- Hirsh, L.; Dantes, A.; Suh, B.S.; Yoshida, Y.; Hosokawa, K.; Tajima, K.; Kotsuji, F.; Merimsky, O.; Amsterdam, A. Phosphodiesterase inhibitors as anti-cancer drugs. Biochem. Pharmacol. 2004, 68, 981–988. [Google Scholar] [CrossRef]
- Goldhoff, P.; Warrington, N.M.; Limbrick, D.D., Jr.; Hope, A.; Woerner, B.M.; Jackson, E.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes brain tumor regression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7717–7725. [Google Scholar] [CrossRef]
- Russo, P.; Catassi, A.; Cesario, A.; Servent, D. Development of novel therapeutic strategies for lung cancer: Targeting the cholinergic system. Curr. Med. Chem. 2006, 13, 3493–3512. [Google Scholar] [CrossRef]
- Erikstein, B.S.; McCormack, E.; Tronstad, K.J.; Schwede, F.; Berge, R.; Gjertsen, B.T. Protein kinase A activators and the pan-PPAR agonist tetradecylthioacetic acid elicit synergistic anti-leukaemic effects in AML through CREB. Leuk. Res. 2010, 34, 77–84. [Google Scholar] [CrossRef]
- Gao, N.; Hibi, Y.; Cueno, M.; Asamitsu, K.; Okamoto, T. A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J. Biol. Chem. 2010, 285, 28097–28104. [Google Scholar] [CrossRef]
- Mani, S.; Goel, S.; Nesterova, M.; Martin, R.M.; Grindel, J.M.; Rothenberg, M.L.; Zhang, R.; Tortora, G.; Cho-Chung, Y.S. Clinical studies in patients with solid tumors using a second-generation antisense oligonucleotide (GEM 231) targeted against protein kinase A type I. Ann. N. Y. Acad. Sci. 2003, 1002, 252–262. [Google Scholar] [CrossRef]
- Hensley, H.H.; Hannoun-Levi, J.M.; Hachem, P.; Mu, Z.; Stoyanova, R.; Khor, L.Y.; Agrawal, S.; Pollack, A. PKA knockdown enhances cell killing in response to radiation and androgen deprivation. Int. J. Cancer 2011, 128, 962–973. [Google Scholar] [CrossRef]
- Linnerth, N.M.; Baldwin, M.; Campbell, C.; Brown, M.; McGowan, H.; Moorehead, R.A. IGF-II induces CREB phosphorylation and cell survival in human lung cancer cells. Oncogene 2005, 24, 7310–7319. [Google Scholar] [CrossRef]
- Ali, A.M.; Reis, J.M.; Xia, Y.; Rashid, A.J.; Mercaldo, V.; Walters, B.J.; Brechun, K.E.; Borisenko, V.; Josselyn, S.A.; Karanicolas, J.; et al. Optogenetic Inhibitor of the Transcription Factor CREB. Chem. Biol. 2015, 22, 1531–1539. [Google Scholar] [CrossRef]
- Alper, O.; Bergmann-Leitner, E.S.; Abrams, S.; Cho-Chung, Y.S. Apoptosis, growth arrest and suppression of invasiveness by CRE-decoy oligonucleotide in ovarian cancer cells: Protein kinase A downregulation and cytoplasmic export of CRE-binding proteins. Mol. Cell. Biochem. 2001, 218, 55–63. [Google Scholar] [CrossRef]
