
Age-Related Lysosomal Dysfunctions


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Aging and Senescence
2. Lysosomes
2.1. Lysosomal Structure and Components
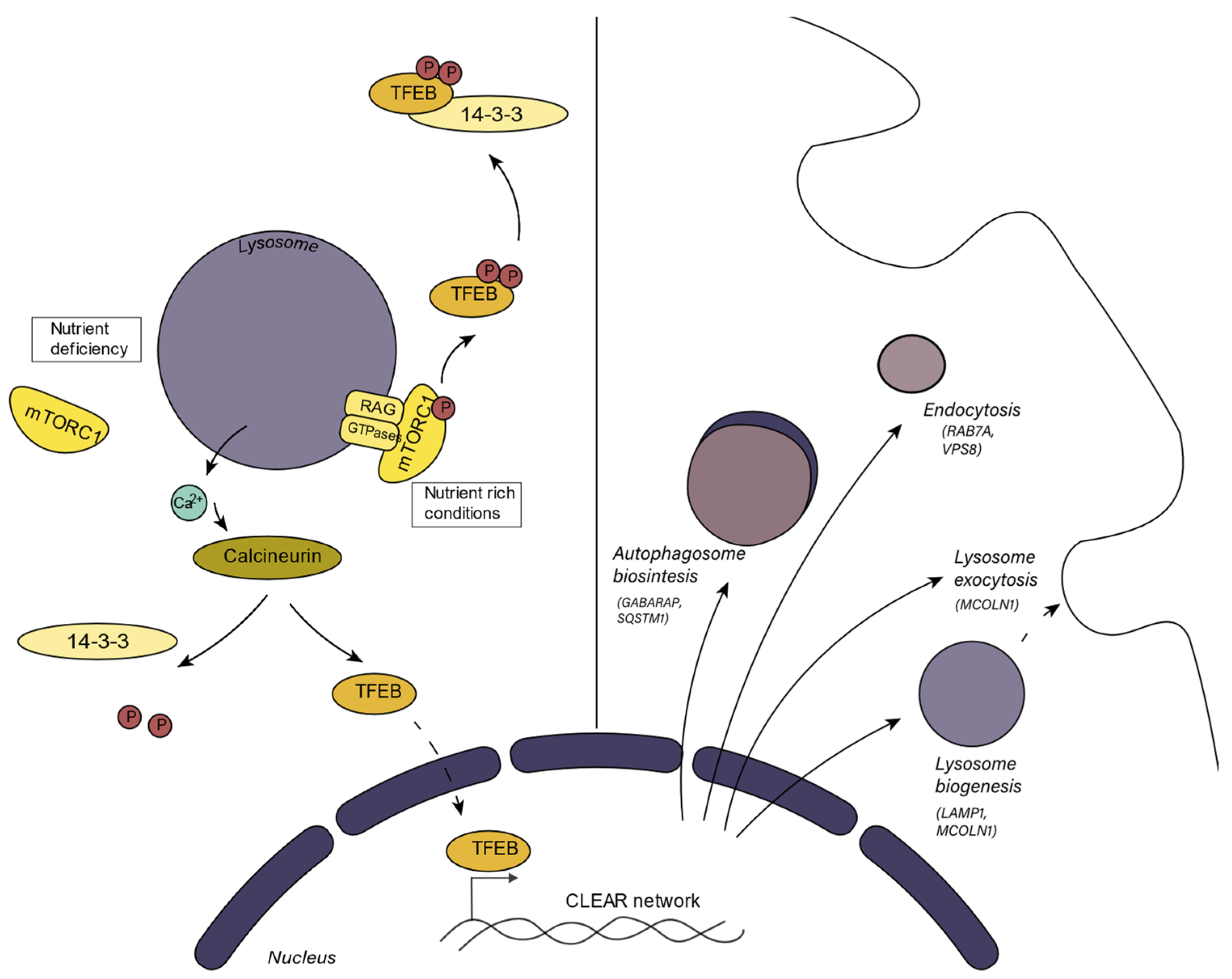
2.2. Lysosomal Biogenetic Pathways and Metabolic Integration
3. Processes in which the Lysosome Participates
3.1. Endocytosis
3.2. Autophagy
3.3. Mitophagy and Mitochondrial Dysfunction
3.4. Lysosomal Exocytosis
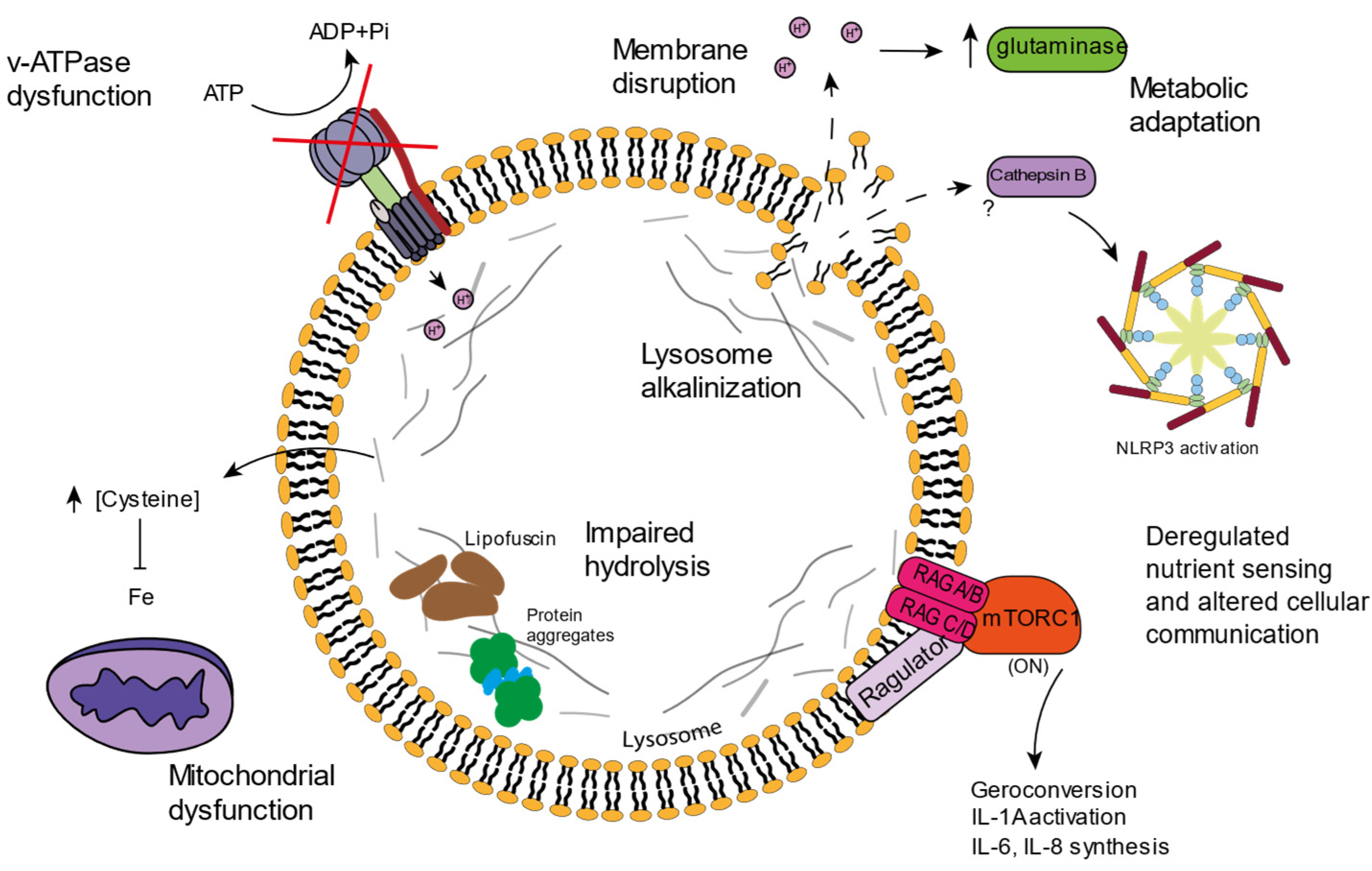
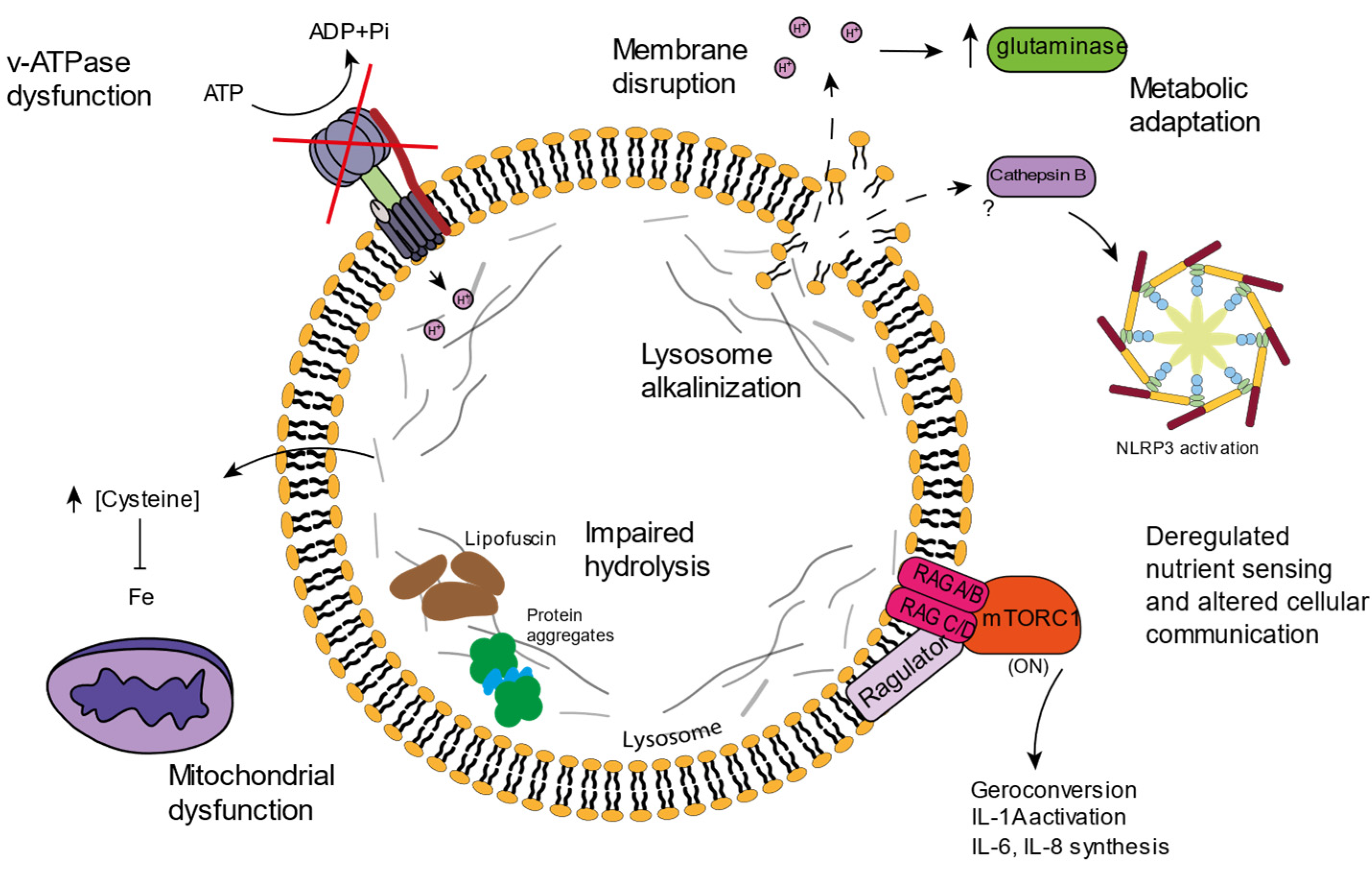
4. Lysosomal Age-Related Dysfunctions
4.1. v-ATPAse Dysfunction and Lysosomal Alkalinization
4.2. Lysosomal Amino Acid Storage and Ion Homeostasis
4.3. Lipofuscin
4.4. Inflammation and Cell Death
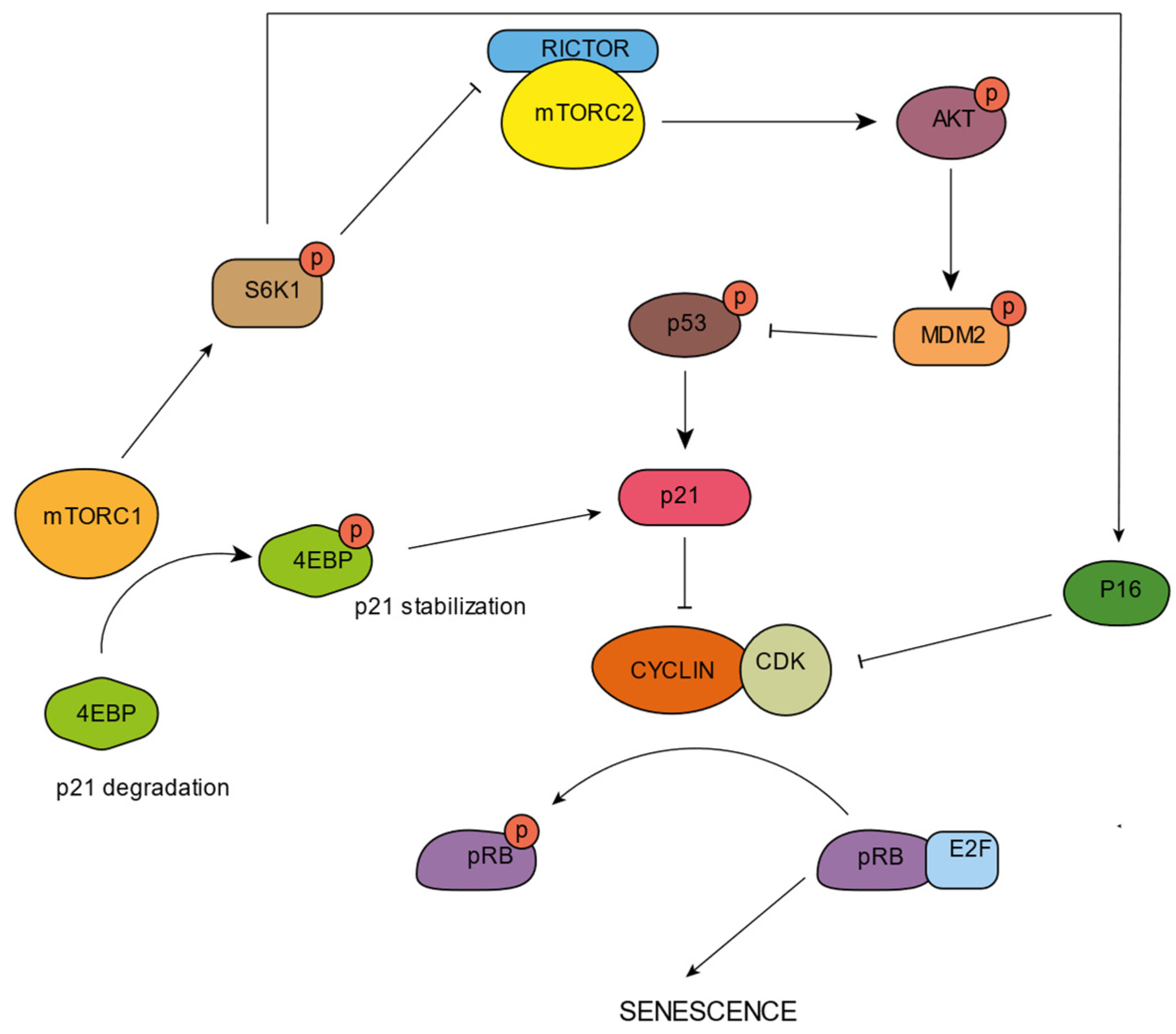
5. mTORC and Senescence
6. Lysosomal Opportunities for Intervention in Aging
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of Senescence and Aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and Molecular Mechanisms of Stress-Induced Premature Senescence (SIPS) of Human Diploid Fibroblasts and Melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-Order Unfolding of Satellite Heterochromatin Is a Consistent and Early Event in Cell Senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-Associated Beta-Galactosidase Is Lysosomal Beta-Galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Hughes, A.L.; Madeo, F.; Ruckenstuhl, C. The Crucial Impact of Lysosomes in Aging and Longevity. Ageing Res. Rev. 2016, 32, 2–12. [Google Scholar] [CrossRef]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic Lysosomal Depletion in Parkinson’s Disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Sintes, R.; Ledesma, M.D.; Boya, P. Lysosomal Cell Death Mechanisms in Aging. Ageing Res. Rev. 2016, 32, 150–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the Lysosome: A Control Centre for Cellular Clearance and Energy Metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Thelen, A.M.; Zoncu, R. Emerging Roles for the Lysosome in Lipid Metabolism. Trends Cell Biol. 2017, 27, 833. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Wada, K.; Kabuta, T. JB Special Review-Recent Topics in Ubiquitin-Proteasome System and Autophagy Lysosomal Degradation of Intracellular Nucleic Acids-Multiple Autophagic Pathways Overview of the Multiple Autophagic Pathways. J. Biochem. 2017, 161, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stütz, A.E.; Wrodnigg, T.M. Carbohydrate-Processing Enzymes of the Lysosome: Diseases Caused by Misfolded Mutants and Sugar Mimetics as Correcting Pharmacological Chaperones. Adv. Carbohydr. Chem. Biochem. 2016, 73, 225–302. [Google Scholar] [CrossRef]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, T.; Patel, S.; Eden, E.R. Calcium Signaling at ER Membrane Contact Sites. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2015, 1853, 2012–2017. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; di Paola, S.; Peluso, I.; Armani, A.; de Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal Calcium Signalling Regulates Autophagy through Calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Griukova, A.A.; Abushik, P.A.; Antonov, S.M.; Nikolsky, N.N.; Burova, E.B. Calcium Alterations Signal Either to Senescence or to Autophagy Induction in Stem Cells upon Oxidative Stress. Aging 2016, 8, 3400. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.T.Y.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ Signaling Occurs between the Endoplasmic Reticulum and Acidic Organelles. J. Cell Biol. 2013, 200, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; DiBenedetto, J.R.; West, M.; Rowland, A.A.; Voeltz, G.K. Endoplasmic Reticulum–Endosome Contact Increases as Endosomes Traffic and Mature. Mol. Biol. Cell 2013, 24, 1030. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, X. Lysosome Biogenesis: Regulation and Functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Peña-Llopis, S.; Vega-Rubin-De-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.T.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by MTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal V-ATPase-Ragulator Complex Is a Common Activator for AMPK and MTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; de Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Jürgen Klisch, T.; et al. TFEB Controls Cellular Lipid Metabolism through a Starvation-Induced Autoregulatory Loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy–Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein Kinase C Controls Lysosome Biogenesis Independently of MTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of MTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Bonifacino, J.S. Lysosome Positioning Influences MTORC2 and AKT Signaling. Mol. Cell 2019, 75, 26–38.e3. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; van de Vlekkert, D.; Wolf, E.; Finkelstein, D.; Neale, G.; Machado, E.; Mosca, R.; Campos, Y.; Tillman, H.; Roussel, M.F.; et al. MYC Competes with MiT/TFE in Regulating Lysosomal Biogenesis and Autophagy through an Epigenetic Rheostat. Nat. Commun. 2019, 10, 3623. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Elledge, S.J. How Autophagy Both Activates and Inhibits Cellular Senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and Senescence: A New Insight in Selected Human Diseases. J. Cell Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; Mcmahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the Switch in Early-to-Late Endosome Transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.Y.; Park, J.H.; You, S.T.; Lee, C.S.; Won, S.Y.; Park, J.J.; Kim, H.B.; Shim, J.; Soung, N.K.; Lee, O.J.; et al. Integrin-Mediated Adhesions in Regulation of Cellular Senescence. Sci. Adv. 2020, 6, eaay3909. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.Y.; Soung, N.K.; Schwartz, M.A.; Kim, E.G. Altered Endocytosis in Cellular Senescence. Ageing Res. Rev. 2021, 68, 101332. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome Formation from Membrane Compartments Enriched in Phosphatidylinositol 3-Phosphate and Dynamically Connected to the Endoplasmic Reticulum. J. Cell Biol. 2008, 182, 685. [Google Scholar] [CrossRef] [Green Version]
