
Ca2+ Sensors Assemble: Function of the MCU Complex in the Pancreatic Beta Cell



Abstract
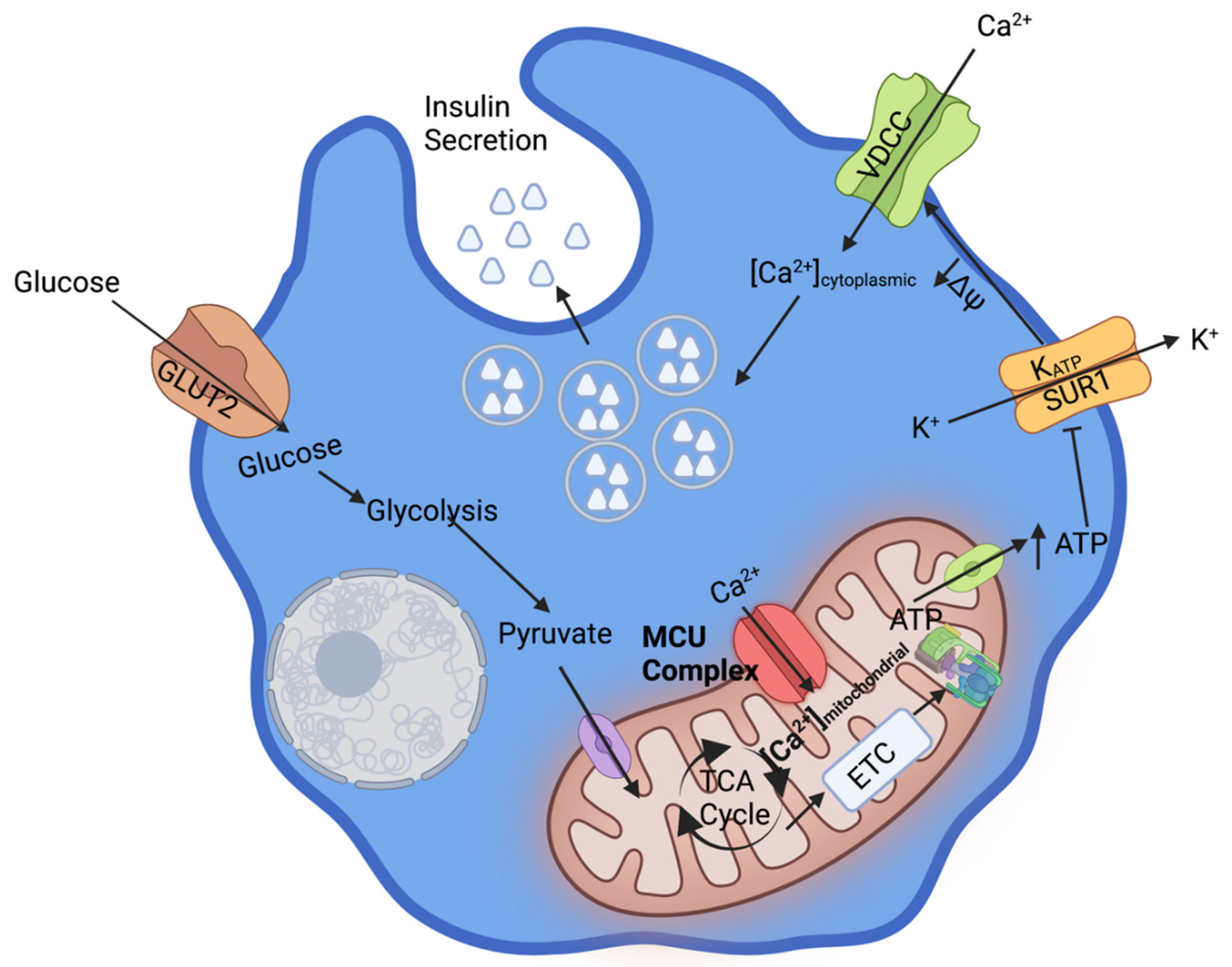
1. Ca2+ Dependent β-Cell Glucose Stimulated Insulin Secretion
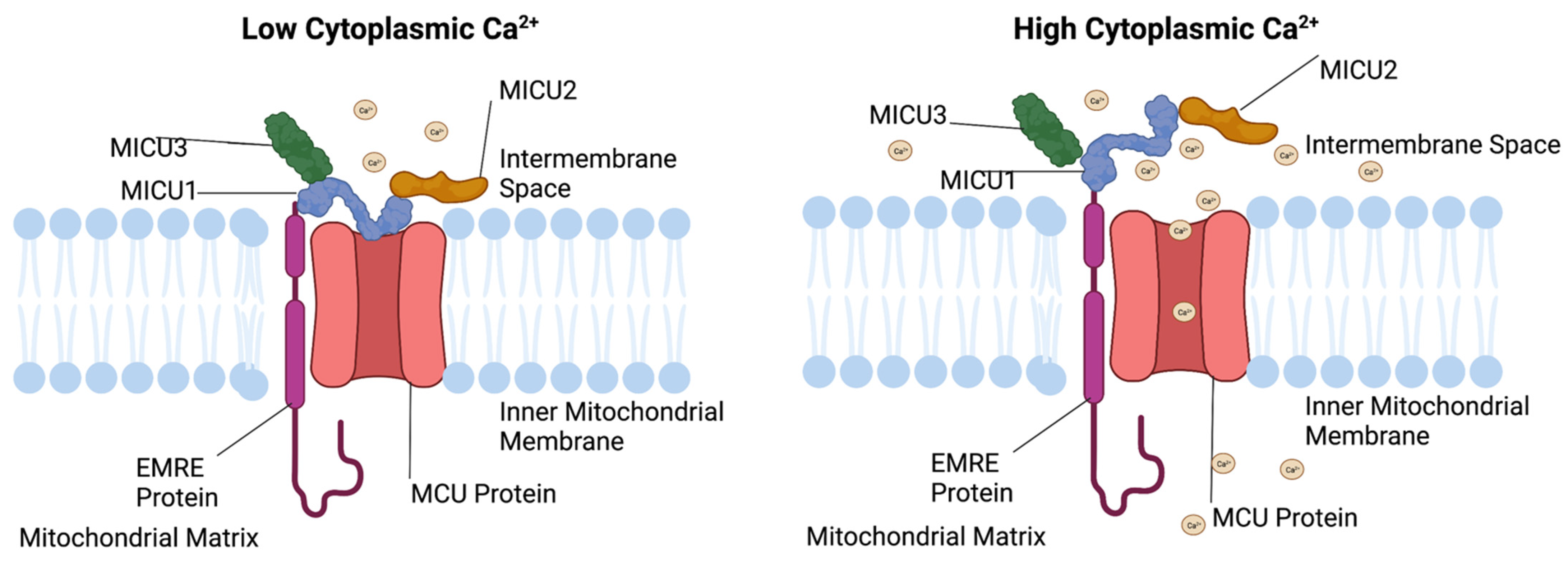
2. Mechanisms of Mitochondrial Ca2+ Uptake
3. MCU Complex in the β-Cell
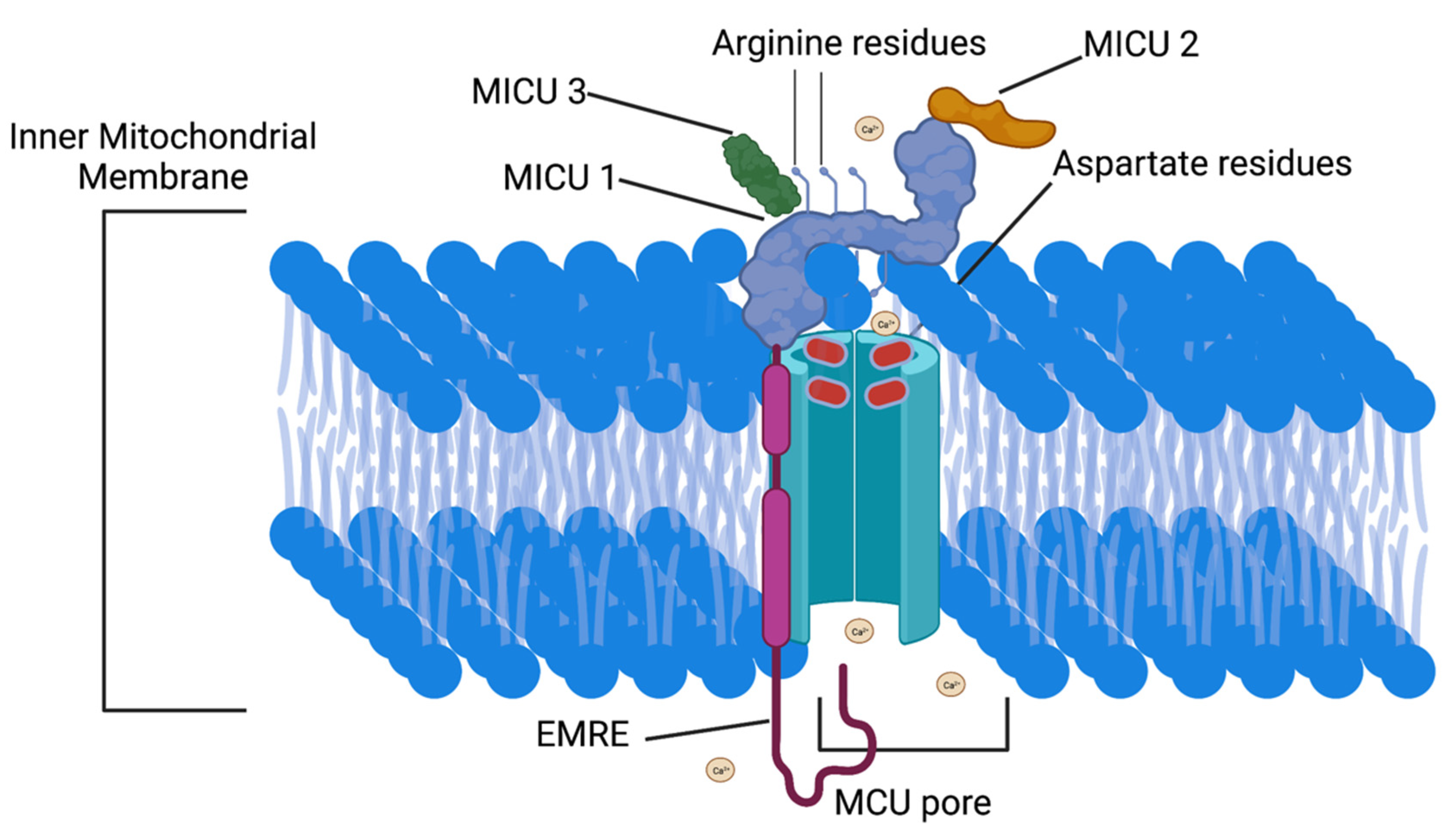
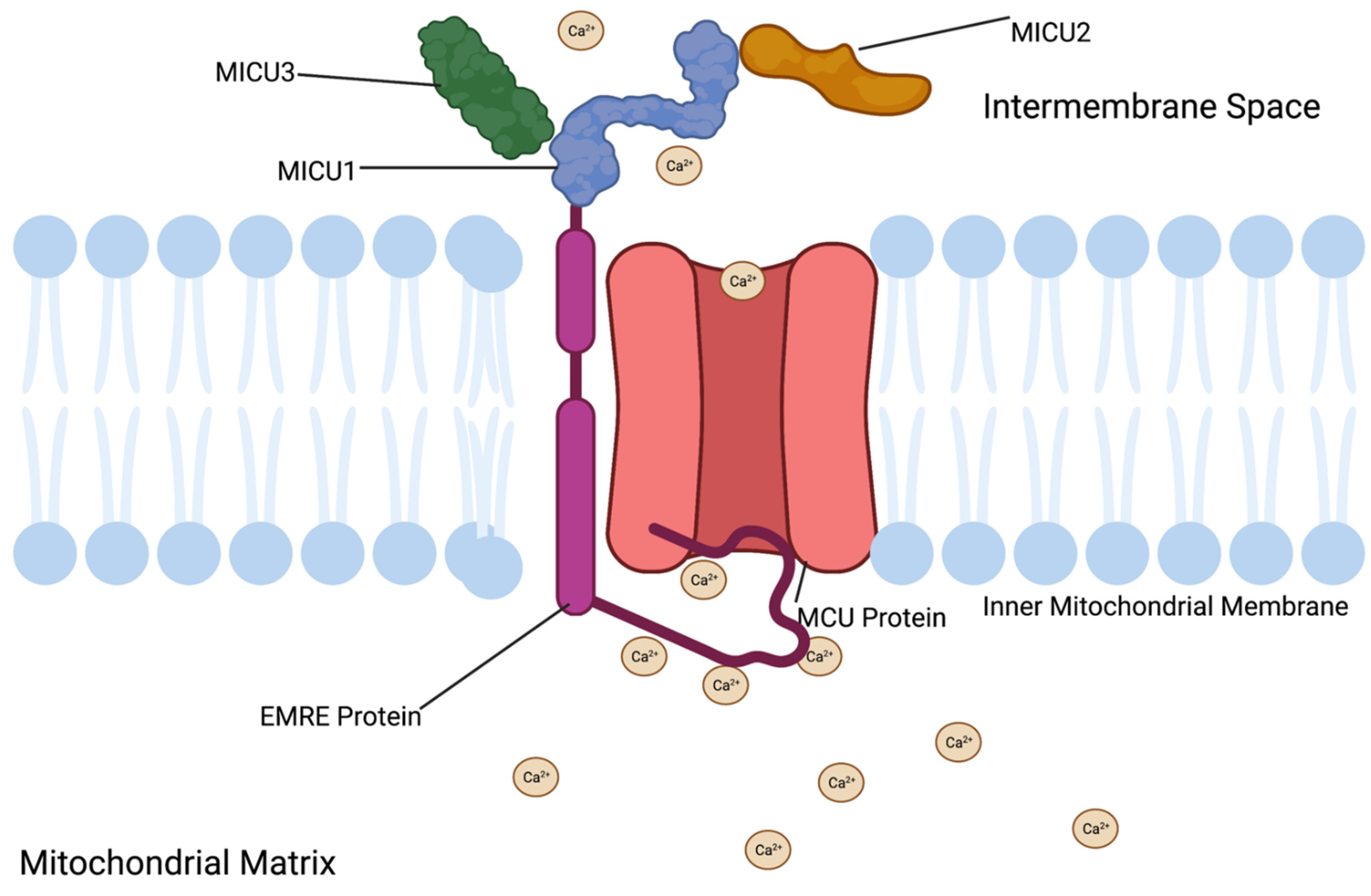
3.1. MCU Protein Function
3.2. MCU Protein Structure
3.3. MICU1 Function
3.4. MICU1 Structure
3.5. MICU2 Function
3.6. MICU2 Location
3.7. MICU3 Function
3.8. MICU3 Location
3.9. EMRE Function
3.10. EMRE Location
4. MCU Complex and Disease
4.1. MCU Mutations
4.2. MICU1 Mutations
4.3. MICU2 Mutations
4.4. EMRE Mutations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sandoval, D.A.; D’Alessio, D.A. Physiology of proglucagon peptides: Role of glucagon and glp-1 in health and disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Barg, S.; Eliasson, L.; Renstrom, E.; Rorsman, P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes 2002, 51 (Suppl. 1), S74–S82. [Google Scholar] [CrossRef] [PubMed]
- Guest, P.C. Biogenesis of the Insulin Secretory Granule in Health and Disease. Adv. Exp. Med. Biol. 2019, 1134, 17–32. [Google Scholar] [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Lewandowski, S.L.; Cardone, R.L.; Foster, H.R.; Ho, T.; Potapenko, E.; Poudel, C.; VanDeusen, H.R.; Sdao, S.M.; Alves, T.C.; Zhao, X.; et al. Pyruvate Kinase Controls Signal Strength in the Insulin Secretory Pathway. Cell Metab. 2020, 32, 736–750.e5. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef]
- Gandasi, N.; Yin, P.; Riz, M.; Chibalina, M.V.; Cortese, G.; Lund, P.-E.; Matveev, V.; Rorsman, P.; Sherman, A.; Pedersen, M.G.; et al. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. J. Clin. Investig. 2017, 127, 2353–2364. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Semplici, F.; Li, D.; Rizzuto, R.; Ravier, M.A.; Gilon, P.; Rutter, G.A. Frequency-dependent mitochondrial Ca2+ accumulation regulates ATP synthesis in pancreatic beta cells. Pflugers Arch. 2013, 465, 543–554. [Google Scholar] [CrossRef]