- Steven, A.; Leisz, S.; Massa, C.; Iezzi, M.; Lattanzio, R.; Lamolinara, A.; Bukur, J.; Müller, A.; Hiebl, B.; Holzhausen, H.J.; et al. HER-2/neu mediates oncogenic transformation via altered CREB expression and function. Mol. Cancer Res. 2013, 11, 1462–1477. [Google Scholar] [CrossRef]
- Best, J.L.; Amezcua, C.A.; Mayr, B.; Flechner, L.; Murawsky, C.M.; Emerson, B.; Zor, T.; Gardner, K.H.; Montminy, M. Identification of small-molecule antagonists that inhibit an activator: Coactivator interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 17622–17627. [Google Scholar] [CrossRef]
- Jiang, M.; Li, B.X.; Xie, F.; Delaney, F.; Xiao, X. Design, synthesis, and biological evaluation of conformationally constrained analogues of naphthol AS-E as inhibitors of CREB-mediated gene transcription. J. Med. Chem. 2012, 55, 4020–4024. [Google Scholar] [CrossRef]
- Li, B.X.; Yamanaka, K.; Xiao, X. Structure-activity relationship studies of naphthol AS-E and its derivatives as anticancer agents by inhibiting CREB-mediated gene transcription. Bioorg. Med. Chem. 2012, 20, 6811–6820. [Google Scholar] [CrossRef]
- Xie, F.; Li, B.X.; Broussard, C.; Xiao, X. Identification, synthesis and evaluation of substituted benzofurazans as inhibitors of CREB-mediated gene transcription. Bioorg. Med. Chem. Lett. 2013, 23, 5371–5375. [Google Scholar] [CrossRef]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem. 2015, 58, 5075–5087. [Google Scholar] [CrossRef]
- Almahariq, M.; Tsalkova, T.; Mei, F.C.; Chen, H.; Zhou, J.; Sastry, S.K.; Schwede, F.; Cheng, X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013, 83, 122–128. [Google Scholar] [CrossRef]
- Almahariq, M.; Mei, F.C.; Wang, H.; Cao, A.T.; Yao, S.; Soong, L.; Sun, J.; Cong, Y.; Chen, J.; Cheng, X. Exchange protein directly activated by cAMP modulates regulatory T-cell-mediated immunosuppression. Biochem. J. 2015, 465, 295–303. [Google Scholar] [CrossRef]
- Vang, A.G.; Housley, W.; Dong, H.; Basole, C.; Ben-Sasson, S.Z.; Kream, B.E.; Epstein, P.M.; Clark, R.B.; Brocke, S. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: A potential role for Epac. Biochem. J. 2013, 456, 463–473. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Huang, R.Y.; Hsieh, K.P.; Huang, W.W.; Yang, Y.H. Use of lithium and cancer risk in patients with bipolar disorder: Population-based cohort study. Br. J. Psychiatry J. Ment. Sci. 2016, 209, 393–399. [Google Scholar] [CrossRef]