- Hilverling, A.; Szegö, E.M.; Dinter, E.; Cozma, D.; Saridaki, T.; Falkenburger, B.H. Maturing Autophagosomes Are Transported Towards the Cell Periphery. Cell Mol. Neurobiol. 2022, 42, 155–171. [Google Scholar] [CrossRef]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome Biogenesis: From Membrane Growth to Closure. J. Cell Biol. 2020, 219, e202002085. [Google Scholar] [CrossRef]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Macroautophagy Is Impaired in Old Murine Brain Tissue as Well as in Senescent Human Fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting Basal Levels of Autophagy in the Nervous System Enhances Longevity and Oxidant Resistance in Adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-Wide Analysis Reveals Mechanisms Modulating Autophagy in Normal Brain Aging and in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a Promoter of Longevity: Insights from Model. Nat. Rev. Mol. Cell Biol. 2018, 19, 579. [Google Scholar] [CrossRef]
- Kuma, A.; Komatsu, M.; Mizushima, N. Autophagy-Monitoring and Autophagy-Deficient Mice. Autophagy 2017, 13, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; de Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.J.; Narita, M. Autophagy Facilitates Oncogene-Induced Senescence. Autophagy 2009, 5, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle Communication between Mitochondria and the Endolysosomal System. Front. Cell Dev. Biol. 2017, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; P’Nguyen, A.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Mosquera, L.; DIogo, C.V.; Yambire, K.F.; Santos, G.L.; Luna Sánchez, M.; Bénit, P.; Rustin, P.; Lopez, L.C.; Milosevic, I.; Raimundo, N. Acute and Chronic Mitochondrial Respiratory Chain Deficiency Differentially Regulate Lysosomal Biogenesis. Sci. Rep. 2017, 7, 45076. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L.; Gottschling, D.E. An Early Age Increase in Vacuolar PH Limits Mitochondrial Function and Lifespan in Yeast. Nature 2012, 492, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Lee, S.H.; Bae, I.H.; Shin, D.W.; Min, D.; Ham, M.; Kim, K.H.; Lee, T.R.; Kim, H.J.; Son, E.D.; et al. Pyruvate Protects against Cellular Senescence through the Control of Mitochondrial and Lysosomal Function in Dermal Fibroblasts. J. Investig. Dermatol. 2018, 138, 2522–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilms, T.; Swinnen, E.; Eskes, E.; Dolz-Edo, L.; Uwineza, A.; van Essche, R.; Rosseels, J.; Zabrocki, P.; Cameroni, E.; Franssens, V.; et al. The Yeast Protein Kinase Sch9 Adjusts V-ATPase Assembly/Disassembly to Control PH Homeostasis and Longevity in Response to Glucose Availability. PLoS Genet. 2017, 13, e1006835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.v. Mitochondria and Iron: Current Questions. Expert Rev. Hematol. 2017, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef] [Green Version]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, H.