- Ricchelli, F.; Šileikytė, J.; Bernardi, P. Shedding light on the mitochondrial permeability transition. Biochim. Biophys. Acta 2011, 1807, 482–490. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Park, K.S.; Dupont, O.; Demaurex, N.; Pozzan, T.; Cline, G.W.; Wollheim, C.B. Matrix alkalinization: A novel mitochondrial signal for sustained pancreatic beta-cell activation. EMBO J. 2009, 28, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Greiser, M.; Lederer, W.J. Calcium influx through the mitochondrial calcium uniporter holocomplex, MCUcx. J. Mol. Cell. Cardiol. 2021, 151, 145–154. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Vasington, F.D.; Murphy, J.V. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962, 237, 2670–2677. [Google Scholar] [CrossRef]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 1966, 41, 445–501. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.M.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Wei, A.-C.; Liu, T.; O’Rourke, B. Dual Effect of Phosphate Transport on Mitochondrial Ca2+ Dynamics. J. Biol. Chem. 2015, 290, 16088–16098. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef]
- Rutter, G.; Denton, R.M. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios. Biochem. J. 1988, 252, 181–189. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Semplici, F.; Ravier, M.A.; Bellomo, E.A.; Pullen, T.J.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef] [PubMed]
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardão, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The mitochondrial permeability transition pore: An evolving concept critical for cell life and death. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2489–2521. [Google Scholar] [CrossRef]