- Peng, Z.; Ji, Z.; Mei, F.; Lu, M.; Ou, Y.; Cheng, X. Lithium inhibits tumorigenic potential of PDA cells through targeting hedgehog-GLI signaling pathway. PLoS ONE 2013, 8, e61457. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, H.; Boulton, S.; Mei, F.; Ye, N.; Melacini, G.; Zhou, J.; Cheng, X. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: Defining the ESI-09 “therapeutic window”. Sci. Rep. 2015, 5, 9344. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases—The lessons from the mouse models: Inhibition ≠ deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Zhao, Y.; Thomas, H.D.; Batey, M.A.; Cowell, I.G.; Richardson, C.J.; Griffin, R.J.; Calvert, A.H.; Newell, D.R.; Smith, G.C.; Curtin, N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006, 66, 5354–5362. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Huston, E.; Lynch, M.J.; Mohamed, A.; Collins, D.M.; Hill, E.V.; MacLeod, R.; Krause, E.; Baillie, G.S.; Houslay, M.D. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc. Natl. Acad. Sci. USA 2008, 105, 12791–12796. [Google Scholar] [CrossRef]
- Rehmann, H. Epac-inhibitors: Facts and artefacts. Sci. Rep. 2013, 3, 3032. [Google Scholar] [CrossRef]
- Maeda, Y.; Kikuchi, R.; Kawagoe, J.; Tsuji, T.; Koyama, N.; Yamaguchi, K.; Nakamura, H.; Aoshiba, K. Anti-cancer strategy targeting the energy metabolism of tumor cells surviving a low-nutrient acidic microenvironment. Mol. Metab. 2020, 42, 101093. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.; Selvaratnam, R.; Ahmed, R.; Van, K.; Cheng, X.; Melacini, G. Mechanisms of Specific versus Nonspecific Interactions of Aggregation-Prone Inhibitors and Attenuators. J. Med. Chem. 2019, 62, 5063–5079. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.X.; Liu, Y.; Zhang, S.M.; Wang, H.F.; Liu, Y.F.; Liu, J.L.; Li, X.H.; Zeng, M.R.; Han, Y.Z.; Liu, F.Y.; et al. Epac activation ameliorates tubulointerstitial inflammation in diabetic nephropathy. Acta Pharmacol. Sin. 2021, 43, 659–671. [Google Scholar] [CrossRef]
- Vliem, M.J.; Ponsioen, B.; Schwede, F.; Pannekoek, W.J.; Riedl, J.; Kooistra, M.R.; Jalink, K.; Genieser, H.G.; Bos, J.L.; Rehmann, H. 8-pCPT-2′-O-Me-cAMP-AM: An improved Epac-selective cAMP analogue. Chembiochem A Eur. J. Chem. Biol. 2008, 9, 2052–2054. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Z.; Chen, H.; Ye, N.; Cheng, X.; Zhou, J. Exchange proteins directly activated by cAMP (EPACs): Emerging therapeutic targets. Bioorg. Med. Chem. Lett. 2017, 27, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Schwede, F.; Bertinetti, D.; Langerijs, C.N.; Hadders, M.A.; Wienk, H.; Ellenbroek, J.H.; de Koning, E.J.; Bos, J.L.; Herberg, F.W.; Genieser, H.G.; et al. Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol. 2015, 13, e1002038. [Google Scholar] [CrossRef]