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy Proteins Regulate the Secretory Component of Osteoclastic Bone Resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, A.K.; Ho, H.; Bodemann, B.; Petersen, S.; Aruri, J.; Koshy, S.; Richardson, Z.; Le, L.Q.; Krasieva, T.; Roth, M.G.; et al. Genome-Wide SiRNA-Based Functional Genomics of Pigmentation Identifies Novel Genes and Pathways That Impact Melanogenesis in Human Cells. PLoS Genet. 2008, 4, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Corrotte, M.; Castro-Gomes, T. Lysosomes and Plasma Membrane Repair. Curr. Top. Membr. 2019, 84, 1–16. [Google Scholar] [CrossRef]
- Tancini, B.; Buratta, S.; Delo, F.; Sagini, K.; Chiaradia, E.; Pellegrino, R.M.; Emiliani, C.; Urbanelli, L. Lysosomal Exocytosis: The Extracellular Role of an Intracellular Organelle. Membranes 2020, 10, 406. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal Accumulation of Anticancer Drugs Triggers Lysosomal Exocytosis. Oncotarget 2017, 8, 45117. [Google Scholar] [CrossRef] [Green Version]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [Green Version]
- Beck, M. The Link between Lysosomal Storage Disorders and More Common Diseases. J. Inborn Errors Metab. Screen. 2016, 4, 232640981668276. [Google Scholar] [CrossRef] [Green Version]
- Toledano-Zaragoza, A.; Ledesma, M.D. Addressing Neurodegeneration in Lysosomal Storage Disorders: Advances in Niemann Pick Diseases. Neuropharmacology 2020, 171, 107851. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of Lysosomal Acidification-the Emerging Role of v-ATPase in Aging and Neurodegenerative Disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.; Hamilton, G.; Wilkes, J.M.; Hutchinson, S.; Barrett, M.P.; Horn, D. Vacuolar ATPase Depletion Affects Mitochondrial ATPase Function, Kinetoplast Dependency, and Drug Sensitivity in Trypanosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 9112–9117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired Lysosomal Acidification Triggers Iron Deficiency and Inflammation in Vivo. eLife 2019, 8, e51031. [Google Scholar] [CrossRef]
- Sun, Y.; Li, M.; Zhao, D.; Li, X.; Yang, C.; Wang, X. Lysosome Activity Is Modulated by Multiple Longevity Pathways and Is Important for Lifespan Extension in C. elegans. eLife 2020, 9, e55745. [Google Scholar] [CrossRef]
- Baxi, K.; Ghavidel, A.; Waddell, B.; Harkness, T.A.; de Carvalho, C.E. Regulation of Lysosomal Function by the DAF-16 Forkhead Transcription Factor Couples Reproduction to Aging in Caenorhabditis Elegans. Genetics 2017, 207, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Folick, A.; Oakley, H.D.; Yu, Y.; Armstrong, E.H.; Kumari, M.; Sanor, L.; Moore, D.D.; Ortlund, E.A.; Zechner, R.; Wang, M.C. Lysosomal Signaling Molecules Regulate Longevity in Caenorhabditis Elegans. Science 2015, 347, 83. [Google Scholar] [CrossRef] [Green Version]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by Glutaminolysis Inhibition Ameliorates Various Age-Associated Disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef]
