
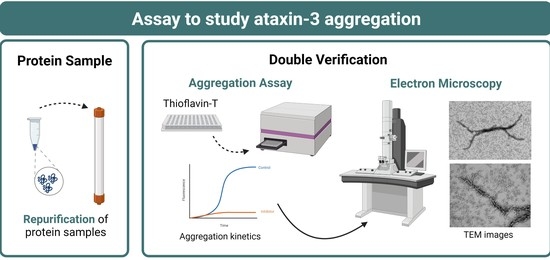
A Robust Assay to Monitor Ataxin-3 Amyloid Fibril Assembly

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Expression Plasmids
2.2. Ataxin-3 Expression and Purification
2.3. HsUBB Expression and Purification
2.4. Thioflavin-T Aggregation Assay
2.5. Transmission Electron Microscopy
2.6. Filter Retardation Assay
2.7. Native Gel Assay
2.8. Ataxin-3 13Q and Ataxin-3 JD Oligomerization Kinetics Measured via SwitchSENSE
3. Results
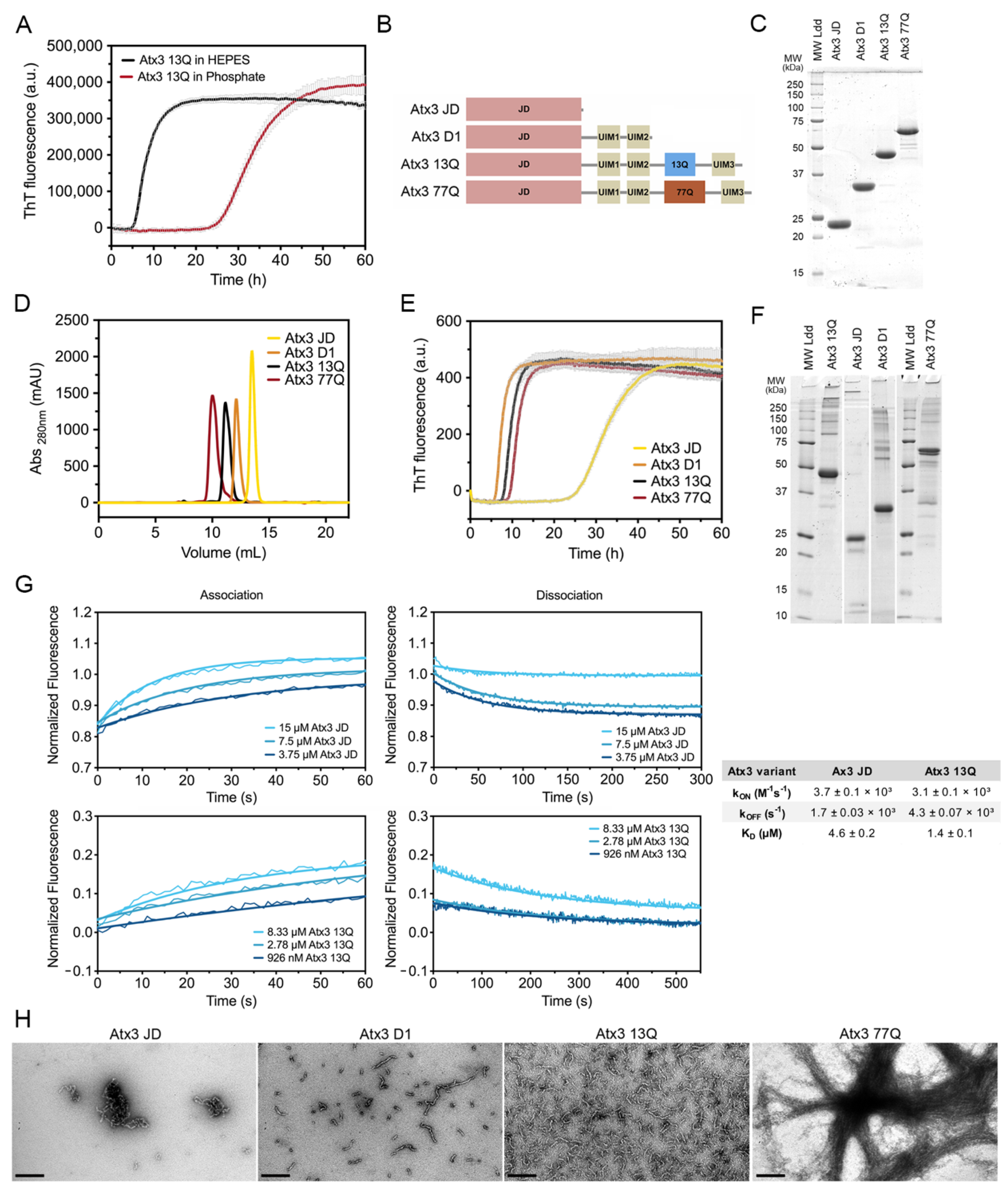
3.1. Development of a Miniaturized Ataxin-3 Aggregation Assay
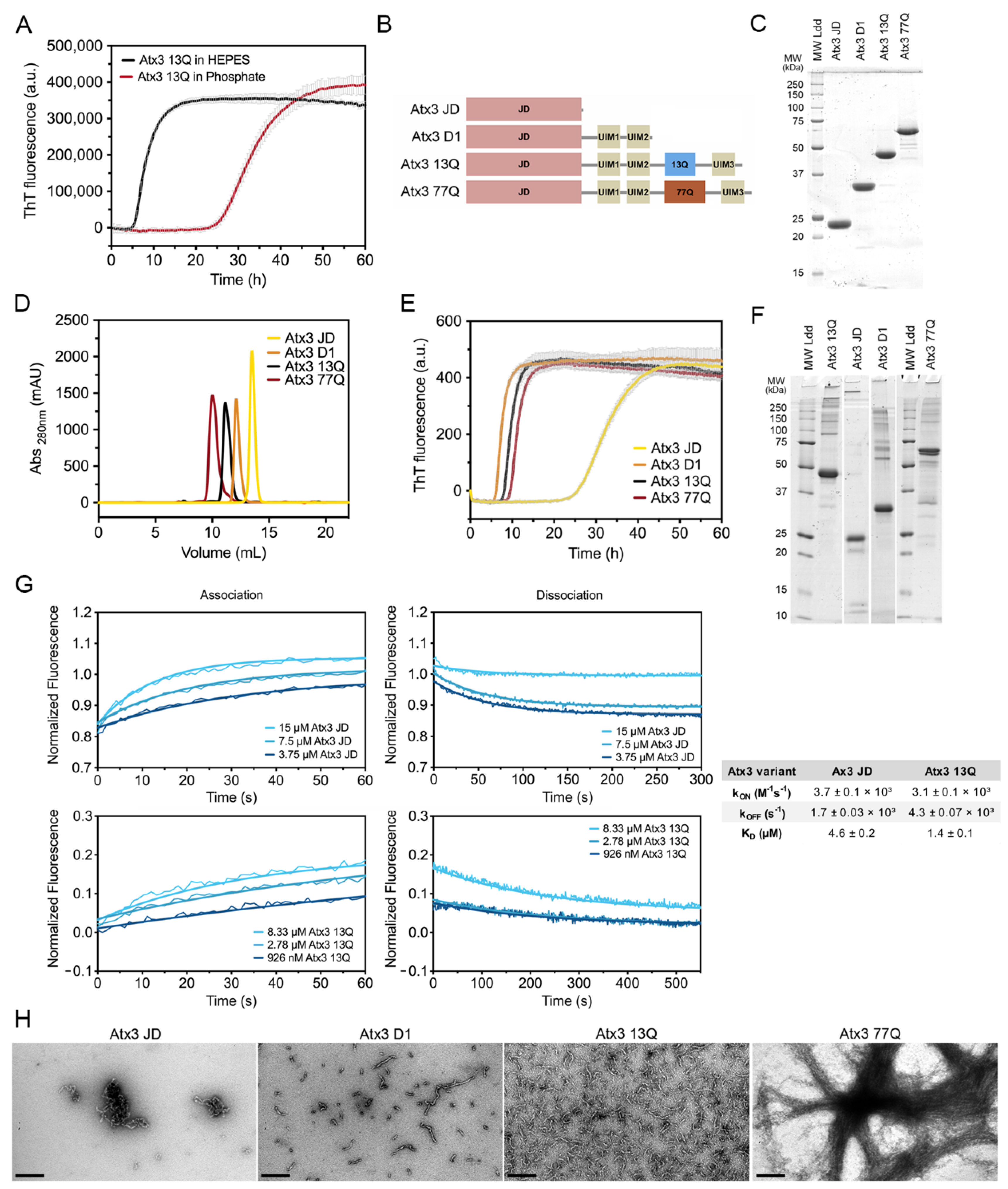
3.2. Analysis of the Aggregation Kinetics of Natural Ataxin-3 Variants
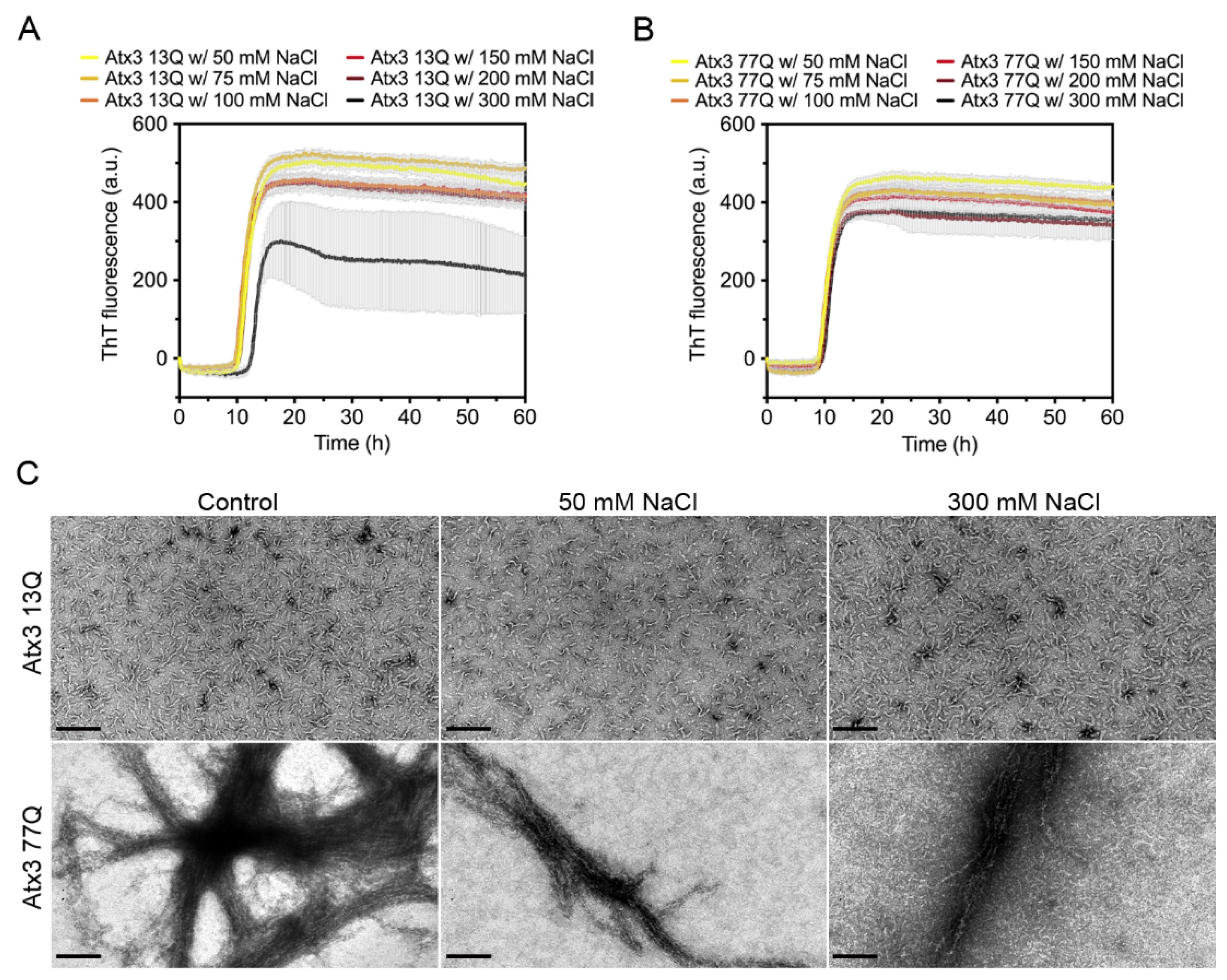
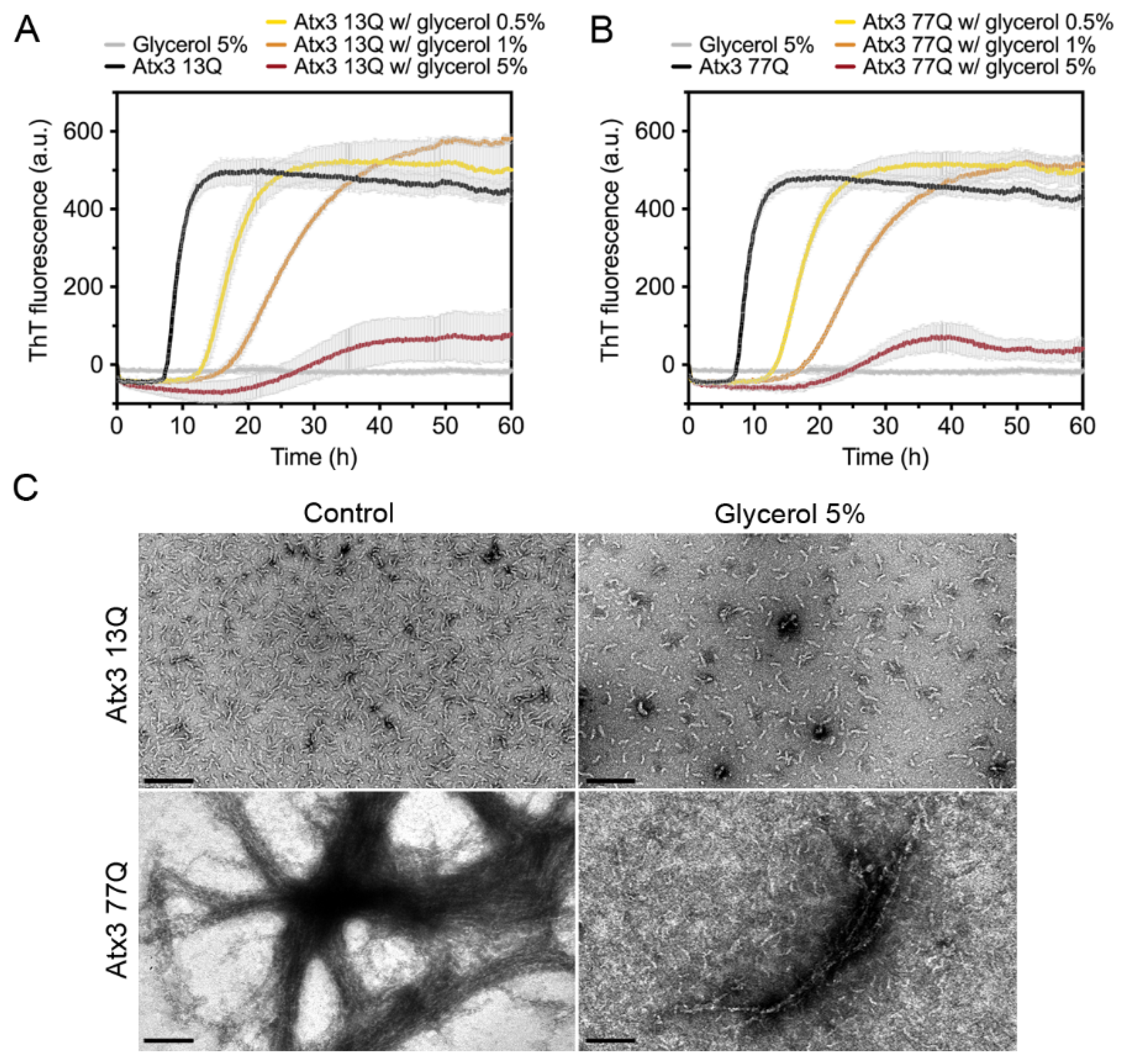
3.3. The Influence of the Buffer Components on Ataxin-3 Aggregation
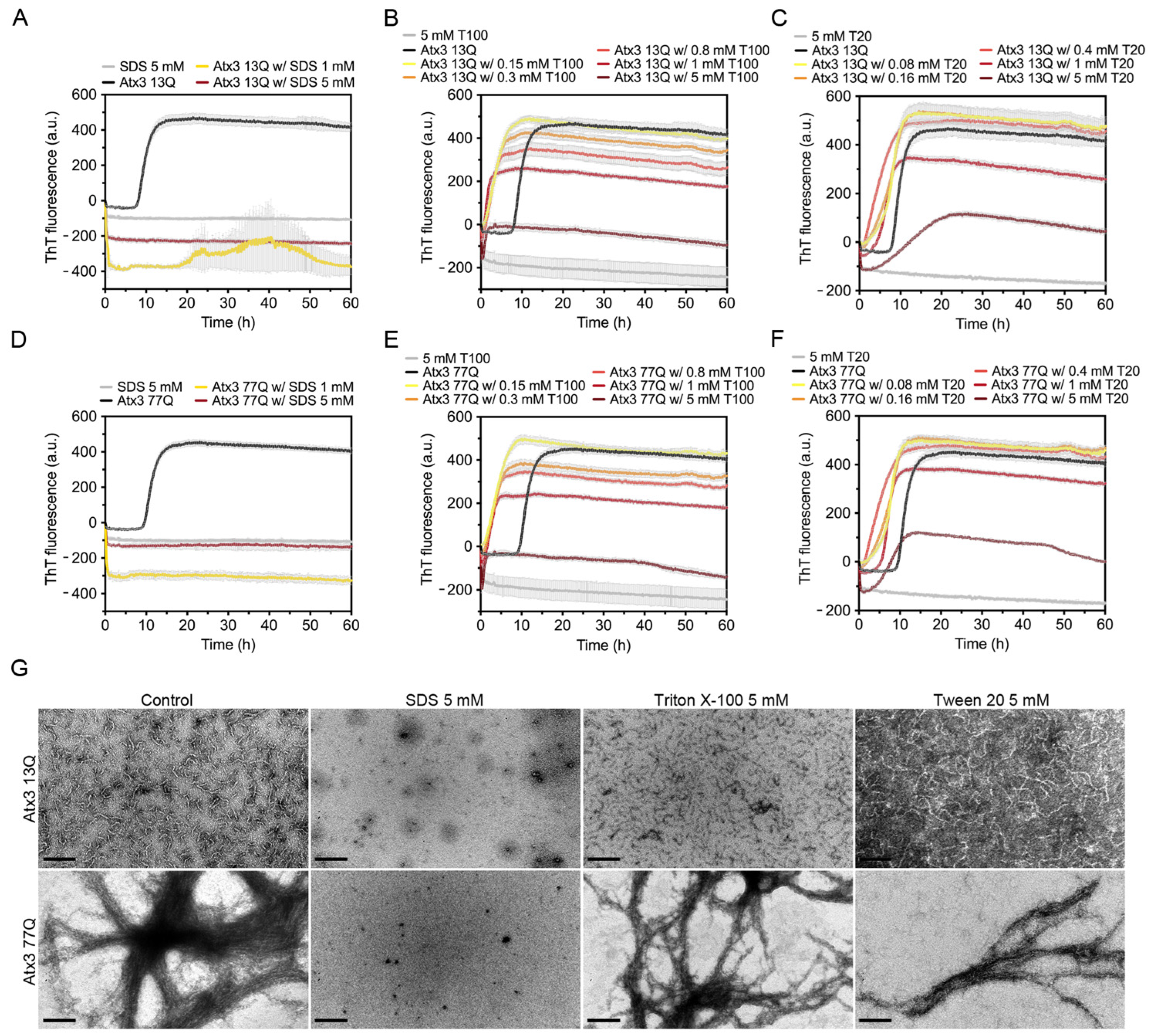
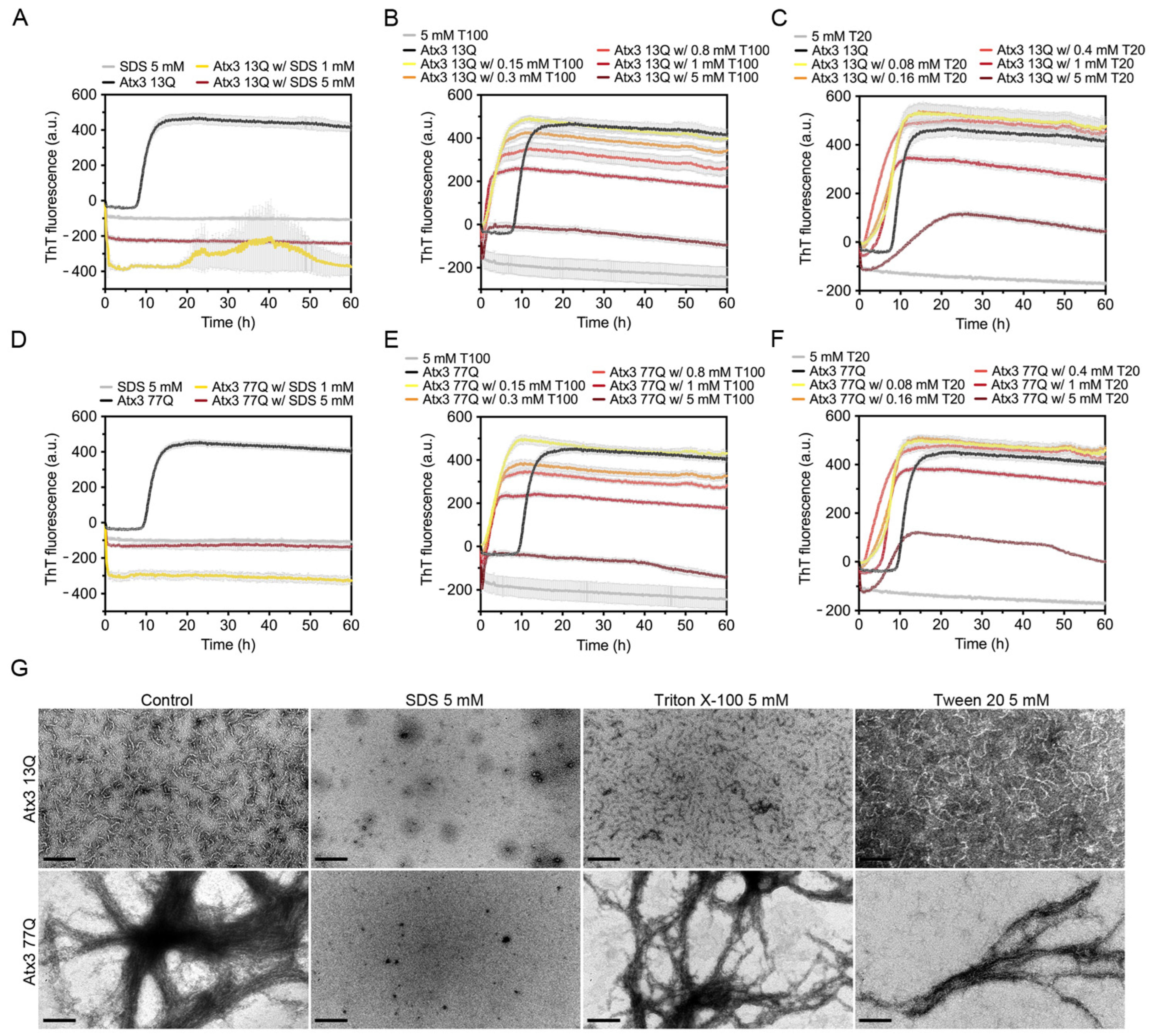
3.4. The Influence of Detergents on Ataxin-3 Aggregation
3.5. The Influence of Molecular Crowders on Ataxin-3 Aggregation
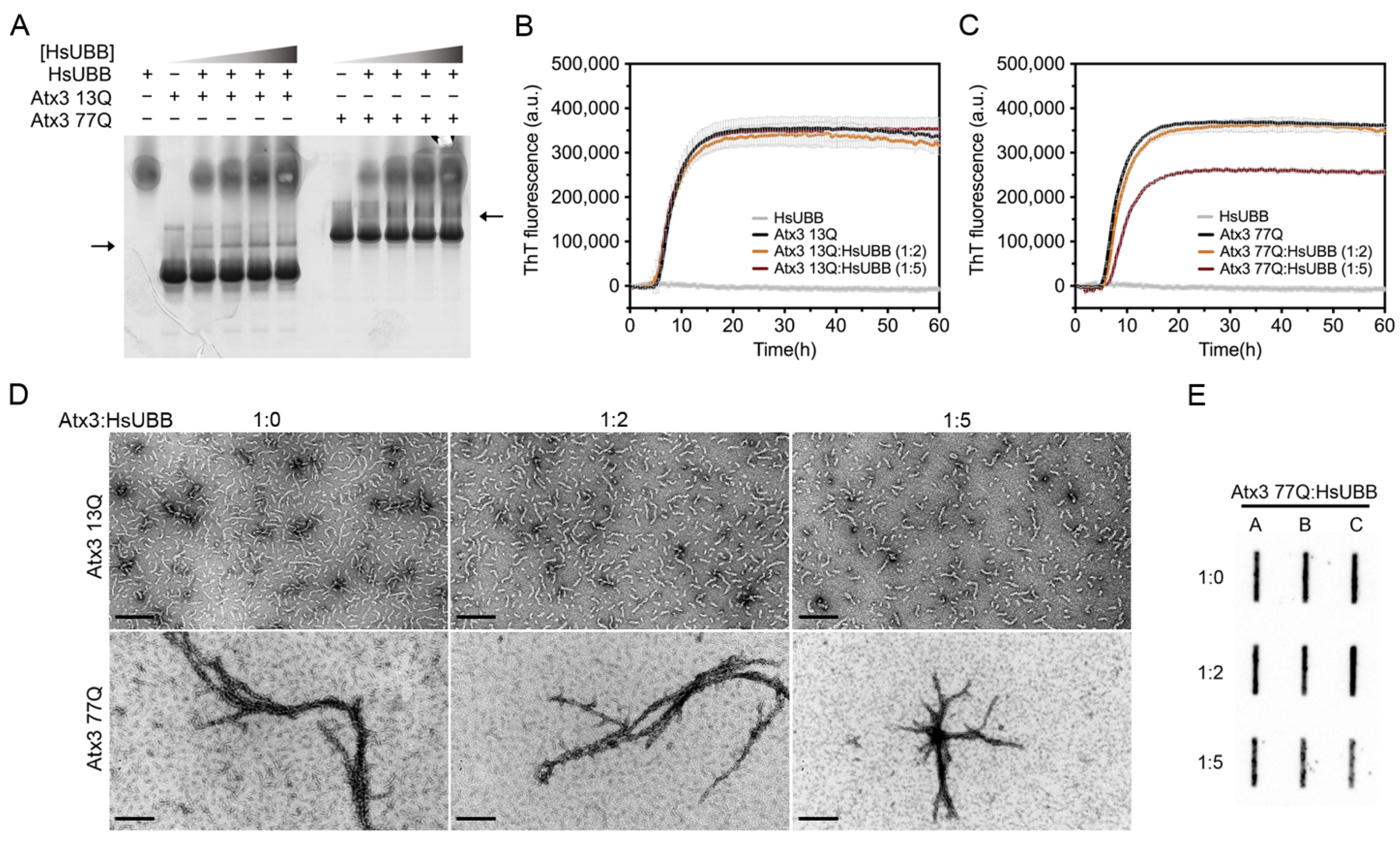
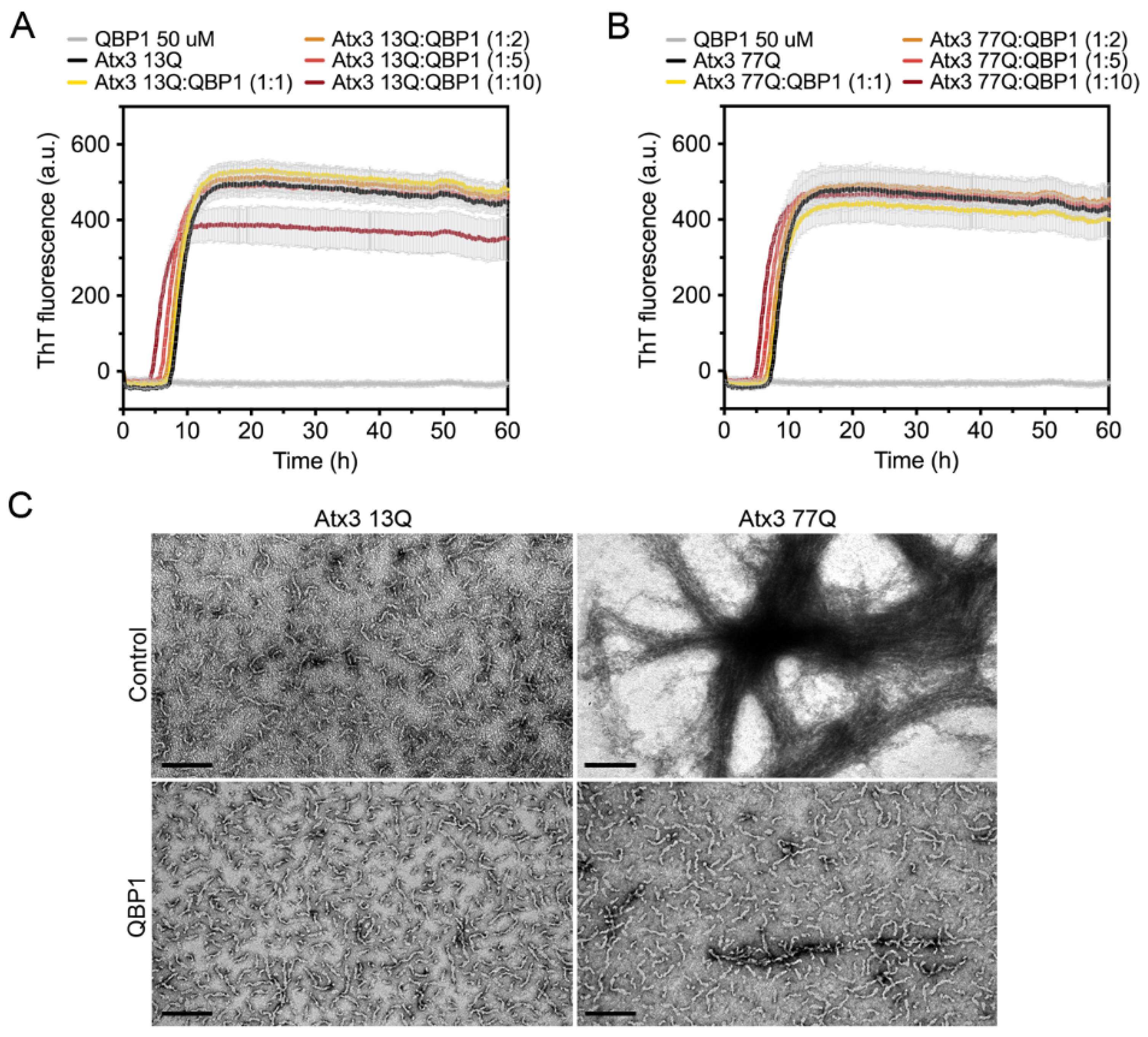
3.6. The Influence of Ataxin-3 Interactors on Ataxin-3 Aggregation
4. Discussion
4.1. The Relevance of the Buffer System for Ataxin-3 Aggregation
4.2. The Role of Additives on Ataxin-3 Aggregation
4.3. The Effect of Detergents on Ataxin-3 Aggregation Monitored by ThT
4.4. Molecular Crowders and Ataxin-3 Aggregation
4.5. The Effect of Ataxin-3-Interacting Proteins in Amyloid Assembly
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGowan, D.; Van Roon-Mom, W.; Holloway, H.; Bates, G.; Mangiarini, L.; Cooper, G.; Faull, R.; Snell, R. Amyloid-like inclusions in Huntington’s disease. Neuroscience 2000, 100, 677–680. [Google Scholar] [CrossRef]
- Paulson, H.L.; Perez, M.; Trottier, Y.; Trojanowski, J.; Subramony, S.; Das, S.; Vig, P.; Mandel, J.-L.; Fischbeck, K.; Pittman, R. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997, 19, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Seidel, K.; Meister, M.; Dugbartey, G.J.; Zijlstra, M.P.; Vinet, J.; Brunt, E.R.; Van Leeuwen, F.W.; Rub, U.; Kampinga, H.H.; Den Dunnen, W.F. Cellular protein quality control and the evolution of aggregates in spinocerebellar ataxia type 3 (SCA3). Neuropatho. Appl. Neurobiol. 