- Herbst, K.J.; Coltharp, C.; Amzel, L.M.; Zhang, J. Direct activation of Epac by sulfonylurea is isoform selective. Chem. Biol. 2011, 18, 243–251. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S.; Nesterova, M.V. Tumor reversion: Protein kinase A isozyme switching. Ann. N. Y. Acad. Sci. 2005, 1058, 76–86. [Google Scholar] [CrossRef]
- Schwede, F.; Maronde, E.; Genieser, H.; Jastorff, B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 2000, 87, 199–226. [Google Scholar] [CrossRef]
- Choi, K.Y.; Ahn, Y.H.; Ahn, H.W.; Cho, Y.J.; Hong, S.H. Involvement of Akt2/protein kinase B β (PKBβ) in the 8-Cl-cAMP-induced cancer cell growth inhibition. J. Cell. Physiol. 2013, 228, 890–902. [Google Scholar] [CrossRef]
- Robinson-White, A.J.; Hsiao, H.P.; Leitner, W.W.; Greene, E.; Bauer, A.; Krett, N.L.; Nesterova, M.; Stratakis, C.A. Protein kinase A-independent inhibition of proliferation and induction of apoptosis in human thyroid cancer cells by 8-Cl-adenosine. J. Clin. Endocrinol. Metab. 2008, 93, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Matera, M.G.; Page, C.; Cazzola, M. PDE inhibitors currently in early clinical trials for the treatment of asthma. Expert Opin. Investig. Drugs 2014, 23, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Ragazzon, B.; Rizk-Rabin, M.; Bertherat, J. Protein kinase A alterations in endocrine tumors. Horm. Metab. Res. 2012, 44, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Omar, F.; Findlay, J.E.; Carfray, G.; Allcock, R.W.; Jiang, Z.; Moore, C.; Muir, A.L.; Lannoy, M.; Fertig, B.A.; Mai, D.; et al. Small-molecule allosteric activators of PDE4 long form cyclic AMP phosphodiesterases. Proc. Natl. Acad. Sci. USA 2019, 116, 13320–13329. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S. Antisense protein kinase A RI alpha-induced tumor reversion: Portrait of a microarray. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2004, 1697, 71–79. [Google Scholar] [CrossRef]
- Goel, S.; Desai, K.; Macapinlac, M.; Wadler, S.; Goldberg, G.; Fields, A.; Einstein, M.; Volterra, F.; Wong, B.; Martin, R.; et al. A phase I safety and dose escalation trial of docetaxel combined with GEM231, a second generation antisense oligonucleotide targeting protein kinase A R1alpha in patients with advanced solid cancers. Investig. New Drugs 2006, 24, 125–134. [Google Scholar] [CrossRef]
- Mitton, B.; Chae, H.D.; Hsu, K.; Dutta, R.; Aldana-Masangkay, G.; Ferrari, R.; Davis, K.; Tiu, B.C.; Kaul, A.; Lacayo, N.; et al. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia 2016, 30, 2302–2311. [Google Scholar] [CrossRef]
- Chae, H.D.; Cox, N.; Dahl, G.V.; Lacayo, N.J.; Davis, K.L.; Capolicchio, S.; Smith, M.; Sakamoto, K.M. Niclosamide suppresses acute myeloid leukemia cell proliferation through inhibition of CREB-dependent signaling pathways. Oncotarget 2018, 9, 4301–4317. [Google Scholar] [CrossRef]