- Simonaro, C.M. Lysosomes, Lysosomal Storage Diseases, and Inflammation. J. Inborn Errors Metab. Screen. 2016, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.E.; Coody, T.K.; Jeong, M.Y.; Berg, J.A.; Winge, D.R.; Hughes, A.L. Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 2020, 180, 296–310.e18. [Google Scholar] [CrossRef]
- Rizzollo, F.; More, S.; Vangheluwe, P.; Agostinis, P. The Lysosome as a Master Regulator of Iron Metabolism. Trends Biochem. Sci. 2021, 46, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, R.S.; Han, S.M.; Leeuwenburgh, C.; Xiao, R. Iron Homeostasis and Organismal Aging. Ageing Res. Rev. 2021, 72, 101510. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Adourian, A.S.; Freiberg, J.J.; Guo, Y.; Muntendam, P.; Falk, E. Risk Factors for Near-Term Myocardial Infarction in Apparently Healthy Men and Women. Clin. Chem. 2010, 56, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, S.L.; Ritz, B. Genetics of Iron Regulation and the Possible Role of Iron in Parkinson’s Disease. Neurobiol. Dis. 2008, 32, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Lei, P. Nigral Iron Elevation Is an Invariable Feature of Parkinson’s Disease and Is a Sufficient Cause of Neurodegeneration. BioMed Res. Int. 2014, 2014, 581256. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Killilea, D.W.; Atamna, H.; Liao, C.; Ames, B.N. Iron Accumulation during Cellular Senescence in Human Fibroblasts in Vitro. Antioxid. Redox Signal. 2003, 5, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron Accumulation in Senescent Cells Is Coupled with Impaired Ferritinophagy and Inhibition of Ferroptosis. Redox Biol. 2018, 14, 100. [Google Scholar] [CrossRef]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of Ferroptosis in Aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef]
- Sasikumar, A.N.; Killilea, D.W.; Kennedy, B.K.; Brem, R.B. Potassium Restriction Boosts Vacuolar Acidity and Extends Lifespan in Yeast. Exp. Gerontol. 2019, 120, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-Bound Iron Is a Major Intracellular Source of Oxidants: Role in Senescent Cells. Free Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Grune, T. Lipofuscin: Formation, Effects and Role of Macroautophagy. Redox Biol. 2013, 1, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid. Redox Signal. 2015, 23, 239. [Google Scholar] [CrossRef] [Green Version]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef]
- Pan, C.; Banerjee, K.; Lehmann, G.L.; Almeida, D.; Hajjar, K.A.; Benedicto, I.; Jiang, Z.; Radu, R.A.; Thompson, D.H.; Rodriguez-Boulan, E.; et al. Lipofuscin Causes Atypical Necroptosis through Lysosomal Membrane Permeabilization. Proc. Natl. Acad. Sci. USA 2021, 118, e2100122118. [Google Scholar] [CrossRef]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Front. Cell Dev. Biol. 2020, 8, 167. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Yao, R.Q.; Li, Y.; Zhao, P.; Ren, C.; Du, X.; Yao, Y.-M. Lysosomal Quality Control of Cell Fate: A Novel Therapeutic Target for Human Diseases. Cell Death Dis. 2020, 11, 817. [Google Scholar] [CrossRef]