- Chen, W.; Yang, J.; Chen, S.; Xiang, H.; Liu, H.; Lin, D.; Zhao, S.; Peng, H.; Chen, P.; Chen, A.F.; et al. Importance of mitochondrial calcium uniporter in high glucose–induced endothelial cell dysfunction. Diabetes Vasc. Dis. Res. 2017, 14, 494–501. [Google Scholar] [CrossRef]
- Quan, X.; Nguyen, T.T.; Choi, S.-K.; Xu, S.; Das, R.; Cha, S.-K.; Kim, N.; Han, J.; Wiederkehr, A.; Wollheim, C.B.; et al. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2015, 290, 4086–4096. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Tengholm, A. Metabolic regulation of calcium signaling in beta cells. Semin. Cell Dev. Biol. 2020, 103, 20–30. [Google Scholar] [CrossRef]
- Rutter, G.A.; Hodson, D.; Chabosseau, P.; Haythorne, E.; Pullen, T.; Leclerc, I. Local and regional control of calcium dynamics in the pancreatic islet. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 30–41. [Google Scholar] [CrossRef] [PubMed]
- Georgiadou, E.; Haythorne, E.; Dickerson, M.T.; Lopez-Noriega, L.; Pullen, T.J.; Xavier, G.D.S.; Davis, S.P.X.; Martinez-Sanchez, A.; Semplici, F.; Rizzuto, R.; et al. The pore-forming subunit MCU of the mitochondrial Ca2+ uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020, 63, 1368–1381. [Google Scholar] [CrossRef]
- Weiser, A.; Feige, J.N.; de Marchi, U. Mitochondrial Calcium Signaling in Pancreatic beta-Cell. Int. J. Mol. Sci. 2021, 22, 2515. [Google Scholar] [CrossRef] [PubMed]
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.-C.; Feige, J.N.; De Marchi, U. Targeting Mitochondrial Calcium Uptake with the Natural Flavonol Kaempferol, to Promote Metabolism/Secretion Coupling in Pancreatic β-cells. Nutrients 2020, 12, 538. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef]