- Illiano, M.; Conte, M.; Salzillo, A.; Ragone, A.; Spina, A.; Nebbioso, A.; Altucci, L.; Sapio, L.; Naviglio, S. The KDM Inhibitor GSKJ4 Triggers CREB Downregulation via a Protein Kinase A and Proteasome-Dependent Mechanism in Human Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 799. [Google Scholar] [CrossRef]
- Kang, X.; Cui, C.; Wang, C.; Wu, G.; Chen, H.; Lu, Z.; Chen, X.; Wang, L.; Huang, J.; Geng, H.; et al. CAMKs support development of acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 30. [Google Scholar] [CrossRef]
- Almahariq, M.; Mei, F.C.; Cheng, X. Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol. Metab. 2014, 25, 60–71. [Google Scholar] [CrossRef]
- Aumo, L.; Rusten, M.; Mellgren, G.; Bakke, M.; Lewis, A.E. Functional roles of protein kinase A (PKA) and exchange protein directly activated by 3′,5′-cyclic adenosine 5′-monophosphate (cAMP) 2 (EPAC2) in cAMP-mediated actions in adrenocortical cells. Endocrinology 2010, 151, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Bork, N.I.; Jacquet, E.; Zipfel, S.; Muñoz-Guijosa, C.; Baczkó, I.; Reichenspurner, H.; Donzeau-Gouge, P.; Maier, L.S.; Dobrev, D.; et al. Mapping genetic changes in the cAMP-signaling cascade in human atria. J. Mol. Cell. Cardiol. 2021, 155, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, R.; Fujita, T.; Suita, K.; Nakamura, T.; Cai, W.; Hidaka, Y.; Umemura, M.; Yokoyama, U.; Knollmann, B.C.; Okumura, S.; et al. Usefulness of Exchanged Protein Directly Activated by cAMP (Epac)1-Inhibiting Therapy for Prevention of Atrial and Ventricular Arrhythmias in Mice. Circ. J. Off. J. Jpn. Circ. Soc. 2019, 83, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jin, L.; Jiang, C.C.; Long, G.V.; Scolyer, R.A.; Wu, Q.; Zhang, X.D.; Mei, Y.; Wu, M. AEBP1 upregulation confers acquired resistance to BRAF (V600E) inhibition in melanoma. Cell Death Dis. 2013, 4, e914. [Google Scholar] [CrossRef]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G. Prominent role of cyclic adenosine monophosphate signalling pathway in the sensitivity of (WT)BRAF/(WT)NRAS melanoma cells to vemurafenib. Eur. J. Cancer 2014, 50, 1310–1320. [Google Scholar] [CrossRef]
- Natale, C.A.; Li, J.; Zhang, J.; Dahal, A.; Dentchev, T.; Stanger, B.Z.; Ridky, T.W.J.E. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. eLife 2018, 7, e31770. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Sunahara, R.K.; Johnson, R.A.; Gosselin, G.; Gilman, A.G.; Sprang, S.R. Two-metal-Ion catalysis in adenylyl cyclase. Science 1999, 285, 756–760. [Google Scholar] [CrossRef]