2012, 38, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Seidel, K.; Siswanto, S.; Fredrich, M.; Bouzrou, M.; Den Dunnen, W.F.A.; Ozerden, I.; Korf, H.W.; Melegh, B.; De Vries, J.J.; Brunt, E.R.; et al. On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with spinocerebellar ataxia type 2 and 3. Brain Pathol. 2017, 27, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ferrone, F.A.; Wetzel, R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. USA 2002, 99, 11884–11889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciel, P.; Gaspar, C.; DeStefano, A.L.; Silveira, I.; Coutinho, P.; Radvany, J.; Dawson, D.M.; Sudarsky, L.; Guimarães, J.; Loureiro, J.E. Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am. J. Hum. Genet. 1995, 57, 54. [Google Scholar]
- França, M.C., Jr.; Emmel, V.E.; D’Abreu, A.; Maurer-Morelli, C.V.; Secolin, R.; Bonadia, L.C.; Silva, M.S.; Nucci, A.; Jardim, L.B.; Saraiva-Pereira, M.L. Normal ATXN3 allele but not CHIP polymorphisms modulates age at onset in Machado–Joseph disease. Front. Neurol. 2012, 3, 164. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.D.C. Recent therapeutic prospects for Machado-Joseph disease. Curr. Opin. Neurol. 2020, 33, 519–526. [Google Scholar] [CrossRef]
- Minakawa, E.N.; Nagai, Y. Protein Aggregation Inhibitors as Disease-Modifying Therapies for Polyglutamine Diseases. Front. Neurosci. 2021, 15, 621996. [Google Scholar] [CrossRef]
- Gales, L.; Cortes, L.; Almeida, C.; Melo, C.V.; Do Carmo Costa, M.; Maciel, P.; Clarke, D.T.; Damas, A.M.; Macedo-Ribeiro, S. Towards a structural understanding of the fibrillization pathway in Machado-Joseph’s disease: Trapping early oligomers of non-expanded ataxin-3. J. Mol. Biol. 2005, 353, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Scarff, C.A.; Sicorello, A.; Tomé, R.J.; Macedo-Ribeiro, S.; Ashcroft, A.E.; Radford, S.E. A tale of a tail: Structural insights into the conformational properties of the polyglutamine protein ataxin-3. Int. J. Mass Spectrom. 2013, 345, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masino, L.; Nicastro, G.; De Simone, A.; Calder, L.; Molloy, J.; Pastore, A. The Josephin domain determines the morphological and mechanical properties of ataxin-3 fibrils. Biophys. J. 2011, 100, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masino, L.; Nicastro, G.; Menon, R.P.; Dal Piaz, F.; Calder, L.; Pastore, A. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J. Mol. Biol. 2004, 344, 1021–1035. [Google Scholar] [CrossRef]