- De Marchi, U.; Fernandez-Martinez, S.; de la Fuente, S.; Wiederkehr, A.; Santo-Domingo, J. Mitochondrial ion channels in pancreatic beta-cells: Novel pharmacological targets for the treatment of Type 2 diabetes. Br. J. Pharmacol. 2021, 178, 2077–2095. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef]
- Vishnu, N.; Hamilton, A.; Bagge, A.; Wernersson, A.; Cowan, E.; Barnard, H.; Sancak, Y.; Kamer, K.; Spégel, P.; Fex, M.; et al. Mitochondrial clearance of calcium facilitated by MICU2 controls insulin secretion. Mol. Metab. 2021, 51, 101239. [Google Scholar] [CrossRef]
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X.-C.; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, 1252–1261.e13. [Google Scholar] [CrossRef] [PubMed]
- Baeshen, N.A.; Baeshen, M.N.; Sheikh, A.; Bora, R.S.; Ahmed, M.M.M.; Ramadan, H.A.I.; Saini, K.S.; Redwan, E.M. Cell factories for insulin production. Microb. Cell Factories 2014, 13, 141. [Google Scholar] [CrossRef]
- Zhang, Z.; Wakabayashi, N.; Wakabayashi, J.; Tamura, Y.; Song, W.-J.; Sereda, S.; Clerc, P.; Polster, B.M.; Aja, S.M.; Pletnikov, M.V.; et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell 2011, 22, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Maechler, P. Beta-cell mitochondrial carriers and the diabetogenic stress response. Biochim. Biophys. Acta 2016, 1863, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Panfili, E.; Crompton, M.; Sottocasa, G.L. Immunochemical evidence of the independence of the Ca2+/Na2+ antiporter and electrophoretic Ca2+ uniporter in heart mitochondria. FEBS Lett. 1981, 123, 30–32. [Google Scholar] [CrossRef][Green Version]
- Jimenez-Sánchez, C.; Brun, T.; Maechler, P. Mitochondrial Carriers Regulating Insulin Secretion Profiled in Human Islets upon Metabolic Stress. Biomolecules 2020, 10, 1543. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Nam, S.M.; Kim, J.-H.; Das, R.; Choi, S.-K.; Nguyen, T.T.; Quan, X.; Chung, C.H.; Lee, E.Y.; Lee, I.-K.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef]