- Zhu, X.; Sun, Y.; Chen, D.; Li, J.; Dong, X.; Wang, J.; Chen, H.; Wang, Y.; Zhang, F.; Dai, J.; et al. Mastocarcinoma Therapy Synergistically Promoted by Lysosome Dependent Apoptosis Specifically Evoked by 5-Fu@nanogel System with Passive Targeting and PH Activatable Dual Function. J. Control. Release 2017, 254, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ding, Y.; Zhong, L.; Jiang, L.; Geng, C.; Yao, X.; Cao, J. Tacrine Induces Apoptosis through Lysosome- and Mitochondria-Dependent Pathway in HepG2 Cells. Toxicol. Vitr. 2014, 28, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Y.; Choi, H.M.C.; Sarkar, C.; Koh, E.Y.; Wu, J.; Lipinski, M.M. Lysosomal Damage after Spinal Cord Injury Causes Accumulation of RIPK1 and RIPK3 Proteins and Potentiation of Necroptosis. Cell Death Dis. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Kawai, T.; Tsuchida, T.; Kozaki, T.; Tanaka, H.; Shin, K.S.; Kumar, H.; Akira, S. Poly IC Triggers a Cathepsin D- and IPS-1-Dependent Pathway to Enhance Cytokine Production and Mediate Dendritic Cell Necroptosis. Immunity 2013, 38, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Bai, Y.; Jia, Y.; Zhao, Y.; Kang, R.; Tang, D.; Dai, E. Ferroptosis Is a Lysosomal Cell Death Process. Biochem. Biophys. Res. Commun. 2018, 503, 1550–1556. [Google Scholar] [CrossRef]
- Chi, H.; Chang, H.-Y.; Sang, T.-K. Molecular Sciences Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Karch, J. Regulation of Cell Death in the Cardiovascular System. Int. Rev. Cell Mol. Biol. 2020, 353, 153–209. [Google Scholar] [CrossRef]
- Nacarelli, T.; Azar, A.; Sell, C. Aberrant MTOR Activation in Senescence and Aging: A Mitochondrial Stress Response? Exp. Gerontol. 2015, 68, 66. [Google Scholar] [CrossRef] [Green Version]
- Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. MTOR and Aging: An Old Fashioned Dress. Int. J. Mol. Sci. 2019, 20, 2774. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hoff, H.; Marinucci, T.; Cristofalo, V.J.; Sell, C. Mitogen-Independent Phosphorylation of S6K1 and Decreased Ribosomal S6 Phosphorylation in Senescent Human Fibroblasts. Exp. Cell Res. 2000, 259, 284–292. [Google Scholar] [CrossRef]
- Barilari, M.; Bonfils, G.; Treins, C.; Koka, V.; de Villeneuve, D.; Fabrega, S.; Pende, M. ZRF1 Is a Novel S6 Kinase Substrate That Drives the Senescence Programme. EMBO J. 2017, 36, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.M.; Pallafacchina, G.; Paoli, A.; et al. Signalling Pathways Regulating Muscle Mass in Ageing Skeletal Muscle. The Role of the IGF1-Akt-MTOR-FoxO Pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Ji, X.; Mao, X.; Xie, L.; Jia, J.; Galvan, V.; Greenberg, D.A.; Jin, K. Differential Activation of MTOR Complex 1 Signaling in Human Brain with Mild to Severe Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 38, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nature Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.J.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial Coupling of MTOR and Autophagy Augments Secretory Phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [Green Version]
- Khor, E.S.; Wong, P.F. The Roles of MTOR and MiRNAs in Endothelial Cell Senescence. Biogerontology 2020, 21, 517–530. [Google Scholar] [CrossRef]