- Ellisdon, A.M.; Pearce, M.C.; Bottomley, S.P. Mechanisms of ataxin-3 misfolding and fibril formation: Kinetic analysis of a disease-associated polyglutamine protein. J. Mol. Biol. 2007, 368, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Scarff, C.A.; Almeida, B.; Fraga, J.; Macedo-Ribeiro, S.; Radford, S.E.; Ashcroft, A.E. Examination of ataxin-3 (atx-3) aggregation by structural mass spectrometry techniques: A rationale for expedited aggregation upon polyglutamine (polyQ) expansion. Mol. Cell. Proteomics 2015, 14, 1241–1253. [Google Scholar] [CrossRef] [Green Version]
- Martins, P.M.; Navarro, S.; Silva, A.; Pinto, M.F.; Sárkány, Z.; Figueiredo, F.; Pereira, P.J.B.; Pinheiro, F.; Bednarikova, Z.; Burdukiewicz, M. MIRRAGGE–minimum information required for reproducible AGGregation experiments. Front. Mol. Neurosci. 2020, 222, 582488. [Google Scholar] [CrossRef]
- Silva, A.; Almeida, B.; Fraga, J.S.; Taboada, P.; Martins, P.M.; Macedo-Ribeiro, S. Distribution of Amyloid-Like and Oligomeric Species from Protein Aggregation Kinetics. Angew. Chem. Int. Ed. 2017, 56, 14042–14045. [Google Scholar] [CrossRef]
- Knight, P.D.; Karamanos, T.K.; Radford, S.E.; Ashcroft, A.E. Identification of a novel site of interaction between ataxin-3 and the amyloid aggregation inhibitor polyglutamine binding peptide 1. Eur. J. Mass Spectrom. 2018, 24, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armen, R.S.; Bernard, B.M.; Day, R.; Alonso, D.O.; Daggett, V. Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 13433–13438. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Tucker, T.; Ren, H.; Kenan, D.J.; Henderson, B.S.; Keene, J.D.; Strittmatter, W.J.; Burke, J.R. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J. Biol. Chem. 2000, 275, 10437–10442. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Popiel, H.A.; Nagai, Y.; Toda, T.; Yoshimitsu, Y.; Ohno, H.; Oishi, S.; Fujii, N. Structure-activity relationship study on polyglutamine binding peptide QBP1. Bioorg. Med. Chem. 2009, 17, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Carvalho, A.L.; Teixeira-Castro, A.; Almeida, C.; Tomé, R.J.; Cortes, L.; Rodrigues, A.-J.; Logarinho, E.; Sequeiros, J.; Macedo-Ribeiro, S.; et al. NEDD8: A new ataxin-3 interactor. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1619–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grou, C.P.; Pinto, M.P.; Mendes, A.V.; Domingues, P.; Azevedo, J.E. The de novo synthesis of ubiquitin: Identification of deubiquitinases acting on ubiquitin precursors. Sci. Rep. 2015, 5, 12836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rames, M.; Yu, Y.; Ren, G. Optimized negative staining: A high-throughput protocol for examining small and asymmetric protein structure by electron microscopy. JoVE 2014, 15, e51087. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; De Almeida, A.V.; Macedo-Ribeiro, S. Polyglutamine expansion diseases: More than simple repeats. J. Struct. Biol. 2018, 201, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.K.; Paulson, H.L.; Bottomley, S.P. Destabilization of a non-pathological variant of ataxin-3 results in fibrillogenesis via a partially folded intermediate: A model for misfolding in polyglutamine disease. J. Mol. Biol. 2004, 335, 333–341. [Google Scholar] [CrossRef]
- Ellisdon, A.M.; Thomas, B.; Bottomley, S.P. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J. Biol. Chem. 2006, 281, 16888–16896. [Google Scholar] [CrossRef] [Green Version]
- Knezevic, J.; Langer, A.; Hampel, P.A.; Kaiser, W.; Strasser, R.; Rant, U. Quantitation of affinity, avidity, and binding kinetics of protein analytes with a dynamically switchable biosurface. J. Am. Chem. Soc. 2012, 134, 15225–15228. [Google Scholar] [CrossRef] [PubMed]
- Müller-Landau, H.; Varela, P.F. Standard operation procedure for switchSENSE DRX systems. Eur. Biophys. J. 2021, 50, 389–400. [Google Scholar] [CrossRef]
- Santambrogio, C.; Frana, A.M.; Natalello, A.; Papaleo, E.; Regonesi, M.E.; Doglia, S.M.; Tortora, P.; Invernizzi, G.; Grandori, R. The role of the central flexible region on the aggregation and conformational properties of human ataxin-3. FEBS J. 2012, 279, 451–463. [Google Scholar] [CrossRef]
- Lupton, C.J.; Steer, D.L.; Wintrode, P.L.; Bottomley, S.P.; Hughes, V.A.; Ellisdon, A.M. Enhanced molecular mobility of ordinarily structured regions drives polyglutamine disease. J. Biol. Chem. 2015, 290, 24190–24200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, C.; Lopes-Cendes, I.; Hayes, S.; Goto, J.; Arvidsson, K.; Dias, A.; Silveira, I.; Maciel, P.; Coutinho, P.; Lima, M. Ancestral origins of the Machado-Joseph disease mutation: A worldwide haplotype study. Am. J. Hum. Genet. 2001, 68, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Martins, S.; Peng, Y.; Wang, P.; Hou, X.; Chen, Z.; Wang, C.; Tang, Z.; Qiu, R.; Chen, C.; et al. Is the High Frequency of Machado-Joseph Disease in China Due to New Mutational Origins? Front. Genet. 2018, 9, 740. [Google Scholar] [CrossRef] [Green Version]