- Tomar, D.; Jaña, F.; Dong, Z.; Quinn, W.J.; Jadiya, P.; Breves, S.L.; Daw, C.; Srikantan, S.; Shanmughapriya, S.; Nemani, N.; et al. Blockade of MCU-Mediated Ca2+ Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell Rep. 2019, 26, 3709–3725.e7. [Google Scholar] [CrossRef]
- Ly, L.D.; Da Ly, D.; Nguyen, N.T.; Kim, J.-H.; Yoo, H.; Chung, J.; Lee, M.-S.; Cha, S.-K.; Park, K.-S. Mitochondrial Ca2+ Uptake Relieves Palmitate-Induced Cytosolic Ca2+ Overload in MIN6 Cells. Mol. Cells 2020, 43, 66–75. [Google Scholar] [CrossRef]
- Koval, O.M.; Nguyen, E.K.; Santhana, V.; Fidler, T.P.; Sebag, S.C.; Rasmussen, T.P.; Mittauer, D.J.; Strack, S.; Goswami, P.C.; Abel, E.D.; et al. Loss of MCU prevents mitochondrial fusion in G1 -S phase and blocks cell cycle progression and proliferation. Sci. Signal. 2019, 12, eaav1439. [Google Scholar] [CrossRef]
- Zhao, H.; Li, T.; Wang, K.; Zhao, F.; Chen, J.; Xu, G.; Zhao, J.; Li, T.; Chen, L.; Li, L.; et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat. Cell Biol. 2019, 21, 476–486. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A., Jr.; Lander, G.C.; Lee, S.-Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science 2018, 361, 506–511. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ozono, M.; Watanabe, A.; Maeda, K.; Nara, A.; Hashida, M.; Ido, Y.; Hiroshima, Y.; Yamada, A.; Terada, H.; et al. Functional analysis of coiled-coil domains of MCU in mitochondrial calcium uptake. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 148061. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef]
- Bick, A.G.; Calvo, S.E.; Mootha, V.K. Evolutionary diversity of the mitochondrial calcium uniporter. Science 2012, 336, 886. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef]