- Buck, J.; Sinclair, M.L.; Schapal, L.; Cann, M.J.; Levin, L.R. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc. Natl. Acad. Sci. USA 1999, 96, 79–84. [Google Scholar] [CrossRef]
- Kleinboelting, S.; Diaz, A.; Moniot, S.; van den Heuvel, J.; Weyand, M.; Levin, L.R.; Buck, J.; Steegborn, C. Crystal structures of human soluble adenylyl cyclase reveal mechanisms of catalysis and of its activation through bicarbonate. Proc. Natl. Acad. Sci. USA 2014, 111, 3727–3732. [Google Scholar] [CrossRef]
- Watson, E.L.; Jacobson, K.L.; Singh, J.C.; Idzerda, R.; Ott, S.M.; DiJulio, D.H.; Wong, S.T.; Storm, D.R. The type 8 adenylyl cyclase is critical for Ca2+ stimulation of cAMP accumulation in mouse parotid acini. J. Biol. Chem. 2000, 275, 14691–14699. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.B.; Islam, S.U.; Lee, Y.S. PRP4 Promotes Skin Cancer by Inhibiting Production of Melanin, Blocking Influx of Extracellular Calcium, and Remodeling Cell Actin Cytoskeleton. Int. J. Mol. Sci. 2021, 22, 6992. [Google Scholar] [CrossRef] [PubMed]
- Oishi, A.; Makita, N.; Sato, J.; Iiri, T. Regulation of RhoA signaling by the cAMP-dependent phosphorylation of RhoGDIα. J. Biol. Chem. 2012, 287, 38705–38715. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.U.; Ahmed, M.B.; Lee, S.J.; Shehzad, A.; Sonn, J.K.; Kwon, O.S.; Lee, Y.S. PRP4 kinase induces actin rearrangement and epithelial-mesenchymal transition through modulation of the actin-binding protein cofilin. Exp. Cell Res. 2018, 369, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Zhang, L.; Murray, F.; Yokouchi, H.; Zambon, A.C. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. 2012, 204, 277–287. [Google Scholar] [CrossRef]
- Wilderman, A.; Guo, Y.; Divakaruni, A.S.; Perkins, G.; Zhang, L.; Murphy, A.N.; Taylor, S.S.; Insel, P.A. Proteomic and Metabolic Analyses of S49 Lymphoma Cells Reveal Novel Regulation of Mitochondria by cAMP and Protein Kinase A. J. Biol. Chem. 2015, 290, 22274–22286. [Google Scholar] [CrossRef]
- Zambon, A.C.; Wilderman, A.; Ho, A.; Insel, P.A. Increased expression of the pro-apoptotic protein BIM, a mechanism for cAMP/protein kinase A (PKA)-induced apoptosis of immature T cells. J. Biol. Chem. 2011, 286, 33260–33267. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Jewell, J.L.; Fu, V.; Hong, A.W.; Yu, F.X.; Meng, D.; Melick, C.H.; Wang, H.; Lam, W.M.; Yuan, H.X.; Taylor, S.S.; et al. GPCR signaling inhibits mTORC1 via PKA phosphorylation of Raptor. eLife 2019, 8, e4303. [Google Scholar] [CrossRef]







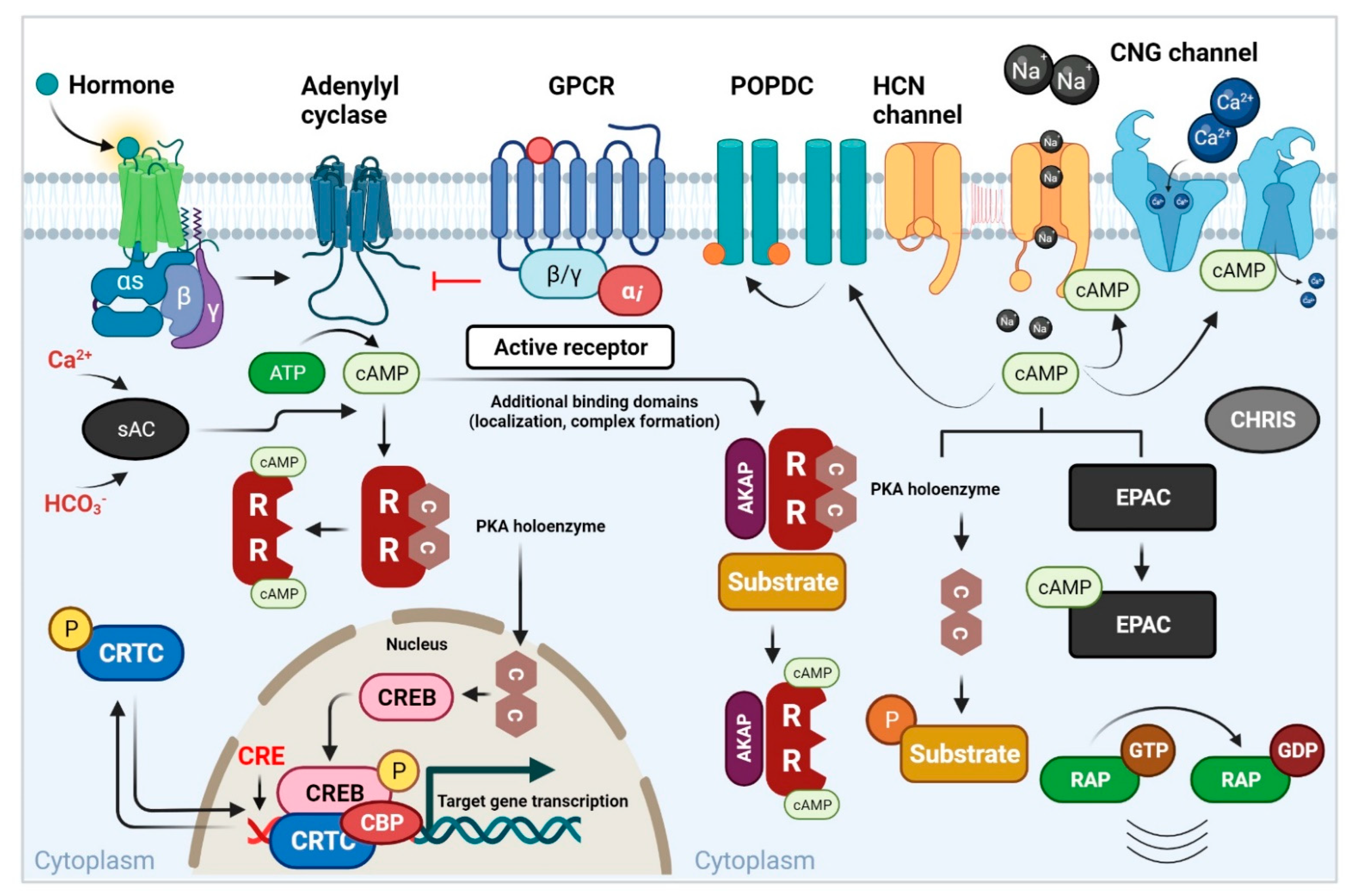{kind=link}
{kind=link}
{kind=link}
{kind=link}