- Sharony, R.; Martins, S.; Costa, I.P.D.; Zaltzman, R.; Amorim, A.; Sequeiros, J.; Gordon, C.R. Yemenite-Jewish families with Machado-Joseph disease (MJD/SCA3) share a recent common ancestor. Eur. J. Hum. Genet. 2019, 27, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Goto, J.; Watanabe, M.; Ichikawa, Y.; Yee, S.-B.; Ihara, N.; Endo, K.; Igarashi, S.; Takiyama, Y.; Gaspar, C.; Maciel, P. Machado–Joseph disease gene products carrying different carboxyl termini. Neurosci. Res. 1997, 28, 373–377. [Google Scholar] [CrossRef]
- Flynn, J.D.; McGlinchey, R.P.; Walker, R.L.; Lee, J.C. Structural features of α-synuclein amyloid fibrils revealed by Raman spectroscopy. J. Biol. Chem. 2018, 293, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, J.; Schroer, M.A.; Erlkamp, M.; Grobelny, S.; Paulus, M.; Tiemeyer, S.; Wirkert, F.J.; Tolan, M.; Winter, R. The effect of ionic strength, temperature, and pressure on the interaction potential of dense protein solutions: From nonlinear pressure response to protein crystallization. Biophys. J. 2012, 102, 2641–2648. [Google Scholar] [CrossRef] [Green Version]
- Munishkina, L.A.; Henriques, J.; Uversky, V.N.; Fink, A.L. Role of protein− water interactions and electrostatics in α-synuclein fibril formation. Biochemistry 2004, 43, 3289–3300. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int. J. Mol. Sci. 2021, 22, 12382. [Google Scholar] [CrossRef]
- Campos-Ramirez, A.; Marquez, M.; Quintanar, L.; Rojas-Ochoa, L.F. Effect of ionic strength on the aggregation kinetics of the amidated amyloid beta peptide Abeta (1-40) in aqueous solutions. Biophys. Chem. 2017, 228, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Jarvet, J.; Barth, A.; Graslund, A.; Danielsson, J. Ionic Strength Modulation of the Free Energy Landscape of Abeta40 Peptide Fibril Formation. J. Am. Chem. Soc. 2016, 138, 6893–6902. [Google Scholar] [CrossRef] [PubMed]
- Marek, P.J.; Patsalo, V.; Green, D.F.; Raleigh, D.P. Ionic strength effects on amyloid formation by amylin are a complicated interplay among Debye screening, ion selectivity, and Hofmeister effects. Biochemistry 2012, 51, 8478–8490. [Google Scholar] [CrossRef]
- Gekko, K.; Timasheff, S.N. Mechanism of protein stabilization by glycerol: Preferential hydration in glycerol-water mixtures. Biochemistry 1981, 20, 4667–4676. [Google Scholar] [CrossRef] [PubMed]