- Phillips, C.B.; Tsai, C.-W.; Tsai, M.-F. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. Elife 2019, 8, e41112. [Google Scholar] [CrossRef]
- Paillard, M.; Csordás, G.; Huang, K.-T.; Várnai, P.; Joseph, S.K.; Hajnóczky, G. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca2+ Flux and Sensitivity to Ru360. Mol. Cell 2018, 72, 778–785.e3. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Malli, R.; Parichatikanond, W.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Klec, C.; Rost, R.; Graier, W.F. Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca2+. Sci. Rep. 2015, 5, 15602. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.I.; Ullah, G. The Function of Mitochondrial Calcium Uniporter at the Whole-Cell and Single Mitochondrion Levels in WT, MICU1 KO, and MICU2 KO Cells. Cells 2020, 9, 1520. [Google Scholar] [CrossRef]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Reane, D.V.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Yang, W.; Liao, Z.-Y.; Wu, Y.-X.; Fan, Z.; Guo, A.; Yu, J.; Chen, Q.-N.; Wu, J.-H.; Zhou, J.; et al. MICU3 regulates mitochondrial Ca2+-dependent antioxidant response in skeletal muscle aging. Cell Death Dis. 2021, 12, 1115. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef]
- Puente, B.N.; Sun, J.; Parks, R.J.; Fergusson, M.M.; Liu, C.; Springer, D.A.; Aponte, A.M.; Liu, J.C.; Murphy, E. MICU3 Plays an Important Role in Cardiovascular Function. Circ. Res. 2020, 127, 1571–1573. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef]
- Vais, H.; Mallilankaraman, K.; Mak, D.-O.D.; Hoff, H.; Payne, R.; Tanis, J.E.; Foskett, J.K. EMRE Is a Matrix Ca2+ Sensor that Governs Gatekeeping of the Mitochondrial Ca2+ Uniporter. Cell Rep. 2016, 14, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Williams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 2016, 5, e15545. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Murphy, E.; Pan, X.; Nguyen, T.; Liu, J.; Holmström, K.; Finkel, T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 2014, 449, 384–385. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- König, T.; Tröder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Mühlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef]
- Li, S.; Chen, J.; Liu, M.; Chen, Y.; Wu, Y.; Li, Q.; Ma, T.; Gao, J.; Xia, Y.; Fan, M.; et al. Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia—Reperfusion injury. Basic Res. Cardiol. 2021, 116, 65. [Google Scholar] [CrossRef]
- Morciano, G.; Rimessi, A.; Patergnani, S.; Vitto, V.A.; Danese, A.; Kahsay, A.; Palumbo, L.; Bonora, M.; Wieckowski, M.R.; Giorgi, C.; et al. Calcium dysregulation in heart diseases: Targeting calcium channels to achieve a correct calcium homeostasis. Pharmacol. Res. 2022, 177, 106119. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Smith, D.; Kamer, K.J.; Griffin, H.; Childs, A.-M.; Pysden, K.; Titov, D.; Duff, J.; Pyle, A.; Taylor, R.W.; Yu-Wai-Man, P.; et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet. 2016, 2, e59. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.-M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Alasmari, A.; Salih, M.A.; Samman, M.M.; Mian, S.A.; AlShidi, T.; Ibrahim, N.; Hashem, M.; Faqeih, E.; Al-Mohanna, F.; et al. A null mutation in MICU2 causes abnormal mitochondrial calcium homeostasis and a severe neurodevelopmental disorder. Brain 2017, 140, 2806–2813. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Syder, N.C.; Ghorashi, N.S.; Willingham, T.B.; Parks, R.J.; Sun, J.; Fergusson, M.M.; Liu, J.; Holmström, K.M.; Menazza, S.; et al. EMRE is essential for mitochondrial calcium uniporter activity in a mouse model. JCI Insight 2020, 5, e134063. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- Carafoli, E.; Lehninger, A.L. A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem. J. 1971, 122, 681–690. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCU Complex Component | Function in the β-Cell |