- Lee, C.H.; Inoki, K.; Karbowniczek, M.; Petroulakis, E.; Sonenberg, N.; Henske, E.P.; Guan, K.L. Constitutive MTOR Activation in TSC Mutants Sensitizes Cells to Energy Starvation and Genomic Damage via P53. EMBO J. 2007, 26, 4812–4823. [Google Scholar] [CrossRef] [Green Version]
- Khor, E.S.; Wong, P.F. Endothelial Replicative Senescence Delayed by the Inhibition of MTORC1 Signaling Involves MicroRNA-107. Int. J. Biochem. Cell Biol. 2018, 101, 64–73. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A Novel Type of Cellular Senescence That Can Be Enhanced in Mouse Models and Human Tumor Xenografts to Suppress Prostate Tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; Jeong, D.; Lee, K.; Park, H.J.; Ko, Y.G.; Lee, J.S. mTOR Kinase Leads to PTEN-Loss-Induced Cellular Senescence by Phosphorylating P53. Oncogene 2019, 38, 1639–1650. [Google Scholar] [CrossRef]
- Lai, K.P.; Leong, W.F.; Chau, J.F.L.; Jia, D.; Zeng, L.; Liu, H.; He, L.; Hao, A.; Zhang, H.; Meek, D.; et al. S6K1 Is a Multifaceted Regulator of Mdm2 That Connects Nutrient Status and DNA Damage Response. EMBO J. 2010, 29, 2994–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, L.-A.; Carriere, A.; Moreau, J.; Roux, P.P. MTORC1-Activated S6K1 Phosphorylates Rictor on Threonine 1135 and Regulates MTORC2 Signaling. Mol. Cell Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt Enhances Mdm2-Mediated Ubiquitination and Degradation of P53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanos, S.; García-Pedrero, J.M.; Morgado-Palacin, L.; Rodrigo, J.P.; Serrano, M. ARTICLE Stabilization of P21 by MTORC1/4E-BP1 Predicts Clinical Outcome of Head and Neck Cancers. Nat. Commun. 2016, 7, 10438. [Google Scholar] [CrossRef]
- Nakano, M.; Nakashima, A.; Nagano, T.; Ishikawa, S.; Kikkawa, U. Branched-Chain Amino Acids Enhance Premature Senescence through Mammalian Target of Rapamycin Complex I-Mediated Upregulation of P21 Protein. PLoS ONE 2013, 8, e80411. [Google Scholar] [CrossRef] [Green Version]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT Induces Senescence in Human Cells via MTORC1 and P53 in the Absence of DNA Damage: Implications for Targeting MTOR during Malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef] [Green Version]
- Kucheryavenko, O.; Nelson, G.; von Zglinicki, T.; Korolchuk, V.I.; Carroll, B. The MTORC1-Autophagy Pathway Is a Target for Senescent Cell Elimination. Biogerontology 2019, 20, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef]
- Demidenko, Z.N.; Zubova, S.G.; Bukreeva, E.I.; Pospelov, V.A.; Pospelova, T.V.; Blagosklonny, M.v. Rapamycin Decelerates Cellular Senescence. Cell Cycle 2009, 8, 1888–1895. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Demidenko, Z.N.; Blagosklonny, M.v. Contact Inhibition and High Cell Density Deactivate the Mammalian Target of Rapamycin Pathway, Thus Suppressing the Senescence Program. Proc. Natl. Acad. Sci. USA 2014, 111, 8832–8837. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.E.; Strong, R.; Dave Sharp, Z.