- Vagenende, V.; Yap, M.G.; Trout, B.L. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef] [PubMed]
- Saunders, H.M.; Hughes, V.A.; Cappai, R.; Bottomley, S.P. Conformational behavior and aggregation of ataxin-3 in SDS. PLoS ONE 2013, 8, e69416. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Singh, A.K.; Krishnamoorthy, G.; Swaminathan, R. Thioflavin T displays enhanced fluorescence selectively inside anionic micelles and mammalian cells. J. Fluoresc. 2008, 18, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, J.; Werkmuller, A.; Winter, R. Macromolecular crowding as a suppressor of human IAPP fibril formation and cytotoxicity. PLoS ONE 2013, 8, e69652. [Google Scholar] [CrossRef]
- Nicastro, G.; Masino, L.; Esposito, V.; Menon, R.P.; De Simone, A.; Fraternali, F.; Pastore, A. Josephin domain of ataxin-3 contains two distinct ubiquitin-binding sites. Biopolymers 2009, 91, 1203–1214. [Google Scholar] [CrossRef]
- Nicastro, G.; Menon, R.P.; Masino, L.; Knowles, P.P.; McDonald, N.Q.; Pastore, A. The solution structure of the Josephin domain of ataxin-3: Structural determinants for molecular recognition. Proc. Natl. Acad. Sci. USA 2005, 102, 10493–10498. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.; Berke, S.S.; Cohen, R.E.; Paulson, H.L. Poly-ubiquitin binding by the polyglutamine disease protein ataxin-3 links its normal function to protein surveillance pathways. J. Biol. Chem. 2004, 279, 3605–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicastro, G.; Todi, S.V.; Karaca, E.; Bonvin, A.M.; Paulson, H.L.; Pastore, A. Understanding the role of the Josephin domain in the PolyUb binding and cleavage properties of ataxin-3. PLoS ONE 2010, 5, e12430. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef] [Green Version]
- Masino, L.; Nicastro, G.; Calder, L.; Vendruscolo, M.; Pastore, A. Functional interactions as a survival strategy against abnormal aggregation. FASEB J. 2011, 25, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvey, M.; Tepper, K.; Haupt, C.; Knüpfer, U.; Klement, K.; Meinhardt, J.; Horn, U.; Balbach, J.; Fändrich, M. Phosphate and HEPES buffers potently affect the fibrillation and oligomerization mechanism of Alzheimer’s Aβ peptide. Biochem. Biophys. Res. Commun. 2011, 409, 385–388. [Google Scholar] [CrossRef]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef]