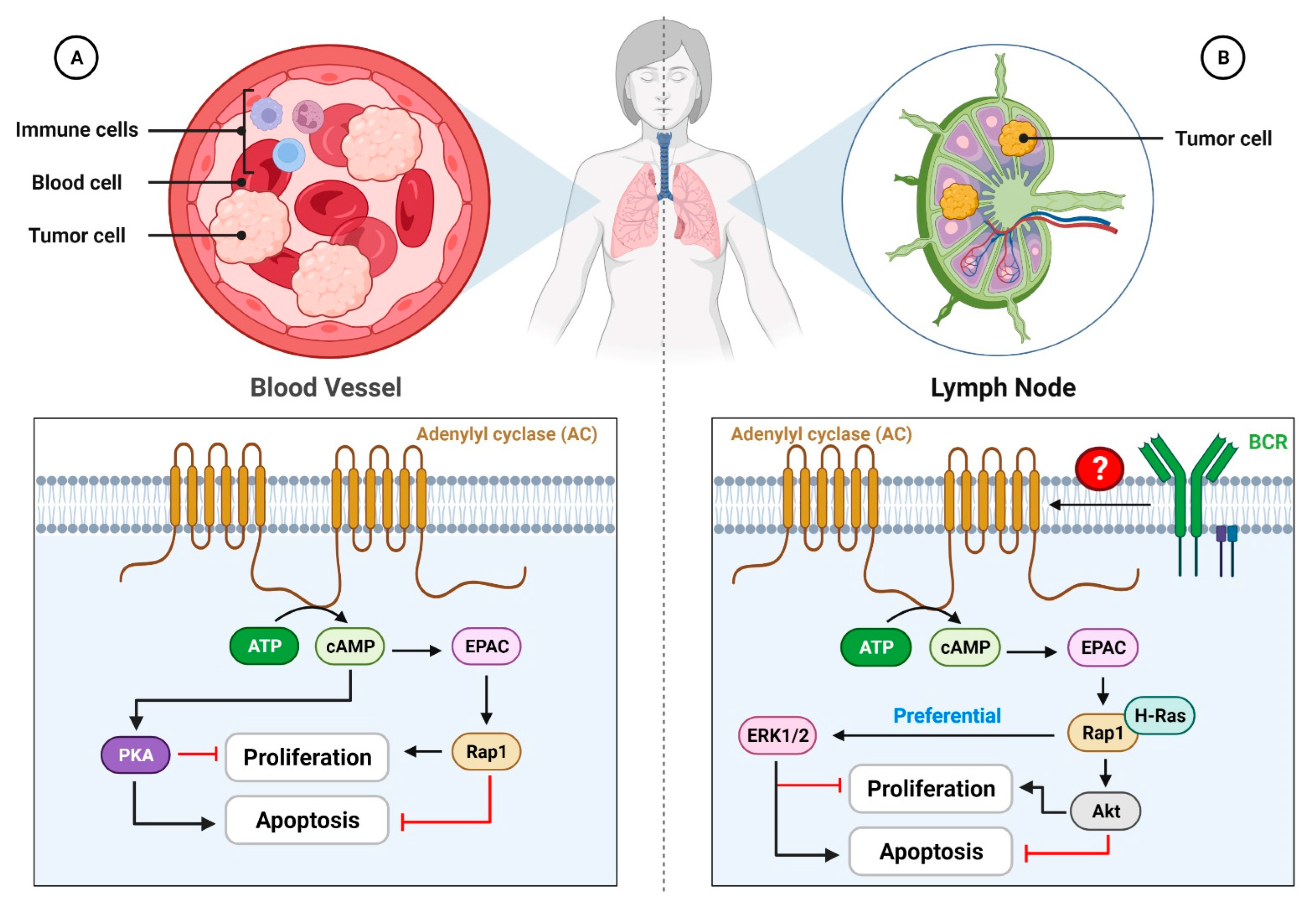{kind=link}
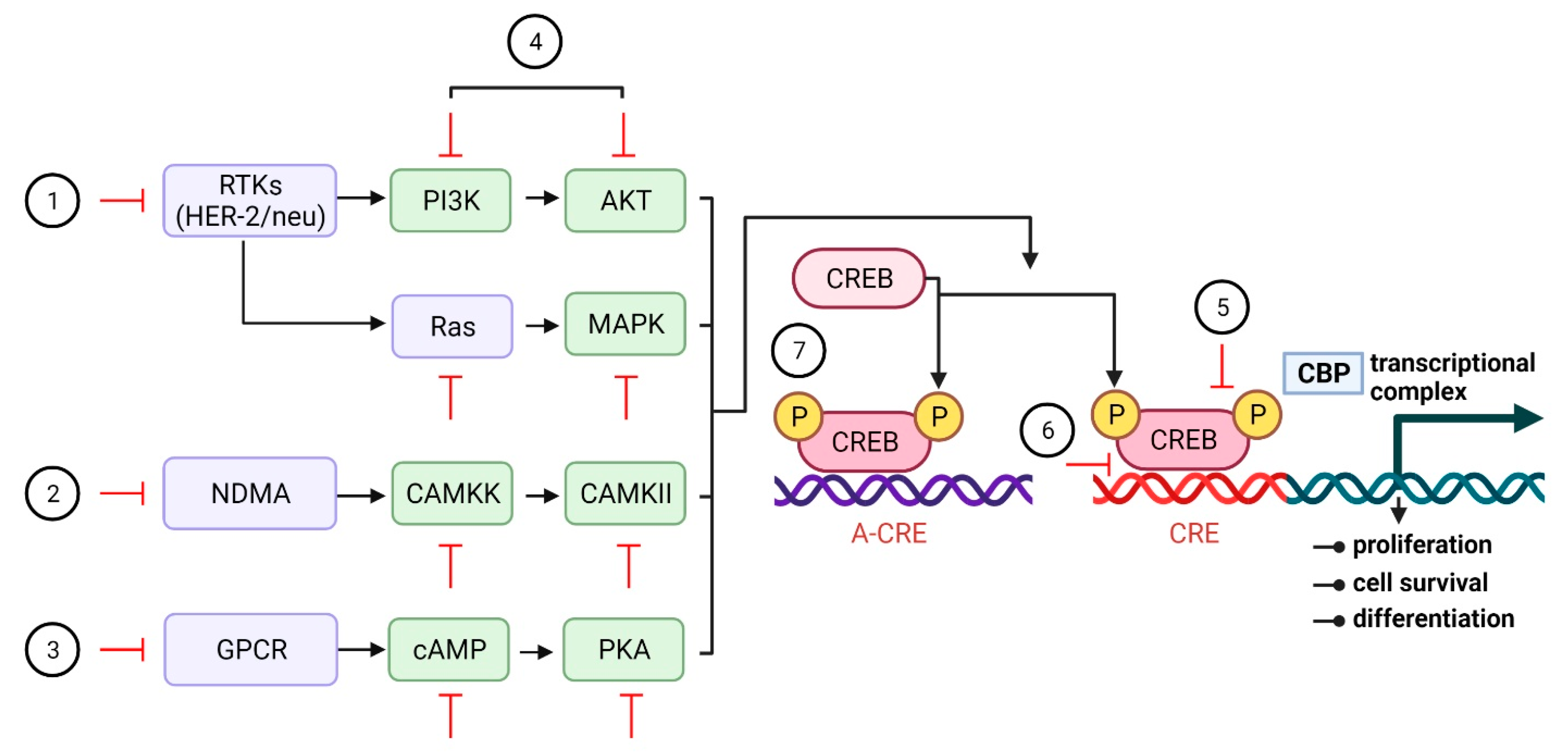{kind=link}
| Type of Cancer | cAMP/PKA Functions | Popeye Domain Containing Protein (POPDC) Cancer Types | Mechanisms and Roles of POPDC Proteins | POPDC Downstream Targets in Cancer Signaling Pathways and Protein Interactions |
|---|---|---|---|---|
| Squamous cell carcinoma ↑ | Increasing the invasion and metastasis in the esophagus by PKA phosphorylating vasodilator-stimulated-phosphoprotein (VASP) [49]. | POPDC1 in CRC, PC, BC, NSCLC, glioma, HNSCC, GC | Promoter hypermethylation [191,192,193,194] | POPDC1/ZO-1 protein interaction in trabecular meshwork cells, HCE, uveal melanoma prevents ZONAB-induced entry to cell cycle and translation of proliferative genes [195]. |
| Lymphoblastic leukemia ↓ | Autophagy, aided by cAMP-induced poly [ADP-ribose] polymerase 1 (PARP1) activation, may treat acute lymphoblastic leukemia [52]. | POPDC1 in HCC | Underexpression of miRNA-122 [188] and overexpression of netrin-1 [189]. | Occludin in HCE and uveal melanoma maintains tight junction formation [182,195]. |
| Liver cancer | PKA phosphorylates many substrates, including CIP4, facilitating HCC invasion and metastasis [58]. | POPDC2 in ductal breast carcinoma (especially HER2+ subtype) | Overexpressed at all clinical stages. Possibly implicated in cancer initiation and sustenance [190]. | LRP6 (Wnt/βcatenin pathway) in HEK293 cells, human colonoids, murine adenoma tumoroids prevents β-catenin activation by inhibition of LRP6 [196]. |
| The vasoactive intestinal peptide lowered cAMP levels, CREB expression, and phospho-CREB (Ser133) phosphorylation via inhibiting B-cell lymphoma-extra-large (Bcl-Xl) expression [59]. | POPDC3 in ductal breast carcinoma (especially HER2+ subtype) | Overexpressed at early clinical stages [190]. | PR61α (c-Myc pathway) in murine colitis-associated cancer cells promotes c-Myc ubiquitination/ degradation [193]. | |
| The catalytic subunit of PKA C (DNAJB1-protein kinase cAMP-activated catalytic subunit alpha (PRKACA)) was overexpressed, PKA activity increased [61]. | POPDC3 in head and neck squamous cell carcinoma (HNSCC) | Overexpression correlates with low patient survival. Potential biomarker for radiotherapy resistance [197]. | ||
| Prostate cancer | The high PKA expression promotes cell proliferation and carcinogenesis [71]. | POPDC3 in gastric cancer | Underexpression due to promoter hypermethylation. Lower POPDC3 levels correlate with increased depth of invasion and metastasis [192]. | |