|---|---|
| MCU protein | Essential for GSIS [35] May alleviate T2D symptoms [36] |
| MICU1 | Promotes mitochondrial Ca2+ influx [35], May play a role in apoptosis regulation [37] |
| MICU2 | Essential for sustained GSIS [38] |
| MICU3 | N/A |
| EMRE | Positioning facilitates rapid GSIS following an excitatory stimulus [39] |
| Mutated Protein | Molecular Effect | Clinical Phenotype |
|---|---|---|
| MCU | Decreased apoptosis [35], decreased pyruvate dehydrogenase activity [77] | Increased levels of spontaneous abortion [79], cardioprotective effects [80] |
| MICU1 | Decreased pyruvate dehydrogenase activity [82] | Decreased insulin secretion [82] extrapyramidal proximal myopathy [83] |
| MICU2 | No known molecular effect | Severe neurological disorder and cognitive impairment [84] |
| MICU3 | No known molecular effect | No known clinical phenotype |
| EMRE | No known molecular effect | probable spontaneous abortion [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, J.G.; Tessem, J.S. Ca2+ Sensors Assemble: Function of the MCU Complex in the Pancreatic Beta Cell. Cells 2022, 11, 1993. https://doi.org/10.3390/cells11131993
Allen JG, Tessem JS. Ca2+ Sensors Assemble: Function of the MCU Complex in the Pancreatic Beta Cell. Cells. 2022; 11(13):1993. https://doi.org/10.3390/cells11131993
Chicago/Turabian StyleAllen, Jack G., and Jeffery S. Tessem. 2022. "Ca2+ Sensors Assemble: Function of the MCU Complex in the Pancreatic Beta Cell" Cells 11, no. 13: 1993. https://doi.org/10.3390/cells11131993
APA StyleAllen, J. G., & Tessem, J. S. (2022). Ca2+ Sensors Assemble: Function of the MCU Complex in the Pancreatic Beta Cell. Cells, 11(13), 1993. https://doi.org/10.3390/cells11131993