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Erby Wilkinson, J.; Frenkel, K.; Carter, C.S.; et al. Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR Kinase on Lifespan in C. Elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Withers, D.J. Mammalian Models of Extended Healthy Lifespan. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.I.; Chen, W.; Gao, Q.I.; Yang, J.I.; Yan, X.; Zhao, H.; Su, L.; Yang, M.; GaoID, C.; Yao, Y.; et al. Rapamycin Directly Activates Lysosomal Mucolipin TRP Channels Independent of MTOR. PLoS Biol. 2019, 17, e3000252. [Google Scholar] [CrossRef] [PubMed]
- Boudewyn, L.C.; Walkley, S.U. Current Concepts in the Neuropathogenesis of Mucolipidosis Type IV. J. Neurochem. 2019, 148, 669–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.V.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; la Spada, A.R. PGC-1α Rescues Huntington’s Disease Proteotoxicity by Preventing Oxidative Stress and Promoting TFEB Function. Sci. Transl. Med. 2012, 4, 142ra97. [Google Scholar] [CrossRef] [Green Version]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Björklund, A. TFEB-Mediated Autophagy Rescues Midbrain Dopamine Neurons from α-Synuclein Toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A Novel Curcumin Analog Binds to and Activates TFEB in Vitro and in Vivo Independent of MTOR Inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Kilpatrick, K.; Zeng, Y.; Hancock, T.; Segatori, L. Genetic and Chemical Activation of TFEB Mediates Clearance of Aggregated α-Synuclein. PLoS ONE 2015, 10, e0120819. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hodge, J.; Liu, Q.; Wang, J.; Wang, Y.; Evans, T.D.; Altomare, D.; Yao, Y.; Murphy, E.A.; Razani, B.; et al. TFEB Is a Master Regulator of Tumor-Associated Macrophages in Breast Cancer. J. Immunother. Cancer 2020, 8, 543. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Cho, Y.R.; Lee, S.Y.; Sung, G.J.; Shin, D.M.; Choi, K.C.; Son, J. TFEB Supports Pancreatic Cancer Growth through the Transcriptional Regulation of Glutaminase. Cancers 2021, 13, 483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhuo, Y.; Wu, S.; Chen, Y.; Ye, J.; Deng, Y.; Feng, Y.; Liu, R.; Cai, S.; Zou, Z.; et al. TFEB Promotes Prostate Cancer Progression via Regulating ABCA2-Dependent Lysosomal Biogenesis. Front. Oncol. 2021, 11, 236. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of Senescent Cells by β-Galactosidase-Targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero-Navarro, L.; Jansen-Dürr, P.; Cavinato, M. Age-Related Lysosomal Dysfunctions. Cells 2022, 11, 1977. https://doi.org/10.3390/cells11121977
Guerrero-Navarro L, Jansen-Dürr P, Cavinato M. Age-Related Lysosomal Dysfunctions. Cells. 2022; 11(12):1977. https://doi.org/10.3390/cells11121977
Chicago/Turabian StyleGuerrero-Navarro, Lena, Pidder Jansen-Dürr, and Maria Cavinato. 2022. "Age-Related Lysosomal Dysfunctions" Cells 11, no. 12: 1977. https://doi.org/10.3390/cells11121977
APA StyleGuerrero-Navarro, L., Jansen-Dürr, P., & Cavinato, M. (2022). Age-Related Lysosomal Dysfunctions. Cells, 11(12), 1977. https://doi.org/10.3390/cells11121977