| cAMP–PKA signaling pathway is required for high levels of osteocalcin and ostesialin production in androgen-independent prostate cancer [90]. | POPDC3 in esophageal and lung cancer | Overexpression of POPDC3 correlates with greater radiotherapy resistance [197]. | ||
| PKA activity may increase with depressive and behavioral stress [92,93]. | LRP6 (Wnt/βcatenin pathway) interacting with POPDC1 in HEK293 cells, human colonoids, murine adenoma tumoroids | Prevention of β-catenin activation by inhibition of LRP6 [196]. | ||
| Small-cell lung cancer (SCLC) ↓ | Inhibition of PKA activity [73]. | Occludin interacting with POPDC1 in HCE, uveal melanoma | Maintenance of tight junction formation [182,195]. | |
| Brain cancer | Stimulation of the cAMP pathway via PKA RII induces cell differentiation and death [74]. | |||
| The catalytic subunit of PKA was found to be decreased in high-grade gliomas [76]. | ||||
| Increased cAMP levels reduce phosphatidylinositol 3-kinase, which decreases neuroblastoma [77]. | ||||
| Lower AC and cAMP levels in glioblastoma cells [79]. |
| Identifier | Title | Cancer Type | Location |
|---|---|---|---|
| NCT00021268 | Tocladesine in the treatment of progressive or recurrent metastatic colorectal cancer | Colorectal | Jonsson Comprehensive Cancer Center, UCLA Los Angeles, California, United States |
| NCT00004902 | Tocladesine in the treatment of progressive or recurrent multiple myeloma | Multiple myeloma and plasma cell tumor | Robert H. Lurie Comprehensive Cancer Center, Northwestern University Chicago, Illinois, United States |
| NCT00004863 | Paclitaxel and GEM 231 in the treatment of refractory or recurrent solid tumors | Unspecified adult solid tumor | Albert Einstein Comprehensive Cancer Center Bronx, New York, United States |
| NCT00004864 | Docetaxel and GEM 231 in the treatment of refractory or recurrent solid tumors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, M.B.; Alghamdi, A.A.A.; Islam, S.U.; Lee, J.-S.; Lee, Y.-S. cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach. Cells 2022, 11, 2020. https://doi.org/10.3390/cells11132020
Ahmed MB, Alghamdi AAA, Islam SU, Lee J-S, Lee Y-S. cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach. Cells. 2022; 11(13):2020. https://doi.org/10.3390/cells11132020
Chicago/Turabian StyleAhmed, Muhammad Bilal, Abdullah A. A. Alghamdi, Salman Ul Islam, Joon-Seok Lee, and Young-Sup Lee. 2022. "cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach" Cells 11, no. 13: 2020. https://doi.org/10.3390/cells11132020
APA StyleAhmed, M. B., Alghamdi, A. A. A., Islam, S. U., Lee, J.-S., & Lee, Y.-S. (2022). cAMP Signaling in Cancer: A PKA-CREB and EPAC-Centric Approach. Cells, 11(13), 2020. https://doi.org/10.3390/cells11132020