- Groover, S.E.; Adegbuyiro, A.; Fan, C.K.; Hodges, B.L.; Beasley, M.; Taylor, K.; Stonebraker, A.R.; Siriwardhana, C.; Legleiter, J. Macromolecular crowding in solution alters huntingtin interaction and aggregation at interfaces. Colloids Surf. B: Biointerfaces 2021, 206, 111969. [Google Scholar] [CrossRef]
- Uversky, V.N.; Cooper, E.M.; Bower, K.S.; Li, J.; Fink, A.L. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 2002, 515, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Yousaf, M.; Huang, Q.; Yang, Y.; Wang, C. Dual effect of PEG-PE micelle over the oligomerization and fibrillation of human islet amyloid polypeptide. Sci. Rep. 2018, 8, 4463. [Google Scholar] [CrossRef]
- Minton, A.P. The effect of volume occupancy upon the thermodynamic activity of proteins: Some biochemical consequences. Mol. Cell. Biochem. 1983, 55, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.; Pinto, M.F.; Macedo-Ribeiro, S.; Pereira, P.J.B.; Rocha, F.A.; Martins, P.M. Protein crystals as a key for deciphering macromolecular crowding effects on biological reactions. Phys. Chem. Chem. Phys. 2020, 22, 16143–16149. [Google Scholar] [CrossRef] [PubMed]
- Winborn, B.J.; Travis, S.M.; Todi, S.V.; Scaglione, K.M.; Xu, P.; Williams, A.J.; Cohen, R.E.; Peng, J.; Paulson, H.L. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 2008, 283, 26436–26443. [Google Scholar] [CrossRef] [Green Version]
- Pastore, A.; Temussi, P.A. The two faces of Janus: Functional interactions and protein aggregation. Curr. Opin. Struct. Biol. 2012, 22, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Weeks, S.D.; Grasty, K.C.; Hernandez-Cuebas, L.; Loll, P.J. Crystal structures of Lys-63-linked tri-and di-ubiquitin reveal a highly extended chain architecture. Proteins Struct. Funct. Bioinf. 2009, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.; Tsai, H.-F.; Lu, T.-M.; Yang, C.-Y.; Wu, H.-M.; Li, S.-Y. Studies of the CAG repeat in the Machado-Joseph disease gene in Taiwan. Hum. Genet. 1997, 100, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32. 1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [PubMed]
- Gu, W.; Ma, H.; Wang, K.; Jin, M.; Zhou, Y.; Liu, X.; Wang, G.; Shen, Y. The shortest expanded allele of the MJD1 gene in a Chinese MJD kindred with autonomic dysfunction. Eur. Neurol. 2004, 52, 107–111. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Srivastava, A.K.; Roy, S.; Jain, S.; Brahmachari, S.K. Identification of a novel 45 repeat unstable allele associated with a disease phenotype at the MJD1/SCA3 locus. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 133, 124–126. [Google Scholar] [CrossRef]
- Takiyama, Y.; Sakoe, K.; Nakano, I.; Nishizawa, M. Machado-Joseph disease: Cerebellar ataxia and autonomic dysfunction in a patient with the shortest known expanded allele (56 CAG repeat units) of the MJD1 gene. Neurology 1997, 49, 604–606. [Google Scholar] [CrossRef]
- Van Alfen, N.; Sinke, R.J.; Zwarts, M.J.; Gabreëls-Festen, A.; Praamstra, P.; Kremer, B.P.; Horstink, M.W. Intermediate CAG repeat lengths (53, 54) for MJD/SCA3 are associated with an abnormal phenotype. Ann. Neurol. 2001, 49, 805–808. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct Name | Protein Expressed | Original Reference | Addgene ID |
|---|---|---|---|
| Atx3 JD | Ataxin-3 Josephin domain | [10,11] | 184247 |
| Atx3 D1 | Ataxin-3 JD + UIM1-2 | [22] | 184246 |
| Atx3 13Q | Ataxin-3 (13Q) with a glycine residue after the polyQ tract | This work | 184248 |
| Atx3 13Q I77K Q78Q W87K | Ataxin-3 (13Q) with a triple mutation on ubiquitin-binding site 2 | This work | 185908 |
| Atx3 77Q | PolyQ-expanded ataxin-3 (77Q) with an arginine residue after the polyQ tract (UniProt natural variant VAR_013689) | [15] | 184249 |
| Atx3 77Q R388G | PolyQ-expanded ataxin-3 (77Q) with a glycine residue after the polyQ tract | This work | 184251 |
| HsUBB | Homo sapiens linear tri-ubiquitin chain | [23] | 69562 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueiredo, F.; Lopes-Marques, M.; Almeida, B.; Matscheko, N.; Martins, P.M.; Silva, A.; Macedo-Ribeiro, S. A Robust Assay to Monitor Ataxin-3 Amyloid Fibril Assembly. Cells 2022, 11, 1969. https://doi.org/10.3390/cells11121969
Figueiredo F, Lopes-Marques M, Almeida B, Matscheko N, Martins PM, Silva A, Macedo-Ribeiro S. A Robust Assay to Monitor Ataxin-3 Amyloid Fibril Assembly. Cells. 2022; 11(12):1969. https://doi.org/10.3390/cells11121969
Chicago/Turabian StyleFigueiredo, Francisco, Mónica Lopes-Marques, Bruno Almeida, Nena Matscheko, Pedro M. Martins, Alexandra Silva, and Sandra Macedo-Ribeiro. 2022. "A Robust Assay to Monitor Ataxin-3 Amyloid Fibril Assembly" Cells 11, no. 12: 1969. https://doi.org/10.3390/cells11121969
APA StyleFigueiredo, F., Lopes-Marques, M., Almeida, B., Matscheko, N., Martins, P. M., Silva, A., & Macedo-Ribeiro, S. (2022). A Robust Assay to Monitor Ataxin-3 Amyloid Fibril Assembly. Cells, 11(12), 1969. https://doi.org/10.3390/cells11121969